

Abstract
In the hallmark neuritic dystrophy of Alzheimer's disease (AD), autophagic vacuoles containing incompletely digested proteins selectively accumulate in focal axonal swellings, reflecting defects in both axonal transport and autophagy. Here, we investigated the possibility that impaired lysosomal proteolysis could be a basis for both of these defects leading to neuritic dystrophy. In living primary mouse cortical neurons expressing fluorescence-tagged markers, LC3-positive autophagosomes forming in axons rapidly acquired the endo-lysosomal markers Rab7 and LAMP1 and underwent exclusive retrograde movement. Proteolytic clearance of these transported autophagic vacuoles was initiated after fusion with bidirectionally moving lysosomes that increase in number at more proximal axon levels and in the perikaryon. Disrupting lysosomal proteolysis by either inhibiting cathepsins directly or by suppressing lysosomal acidification slowed the axonal transport of autolysosomes, late endosomes, and lysosomes and caused their selective accumulation within dystrophic axonal swellings. Mitochondria and other organelles lacking cathepsins moved normally under these conditions, indicating that the general functioning of the axonal transport system was preserved. Dystrophic swellings induced by lysosomal proteolysis inhibition resembled in composition those in several mouse models of AD and also acquired other AD-like features, including immunopositivity for ubiquitin, amyloid precursor protein, and hyperphosphorylated neurofilament proteins. Restoration of lysosomal proteolysis reversed the affected movements of proteolytic Rab7 vesicles, which in turn essentially cleared autophagic substrates and reversed the axonal dystrophy. These studies identify the AD-associated defects in neuronal lysosomal proteolysis as a possible basis for the selective transport abnormalities and highly characteristic pattern of neuritic dystrophy associated with AD.
Introduction
In Alzheimer's disease (AD), focal swellings develop along the axons and dendrites of neurons throughout affected brain regions. These dystrophic neurites also form a denser spherical plexus intermixed with extracellular deposits of amyloid β, referred to as senile or “neuritic” plaques. Together, neuritic plaques and neurofibrillary tangles within neurons constitute the defining neuropathological hallmarks of AD. The accumulation of organelles within dystrophic neurites has long been believed to reflect a disruption of axonal transport (Morfini et al., 2009). Recent ultrastructural observations have shown that the vast majority of structures present in dystrophic neurites in AD brain are organelles specifically related to the autophagic–lysosomal pathway [autophagic vacuoles (AVs)] (Nixon et al., 2005), suggesting that transport of this class of organelles is selectively disrupted. In addition, the remarkable abundance of undigested protein within accumulated AVs, many of which contain lysosomal proteases, strongly implies a defect in the proteolytic clearance of autophagy substrates by lysosomes. In this regard, comparable levels of protein “storage” in brain are most often observed in certain primary lysosomal storage diseases associated with severe cognitive disabilities and neurodegenerative phenotypes that share key neuropathological features of AD (Bahr and Bendiske, 2002; Ohm et al., 2003; Ballabio and Gieselmann, 2009). Further implicating primary dysfunction of the lysosomal system in AD is evidence for a continuum of abnormalities within endocytic and autophagic pathways connected to lysosomes, some linked directly to genes causing early-onset AD (Nixon et al., 2000; Nixon and Cataldo, 2006; Lee et al., 2010).
Autophagy is the cell's sole mechanism for the degradation of organelles and a major route for the bulk degradation of cytoplasm, especially of long-lived proteins (Dunn, 1994). Its activation by nutrient deprivation or the presence of protein aggregates promotes survival (Shintani and Klionsky, 2004). After the highly regulated sequestration of organelles and cytoplasm within a double-membrane vacuole, the autophagosome (Nakatogawa et al., 2009; Inoue and Klionsky, 2010), the rate-limiting step of substrate digestion occurs when autophagosomes fuse with lysosomes to form autolysosomes (Tanida et al., 2005). Activation of acid hydrolases, including proteases (cathepsins) within the lysosomes/autolysosomes, requires acidification of the intralumenal environment by the vacuolar ATPase (v-ATPase) proton pump (Sun-Wada et al., 2003).
In this study, we investigated the possible relationship between lysosomal dysfunction, neuritic dystrophy, and selective accumulation of AVs and lysosomes. Using live imaging of fluorescently tagged compartmental markers in primary cortical neurons, we characterized the fate of autophagosomes in axons and compared the axonal transport of specific organelles after disrupting lysosomal proteolysis. Our findings demonstrate that lysosomal proteolysis inhibition, but not autophagy activation, another potential basis for promoting axonal dystrophy, slowed the axonal transport of autophagy-related organelles, without affecting transport of other organelles. Moreover, the same class of lysosomal system cargoes accumulated within axonal swellings that also acquired additional biomarkers characteristic of dystrophic neurites in AD brains. These results directly link lysosomal proteolysis dysfunction to two additional key aspects of AD pathogenesis, underscoring the importance of this deficit as a target for AD therapies (Sun et al., 2008; Yang et al., 2011).
Materials and Methods
Antibodies and plasmids.
Mouse monoclonal antibodies to LC3 (NanoTools), phosphorylated neurofilament (NF)-M/H (SMI-31) and unphosphorylated NF-M/H (SMI-32; Sternberger Monoclonal), dynein intermediate chain (DIC; Sigma), β-tubulin (Sigma), p62 (Abnova), ubiquitin (Millipore), microtubule-associated protein 2 (MAP2; Millipore), rat monoclonal antibody to lysosome-associated membrane protein 2 (LAMP2; Hybridoma Bank), rabbit polyclonal antibodies to LC3 (Novus Biologicals), green fluorescent protein (GFP; Abcam), ubiquitin (Dako), glyceraldehyde 3-phosphate dehydrogenase(Santa Cruz Biotechnology), and guinea pig polyclonal antibody to p62 C terminus (Progen Biotechnik) were purchased from commercial sources as listed. Rabbit polyclonal antibodies against NF-L and cathepsin D were generated in our laboratory. Mouse monoclonal anti-amyloid precursor protein (APP) for human and murine APP, referred to as antibody C1/6.1, have been described previously (Mathews et al., 2002), and mouse monoclonal murine-specific APP antibody M3.2 that recognizes APP holoprotein, sAPPα, β-CTF (carboxy-terminal fragment), and Aβ was provided by P. Mathews (Nathan Kline Institute, New York University, New York, NY).
Enhanced GFP (EGFP)–LC3 was a gift from Dr. N. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan); GFP-RFP-LC3 (Kimura et al., 2007) was generously provided by Dr. T. Yoshimori (Osaka University, Osaka, Japan). Yellow fluorescent protein (YFP)– LAMP1 was provided by Dr. J. Swanson (University of Michigan, Ann Arbor, MI). GFP-Rab7 was a gift from Dr. A. Cataldo (McLean Hospital, Belmont, MA). DsRed-LC3 was made as described previously (Boland et al. 2008). pDsRed2-Mito and pAcGFP1-Endo were purchased from Clontech.
Cell culture and transfection.
Embryonic mouse cortical neurons from C57BL/6J mice were cultured as described previously (Boland et al., 2008). Primary cortical neurons were harvested from embryonic day 16–17 pups. Pups were decapitated, and cerebral cortices were removed and dissociated by incubation in hibernating medium (BrainBit) containing papain (10 mg/ml) and DNase for 15 min at 37°C. After digestion, cells were centrifuged at 1000 rpm for 3 min, and the pellet was resuspended in modified Eagle's medium with 10% FBS (HyClone Laboratories) for counting and plating. Cells were plated (0.75 × 105 cells/cm2) on glass-bottom chamber dishes (BD Biocoat) or on circular coverslips (1.5 × 105 cells/cm2), both coated with poly-d-lysine (200 μg/ml) and incubated at 37°C for at least 4 h. After the cells attached, the plating medium was changed to Neurobasal medium containing B27, penicillin/streptomycin, and Glutamax. Half the volume of culture medium was exchanged every 3 d. Penicillin/streptomycin was removed from the medium after the first feeding. Neurons were transfected on 3–4 d in vitro (DIV) using Lipofectamine 2000 (Invitrogen), based on the manufacturer's suggestions with minor modifications. Conditioned medium was replaced with fresh Neurobasal medium containing 1–3 μg of DNA/4 μl of Lipofectamine 2000 in 500 ml of Optimem (Invitrogen). The neurons were transfected for 30 min at 37°C, subsequently washed three times with Neurobasal medium to remove DNA, and resuspended in conditioned medium. Neurons were grown for 24–48 h after transfection before treatments or time-lapse imaging.
For lysosomal inhibition or autophagy activation, cells were treated with leupeptin (20–100 μm; International Peptides), E64 (10 μm; Sigma), pepstatin (20 μm; Sigma), or bafilomycin (10–50 nm; Sigma) or rapamycin (10 nm; Sigma). For leupeptin recovery in GFP-Rab7 neurons, transfected neurons were treated with leupeptin (20 μm) or normal medium (control recovery) for 24 h starting 6 h after transfection and recovered in normal Neurobasal medium for an additional 24 h.
Western blot analysis.
Neurons (5–7 DIV) grown in six-well plastic dishes (BD Biosciences) were washed (three times) in Tris-buffered saline (TBS; pH 7.4) at room temperature and scraped in 200 μl/well of ice-cold lysis buffer (M-PER buffer containing Halt Protease Inhibitor Cocktail (1:100) and 50 mm EDTA; Pierce Chemical). Protein concentration was determined using the Bradford Assay (Pierce Chemical), and samples were standardized to 1 mg/ml using 70% trichloroacetic acid to precipitate cell lysates that were resuspended in equal volumes of lysis buffer. Sample loading buffer (2×; 62.5 mm Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, 0.01% bromophenol blue, 710 mm β-mercaptoethanol) was added to cell lysates at a 1:1 ratio with lysis buffer before heating samples for 5 min at 90°C. Tris-glycine gels (Invitrogen) were loaded with 20 μg/well of protein and separated either on 4–20% gels or 16% gels to separate LC3-I and LC3-II. Separated proteins were transferred onto 0.2 μm nitrocellulose membranes (Protran; Whatman) for 24 h at 10 V. Membranes were rinsed in TBS containing 0.1% Tween 20 (TBS-T) before being blocked at room temperature for 1 h in a 5% nonfat milk/TBS-T solution. Membranes were incubated with a primary antibody overnight at 4°C, washed three times in TBS-T, and incubated for 1 h at room temperature in a secondary antibody solution (3% nonfat milk/TBS-T) containing HRP-conjugated secondary antibodies (Promega). Membranes were washed three times for 10 min in TBS-T before chemiluminescent substrates (ECL; PerkinElmer Life and Analytical Sciences) were applied and membranes were exposed to film. Densitometry of bands representing protein expression was performed using ImageJ software (NIH Image version 1.42l; http://rsb.info.nih.gov/ij). For each immunoblot, the band intensity of each lane was normalized relative to the loading control and compared with the control lane. Subsequently, the percentage change between treatments was calculated based on the normalized values.
Immunocytochemistry.
Neurons were fixed for immunocytochemistry analyses by removing culture medium, washing (three times) in cold TBS, and adding 4% paraformaldehyde/5% sucrose/TBS, pH 7.4, for 15 min at room temperature; washed three times with TBS; and permeabilized for 15 min in 0.02% Triton X-100/TBS. For LC3 and LAMP immunocytochemistry, cells were fixed in −20°C methanol for 5 min, washed three times in TBS, and permeabilized with digitonin/TBS (0.1 mg/ml). Nonspecific antigens were blocked by incubation in 10% NGS/0.02% Triton X-100/TBS blocking solution for 1 h at room temperature. Primary antibodies were diluted in 0.5% BSA/TBS and incubated overnight at 4°C. The next day, cells were washed (three times) in TBS for 10 min before incubation with Alexa-488 or Alexa-546 secondary antibodies (1:1000 in blocking solution; Invitrogen) for 1 h at room temperature. Neurons were washed (three times) in TBS for 10 min before being mounted onto microscope slides with anti-fade Gelmount (Biomeda) and visualized using a Zeiss confocal microscope.
Magic Red, LysoTracker Red, and Bodipy-pepstatin loading.
Neurons plated on 35 mm glass-bottom chamber dishes (BD Biocoat) were incubated with 1 μm of either Bodipy-pepstatin-FL (Invitrogen), Magic Red cathepsin B (Marker Gene Technologies), or LysoTracker Red (Invitrogen) in Neurobasal medium for 30–60 min at 37°C followed by washing with fresh Neurobasal medium (two times). Subsequently, Neurobasal medium was replaced with prewarmed low-fluorescence hibernating medium (Brainbits) to reduce fluorescent background. For live imaging, cultures were placed in a humidified chamber maintained at 37°C and 5% CO2 and mounted on an LSM510 confocal microscope (Zeiss). Z-stacks were acquired using LSM 510 software.
Live imaging.
Thirty-five-millimeter glass bottom dishes (BD Biocoat) containing normal growth medium (NB supplemented with B27) or low fluorescence hibernate medium (Brainbits) supplemented with B27 were mounted in a temperature-controlled stage on the confocal microscope and maintained at 37°C and 5% CO2. The imaging was performed using a Zeiss confocal microscope equipped with the LSM 510 attachment using a 40× oil-immersion lens. Laser lines at 488 nm (GFP-tagged constructs and YFP construct) and 543 nm (DsRed constructs) were used. Time-lapse recordings were acquired by scanning single plane images every 3–5 s for at least 5 min. Our data were collected in the axon (longest process emerging from the cell body) up to ∼400 μm away from the cell body in any area where a substantial length of axon was in focus and appropriate to record movements. Kymographs of time-lapse movies were generated using ImageJ (Multiple Kymograph plugin).
Transport analysis.
Transport parameters of vesicle movement were generated using the ImageJ plugin MtrackJ (www.imagescience.org/meijering/software/mtrackj). For each time-lapse movie, manually tracking individual vesicles using this program generates “points” data (x, y, t coordinates of each point/frame) as well as “tracks” data (duration, minimum/maximum/mean V, etc.) based on an arbitrary reference point representing the most proximal point in the neurite that is closest in proximity to the cell body. After manually tracking vesicles until they were no longer visible, points and tracks data were transferred to Microsoft Excel to calculate net transport direction, relative frequency of velocity, average velocity, and frequency of direction changes. The minimum velocity threshold was set at 0.1 μm/s, where vesicles with a maximum velocity <0.1 μm/s for the entire track were categorized as stationary, whereas vesicles with at least one movement with velocity of at least 0.1 μm/s were categorized as motile (moving). The transport direction was calculated based on the algebraic sign of dx/dt from frame to frame (x1 − x0)/(t1 − t0), where anterograde movement was positive and retrograde movement was negative. Net transport direction was determined by comparing the initial and final (x, y) position of the vesicle and classified as retrograde, anterograde, or stationary (if no movement above 0.1 μm/s occurred despite a net change in position). Motility and net transport direction was expressed as an averaged percentage of total vesicles per axon. The relative frequency of instantaneous velocity was obtained by sorting and binning instantaneous velocity values of each track in intervals of 0.2 μm/s and using the COUNT function of Microsoft Excel for each bin divided by the total number of movements. Frequency of direction changes were calculated using a Microsoft Excel function created by the author to calculate the total number of switches in a given track divided by the total duration of the track. A switch is defined as a different direction of motile behavior from one frame to the next frame. All values represent means with SEM. The Student's t test for statistical analyses (p values) was performed in Microsoft Excel.
Morphometric analysis of GFP-LC3 neurons.
The number of GFP-LC3 neurons containing GFP puncta in the cell body or in neurites, were counted using confocal images of GFP-LC3-transfected neurons, imaged at 40× direct magnification for the various treatment conditions. At least 40 neurons were examined, and values are expressed as a percentage total of counted cells. Based on visual detection of puncta, we categorized neurons as either neurons with vesicular structures or neurons with no puncta (diffuse LC3 only). When punctate LC3 was detectable, the number of cells where puncta was visualized in the cell body, axons, or both was counted. All numerical values are expressed as a mean ± SEM. For counting number of GFP-LC3 vesicles in axons, the number of LC3 vesicles were manually counted and subsequently divided by the total length of the axon as measured using the NIH ImageJ plugin NeuronJ.
The number of GFP-LC3 vesicle swellings per length of neurite and the percentage of GFP-LC3 neurons containing swellings were analyzed by measuring the total length of the GFP-LC3 neurite in the 40× field using the ImageJ plugin NeuronJ and counting the number of visible swellings. At least 30 neurons were examined for each condition.
Electron microscopy.
All animal studies were performed according to the regulations of the Institutional Animal Care and Use Committeeat the Nathan Kline Institute. Transgenic mice expressing either the Swedish mutation of human APP (APPK670M/N671L; referred to as APP) or APPswe/PS1M140L (referred to as PSAPP) at the indicated plaque-bearing ages were anesthetized and transcardially perfused with 4% paraformaldehyde/0.1 m sodium cacodylate buffer, after which brains were dissected and submerged in 2% glutaraldehyde/0.1 m sodium cacodylate buffer overnight at room temperature. Fifty-micrometer coronal sections cut by vibratome were postfixed in 1% osmium tetroxide in Sorensen's phosphate buffer for 1 h at room temperature and dehydrated in a series of increasing concentration of ethyl alcohols (50–100%). For neurons, DIV 4–5 neurons grown on glass coverslips were fixed by removing culture medium, washing once in 37°C supplement-free Neurobasal medium, and adding 4% paraformaldehyde/1% glutaraldehyde/5% sucrose in 0.1 m sodium cacodylate buffer (pH 7.2; Electron Microscopy Sciences) for 24 h at room temperature. After fixation, neurons were washed (three times) in 0.1 m sodium cacodylate buffer, postfixed in 1% osmium tetroxide, and progressively dehydrated in a graded series of ethanols (50–100%) followed by embedding in Epon (Electron Microscopy Sciences) for at least 3 d at room temperature. Tissue was embedded in Epon and cut serially into ultrathin (0.06 μm) sections. Ultrathin sections were stained with uranyl acetate and lead citrate. Ultrathin sections were cut from Epon-embedded blocks and placed on copper grids for structural analysis.
Quantitative analysis of AVs from EM images.
For quantification of organelles in neuritic swellings after leupeptin treatment, 30 randomly selected EM images with accumulated organelles were captured at a final magnification of 10,500×, and the number of each type of AV, mitochondria, and single membrane vesicle was counted using the criteria for AV identification established previously (Nixon et al., 2005). For APP and PSAPP mice, the same procedure was applied, but 50 dystrophic neurites in each of two mice per genotype were used at the indicated ages.
Results
Autophagosome identification in primary cortical neurons
To study the dynamic behavior of autophagosomes in primary cortical neurons (4–5 DIV), we monitored the dynamics of LC3-positive vesicles in the cell bodies and processes after EGFP-LC3 transfection, an established method for autophagosome detection (Kabeya et al., 2000; Bampton et al., 2005; Klionsky et al., 2008). By 24–48 h after transfection, GFP-LC3 distributed uniformly and predominantly as cytosolic LC3-I, and the majority (>90%) of GFP-LC3 neurons exhibited smooth processes and a healthy appearance without evident toxicity (Fig. 1A). The axon of each neuron, the longest projection emerging from the cell body, was clearly identified by its strong immunolabeling for the axonal marker neurofilament (Fig. 1B), but not for MAP2, a marker of dendrites (Fig. 1C). LC3 was also present in MAP2-positive dendrites; however, these dendrites were comparatively much shorter in length than axons (Fig. 1D).
Figure 1.

Identification of axonal AVs using fluorescent LC3. A, GFP-LC3 vesicles (arrows) are detected in the cell body and in neurites. Cell body and boxed area of the neurite are enlarged. B, C, Long projections in GFP-LC3 neurons are immunoreactive for phosphorylated neurofilaments (B) but not MAP2 (C). D, Short dendrites immunoreactive for MAP2 (arrows) also express GFP-LC3. E, F, Live image of DsRed-LC3 (E) or GFP-RFP-LC3 (F) neuron, where LC3 vesicles are present in axons (left) and the cell body (right). G, Time-lapse frames of GFP-LC3 in the cell body demonstrating de novo synthesis (arrows). H, Kymograph of GFP-LC3 in axons and corresponding time-lapse frames illustrating a new LC3 vesicle (arrow) budding off an existing vesicle (arrowhead). All scale bars are as indicated.
Autophagosomes were identified as strongly GFP-LC3-positive, or LC3-immunopositive, vesicular compartments, and nearly all LC3 vesicles fell into a size range of 0.5–1 μm, consistent with the reported size of ∼90% of red fluorescent protein (RFP)-LC3 vesicles in healthy Purkinje neurons, which were smaller than 0.75 μm in diameter (Bains and Heidenreich, 2009). Under normal conditions, GFP-LC3 vesicles distributed throughout the somatodendric region and axons (Fig. 1A, insets). In live imaging analyses, de novo formation of LC3 vesicles in cell bodies could be detected occasionally (Fig. 1G), whereas LC3 vesicles in axons often originated by budding from existing vesicles (Fig. 1H). The dynamic behaviors of these profiles as well as the colocalization of intrinsic membrane proteins in those structures excluded the possibility that they represented artifactual aggregates of overexpressed LC3 (Ciechomska and Tolkovsky, 2007). Of the ∼70% of GFP-LC3 neurons (n = 42) displaying LC3-positive vesicles, LC3 puncta were more frequently found in axons (83 ± 5.29% of neurons contain an average of five puncta per 100 μm of axon length) than in perikarya (33 ± 6.6% of neurons displayed LC3 puncta in their perikarya). Neurons transfected with red LC3 constructs (DsRed-, RFP-) exhibited a similar pattern but displayed more vesicles in cell bodies (Fig. 1E,F), consistent with the relatively greater stability of red fluorescence compared with green in fully acidified autolysosomes after autophagosome–lysosome fusion (Kimura et al., 2007), which is an event that is most active in the cell body (Jahreiss et al., 2008).
Autophagosomes undergo predominantly retrograde transport
To analyze the movement of axonal LC3 vesicles in relation to other organelles, we coexpressed the mitochondrial marker DsRed-Mito with GFP-LC3. The two markers rarely colocalized in axons (Fig. 2A,B) and exhibited very different patterns of transport. LC3 vesicles moved in a processive pattern almost exclusively in the retrograde direction (Fig. 2A). Among over 3500 total individual measurements, there were <50 anterograde movement events. Moreover, the net direction of movement occurred mostly in the retrograde direction (Fig. 2). In contrast to the active retrograde movement of LC3 vesicles, mitochondria were most frequently paused for long periods (≥5 min) and, when moving, traveled in either an anterograde or retrograde direction (Hollenbeck and Saxton, 2005) (Fig. 2B). The relatively uniform distribution of autophagosomes along axons and their strong net movement in the retrograde direction implies that autophagosome formation is an active process in axons.
Figure 2.

LC3 vesicles primarily undergo retrograde movement. A representative live image of an axon coexpressing GFP-LC3 and DsRed-Mito is shown. LC3-positive vesicles (A, arrows) and mitochondria (B, arrowheads) do not colocalize. A 15 min kymograph of LC3 movement (A) demonstrates that LC3 undergoes predominantly retrograde movement with intermittent pausing indicated by vertical segments, whereas mitochondria (B) are frequently stationary (vertical lines), although movement occasionally occurs. All scale bars are as indicated.
Late endosomes and lysosomes undergo bidirectional movement
To investigate the trafficking of lysosomes in axons, we identified compartments that were strongly acidified and contained active cathepsins or LAMP1, a marker of late endosomes and lysosomes. After expression of YFP-tagged LAMP1, LAMP1-positive vesicles were concentrated near the somatodendritic region, although LAMP1 vesicles were also scattered throughout the axon (Fig. 3A,B). YFP-tagged-LAMP1 (Fig. 3A,B) or acidified vesicles identified using LysoTracker Red exhibited a similar distribution pattern (Fig. 3C), as did endogenous lysosomes detected by LAMP1/cathepsin D double immunofluorescence in nontransfected neurons (Fig. 3D).
Figure 3.
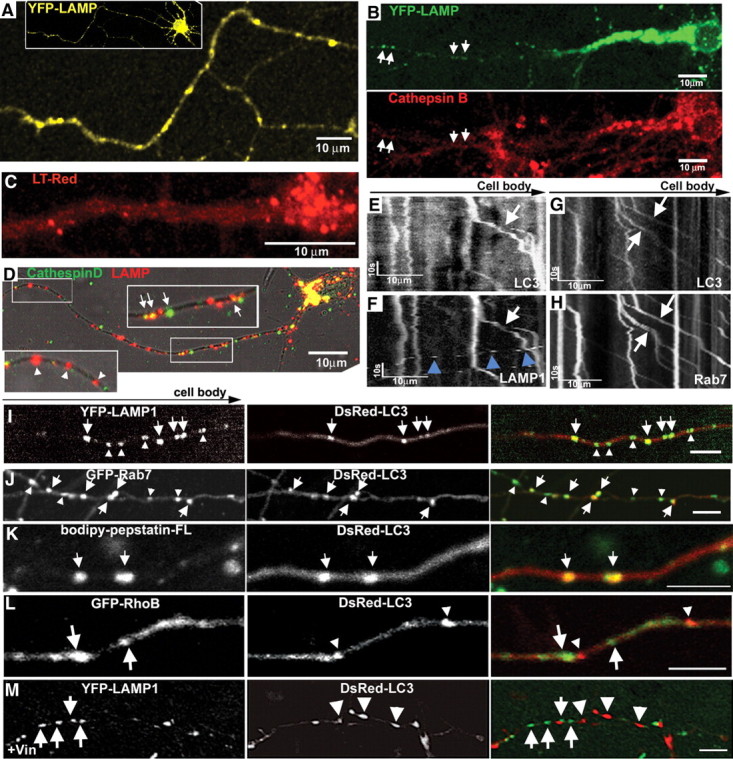
LC3 vesicles undergo maturation during retrograde transport. A, Representative axon image from YFP-LAMP neuron. Inset, Low-magnification image of a neuron. B, YFP-LAMP double labeled with active cathepsin B marker Magic Red. Cell-body LAMP vesicles are enriched with active lysosomal cathepsin B, whereas more distal LAMP vesicles have relatively lower levels of active protease (arrows). C, LysoTracker Red vesicles are concentrated near the somatodendritic area. D, Double labeling of endogenous LAMP and cathepsin D under normal conditions. LAMP vesicles in the perinuclear area contain cathepsins (arrows). The proximal area is enlarged. E, F, Representative kymographs of DsRed-LC3 (E) and YFP-LAMP (F) movement. LC3/LAMP vesicles undergo retrograde movement (arrows), whereas LAMP vesicles not colocalized with LC3 are anterograde (F, arrowheads). G, H, Representative kymographs of DsRed-LC3 (G) and GFP-Rab7 (H) movement. LC3/Rab7 vesicles undergo retrograde movement (arrows). I, Representative axon coexpressing YFP-LAMP and DsRed-LC3. LC3 vesicles are LAMP positive (arrows). J, Representative axon coexpressing DsRed-LC3/GFP-Rab7. Most LC3 vesicles are fused with Rab7 vesicles (arrows), whereas a subset of Rab7 vesicles are not colocalized with LC3 (arrowheads). K, Representative DsRed-LC3 vesicles double labeled with active cathepsin D marker Bodipy-pepstatin-FL. LC3 vesicles contain active cathepsin D. L, Representative axon coexpressing GFP-RhoB and DsRed-LC3. DsRed-LC3 vesicles (arrowheads) do not colocalize with GFP-RhoB (arrows). M, Vinblastine treatment (1 μm, 1 h) prevents colocalization between LC3 and LAMP vesicles. Scale bars: I–M, 5 μm, or as indicated.
To confirm that YFP-LAMP1 vesicles contained active cathepsins, we double labeled YFP-LAMP1 vesicles with a cathepsin B substrate linked to Magic Red, an indicator dye that fluoresces only when the cathepsin B substrate is cleaved. Active cathepsin B was detected in virtually all LAMP1 vesicles in the cell body and within proximal regions of the axon. In contrast, at more distal levels of the axon, the intensity of Magic Red in YFP-LAMP1 vesicles progressively declined (Fig. 3B). This colocalization pattern resembled the distribution pattern of cathepsin D-immunoreactive LAMP vesicles, where <20% of LAMP vesicles in axons were cathepsin D positive (Fig. 3D, inset). LC3 vesicles had matured to mainly autolysosomes, which was confirmed by the detection of Bodipy-pepstatin-FL, a probe for active cathepsin D and a marker for autolysosomes/lysosomes in DsRed-LC3 vesicles (Fig. 3K). Compared with axons, dendrites contained abundant lysosomes, suggesting that lysosomal degradation efficiency in dendrites is more comparable to that in the cell body than in the axon (data not shown).
Compared with the transport of LC3 vesicles, LAMP1 vesicles moved more frequently in the anterograde direction (anterograde LAMP, 37.9 ± 7.4% vs anterograde LC3, <2% total), frequently changed direction (five events per minute, on average), and also exhibited more rapid velocities in either direction (LAMP, 0.5 μm/s vs LC3, 0.3 μm/s) (Fig. 4D). LAMP1 vesicle- and LysoTracker Red-positive vesicles in nontransfected neurons had nearly identical transport behaviors, which were consistent with parameters of LysoTracker-positive vesicles reported in other neuronal culture systems (Lalli and Schiavo, 2002). Thus, the active bidirectional movements of lysosomes in axons are likely to facilitate efficient fusion with LC3 vesicles. As LC3 is delivered by retrograde transport to proximal axons and the perikarya, a higher incidence of lysosomes in the cell body make AV–lysosome fusion (i.e., complete autophagosome clearance) more likely (Kimura et al., 2007, 2008).
Figure 4.

Lysosomal proteolysis inhibition slows LC3 vesicle transport without causing generalized axonal transport defects. A, Representative 5 min kymographs of GFP-LC3 movies after leupeptin (20 μm, 24 h) or bafilomycin A (10 nm, 2 h; compared with controls) (see Fig. 2A). B, Quantification of GFP-LC3 movements after leupeptin (n = 81 vesicles), bafilomycin A (n = 44 vesicles), or controls (n = 96 vesicles). The percentage of moving LC3 vesicles (motility) and LC3 vesicles undergoing net retrograde movement are significantly reduced by leupeptin or bafilomycin treatment. LC3 vesicles also have slower retrograde velocities after leupeptin or bafilomycin, and the frequency of instantaneous retrograde velocities show a depression after leupeptin or bafilomycin. C, DIC immunolabeling on GFP-LC3 vesicles under normal conditions and after treatment with leupeptin. A portion of LC3 vesicles did not colocalize with DIC after leupeptin (arrowheads). D, Quantification of the percentage of LC3 vesicles that colocalized with DIC (per axon) shows reduction after leupeptin (n = 60). E, F, Motility (E) and net direction (F) of DsRed-Mito-positive mitochondria (control, n = 69; leupeptin, n = 78) show that mitochondria movements were not affected by leupeptin (20 μm, 24 h). Values represents means ± SEM. *p < 0.05; **p < 0.01. No Tx, No treatment; BafA, bafilomycin A; Leup or LEU, leupeptin.
Autophagosomes mature to late endosomes and lysosomes during retrograde transport
We confirmed that autophagosomes fuse with lysosomes in axons by finding that over 97% of LC3 vesicles colocalized with GFP-Rab7 (Fig. 3J) or YFP-LAMP (Fig. 3I). We determined whether LC3 vesicles in axons fuse with those containing Rab7 or LAMP after coexpressing DsRed-LC3 with GFP-Rab7 or YFP-LAMP1. Although smaller proportions of LAMP1 vesicles (31 ± 3%, n = 179 vesicles) or Rab7 vesicles (43 ± 5%, n = 159 vesicles) were also LC3 positive, LC3-Rab7/LAMP vesicles retained the transport properties of predominant retrograde transport that characterize GFP-LC3 vesicles (Fig. 3E–H), rather than assuming the predominantly anterograde movement of LAMP-positive vesicles lacking LC3 (Fig. 3F, arrowheads). To eliminate the possibility that combined overexpression of an endosomal protein and LC3 would promote their colocalization, we coexpressed DsRed-LC3 with AcGFP-RhoB, a protein present on endocytic membranes and widely used as an early endosome marker (Rondanino et al., 2007), and found minimal colocalization (Fig. 3L). In further support of this observation, efficient fusion between axonal LC3 and LAMP vesicles required microtubule-dependent transport, as the microtubule-destabilizing agent vinblastine (1 μm, 1 h) abolished colocalization (Fig. 3M). These data combined with the distribution of LAMP/cathepsin-positive vesicles (Fig. 3A–D) indicate that regardless of the proximity to the cell body, autophagosomes in axons acquire Rab7/LAMP rapidly such that LC3 vesicles observed along the axon are almost always observed to be fused with endosomes (to form amphisomes) or with a lysosome. However, given that autophagosomes undergo multiple fusion events with lysosomes for complete degradation (Kimura et al., 2007), full maturation to autolysosomes containing activated lysosomal hydrolases is most likely to occur when AVs are transported to proximal regions of axons or to the cell body where lysosomes are concentrated.
Lysosomal acidification failure or cathepsin inhibition selectively impairs axonal transport of cathepsin-containing vesicles
In light of evidence pointing to significant impairment of both autophagic proteolysis and axonal transport in AD, we investigated the effects on transport dynamics of blocking lysosomal proteolysis by either of two independent mechanisms. The lysosomal inhibitor leupeptin, a cysteine protease inhibitor (20 μm for 24 h), or bafilomycin A, an inhibitor of lysosomal v-ATPase (50 nm for 4 h), effectively blocks lysosomal cysteine protease activity or intralumenal acidification, respectively, resulting in impaired degradation of the autophagic vesicles reflected by the increase in the ratio of LC3-II (autophagosome membrane associated) to LC3-I (cytosolic), an index of autophagy turnover (Klionsky et al., 2008), and higher levels of autophagy substrate p62 (see Fig. 9C). Both treatments significantly inhibited movements of LC3 vesicles based on time-lapse movies of GFP-LC3 taken from axonal regions that did not show obvious changes in axonal diameter or organelle accumulation. Quantification of kymographs from GFP-LC3 time-lapse movies confirmed that the percentage of stationary LC3 vesicles significantly increased from <5% under normal conditions to over 50% after leupeptin and over 75% in bafilomycin (Fig. 4A,B). Furthermore, the mean velocity during retrograde transport was slowed (Fig. 4B) especially after bafilomycin treatment. However, within LC3 vesicle that retained movement after leupeptin, budding events were occasionally observed, suggesting that budding may not be affected by proteolysis inhibition.
Figure 9.
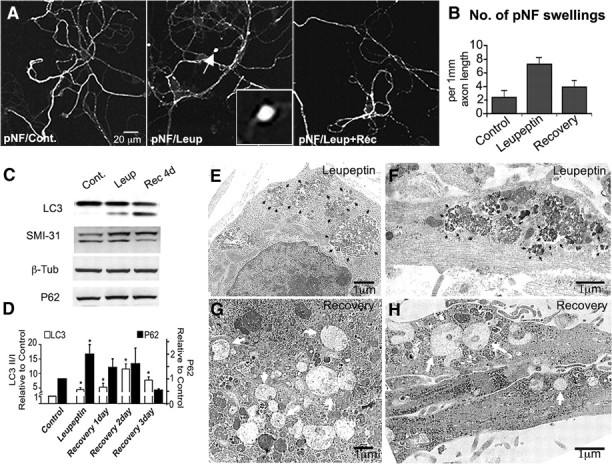
Recovery of lysosomal proteolysis reverses axonal dystrophies and enhances maturation of accumulated AVs. A, Phosphorylated neurofilament immunofluorescence using SMI-31 antibody in neurons after leupeptin (20 μm, 24 h) followed by 24 h in normal medium. B, Quantification of the number of SMI-31-enriched swellings per 103 μm axon length after leupeptin recovery is similar to controls (number of 40× fields quantified: n = 29 control; n = 26 leupeptin; n = 33 recovery). C, Western blots of LC3, phosphorylated neurofilaments (SMI-31), β-tubulin (β-Tub), and P62 in leupeptin (Leup) and leupeptin-recovery (Rec 4d) neurons compared with untreated neurons. Cont., Control. D, Quantification of LC3 and P62 Western blot densitometry for leupeptin-treated and leupeptin-recovered neurons for 1–4 d. n = 4 for each treatment. E, F, Representative ultrastructural images of AVs accumulated in the cell body (E) or neurites (F) after leupeptin treatment (20 μm, 24 h). AVs are filled with undegraded electron-dense material (black arrowheads). G, H, Representative ultrastructural images of AVs in the cell body (G) or neurites (H) after 4 d of recovery in normal medium after 24 h leupeptin treatment (20 μm). Most AVs in the cell body and neurites have a clear lumen (white arrows). Values represents means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To investigate whether or not the higher incidence of stationary LC3 vesicles after proteolysis inhibition may involve detachment from its retrograde transport motor, we probed for the minus-end motor dynein immunoreactivity on LC3 vesicles using a DIC antibody. Consistent with evidence that LC3 retrograde transport is predominantly mediated by dynein (Kimura et al., 2008; Katsumata et al., 2010), we found that 93 ± 3.9% of axonal LC3 vesicles were normally associated with dynein (n = 60 LC3 vesicles for control) (Fig. 4C,D). In contrast, 27 ± 6.3% of LC3 vesicles along axons were not colocalized with DIC after leupeptin treatment (p < 0.05; n = 163 vesicles for leupeptin treatment) (Fig. 4C,D), and this fraction of LC3 vesicles was slightly less than the percentage increase in stationary behavior of LC3 vesicles after leupeptin (∼40%), suggesting that detachment from the retrograde motor may partially account for the increased proportions of stationary LC3 vesicles resulting from cathepsin inhibition.
Because both leupeptin and bafilomycin inhibit lysosomal proteolytic activity and LC3 compartments rapidly acquire lysosomal membrane protein LAMP1, we quantified the movements of LAMP1 and LysoTracker Red vesicles after these treatments. Proteolysis inhibition significantly increased the number of LAMP or LysoTracker Red vesicles that were stationary (Fig. 5A) and reduced the frequency of direction changes and mean anterograde or retrograde velocities of still-moving vesicles (Fig. 5D). Leupeptin or bafilomycin had similar effects on GFP-Rab7-positive late endosomes, another organelle containing cathepsins (Bucci et al., 2000) (Figs. 5B,E). In contrast, lysosomal proteolysis inhibition did not affect the transport behaviors of vesicular organelles lacking cathepsins. AcGFP-RhoB-positive early endosomes displayed a vesicular distribution pattern, and immunofluorescence colabeling with cathepsin D confirmed that these compartments do not contain cathepsin (data not shown). Axonal transport of AcGFP-RhoB vesicles was very active and bidirectional (Fig. 5C) with a bias toward retrograde movement. Unlike protease-containing vesicles, however, GFP-RhoB vesicles in axons were transported normally either before or after treatment with leupeptin or bafilomycin (Fig. 5C,E). Similarly, mitochondrial transport was unaltered by leupeptin treatment (Fig. 4E,F). In contrast to the effects of lysosomal proteolysis inhibition on the transport of LC3, LAMP1-, Rab7-, and LysoTracker-positive vesicles, strong induction of autophagy with rapamycin had no detectable effect on transport (Table 1).
Figure 5.
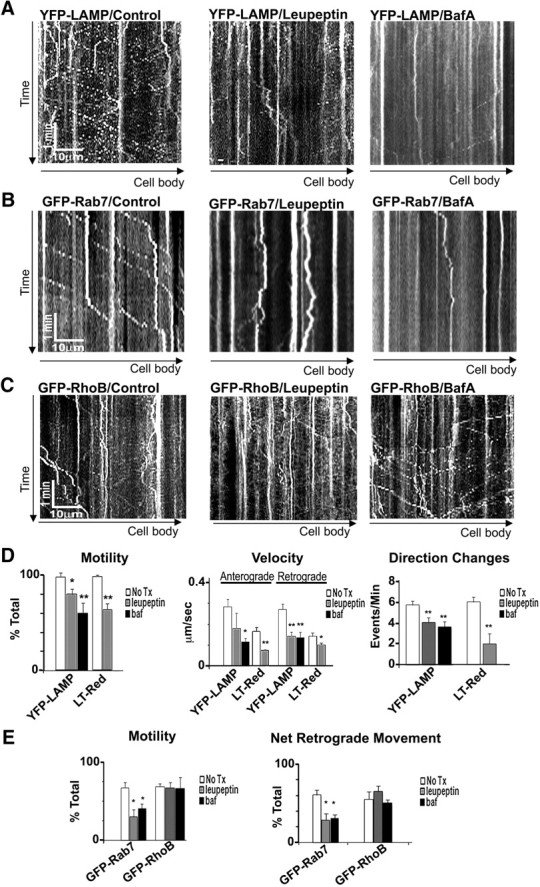
Lysosomal proteolysis inhibition slows endo-lysosome transport. A–C, Representative 5 min kymographs of YFP-LAMP (A), GFP-Rab7 (B), and GFP-RhoB (C) after leupeptin (20 μm, 24 h) or bafilomycin A (10 nm, 2 h) compared with controls. D, Quantification of YPF-LAMP and LysoTracker Red (LT-Red) vesicle motility, net transport direction, and frequency of direction changes (YFP-LAMP: control, n = 82 vesicles; leupeptin, n = 68 vesicles; bafilomycin, n = 44; LT-Red: control, n = 62 vesicles; leupeptin, n = 71 vesicles; see Materials and Methods for details). Both leupeptin and bafilomycin significantly reduce the percentage of moving LAMP/LT vesicles, reduce anterograde and retrograde net movements, and decrease the frequency of direction changes. E, Quantification of GFP-Rab7 and GFP-RhoB movements after leupeptin (Rab7, n = 66 vesicles; RhoB, n = 54 vesicles), bafilomycin A (Rab7, n = 94 vesicles; RhoB, n = 43 vesicles), or controls (Rab7, n = 109 vesicles; RhoB, n = 54). Leupeptin and bafilomycin A reduce the percentage of moving vesicles and the retrograde transport of Rab7 without affecting the retrograde transport of RhoB. Values represents means ± SEM. *p < 0.05; **p < 0.01. No Tx, No treatment; BafA or baf, bafilomycin A.
Table 1.
Time-lapse summary of GFP-LC3, YFP-LAMP, and LysoTracker Red vesicles after rapamycin treatment
| GFP-LC3 |
YFP-LAMP |
LysoTracker Red |
||||
|---|---|---|---|---|---|---|
| Control | RM | Control | RM | Control | RM | |
| Total vesicles | 107 | 64 | 82 | 84 | 62 | 77 |
| Total movements | 3648 | 1074 | 1975 | 3139 | 2039 | 3257 |
| Mean retrograde velocity (μm/s) | 0.32 ± 0.07 | 0.33 ± 0.02 | 0.54 ± 0.05 | 0.47 ± 0.06 | 0.29 ± 0.03 | 0.25 ± 0.02 |
| Net anterograde vesicles | 3 | 2 | 46 | 48 | 31 | 42 |
| Net retrograde vesicles | 95 | 57 | 32 | 31 | 29 | 30 |
| Stationary vesicles | 8 | 5 | 4 | 5 | 2 | 5 |
Axonal transport of GFP-LC3, YFP-LAMP, and LysoTracker Red vesicles under normal conditions (control) or after rapamycin (RM) treatment (10 nm, 5 h). RM does not significantly alter motility, movement direction, or velocity for GFP-LC3, YFP-LAMP, or LysoTracker Red vesicles.
Disrupted lysosomal proteolysis causes AD-like axonal dystrophy
The selective organelle trafficking disruption induced by lysosomal proteolysis inhibition caused LC3 vesicles to accumulate in axons, especially within focal swellings (Fig. 6A,D). The percentage of GFP-LC3 neurons that displayed axonal swelling (Fig. 6F) as well as the frequency of swellings (Fig. 6G) increased approximately threefold after leupeptin, E64 (a more selective cysteine protease inhibitor), pepstatin (an aspartic protease inhibitor), or bafilomycin (Fig. 6F) treatment. The frequency of GFP-LC3 vesicles between axonal swellings was also increased over twofold by leupeptin and bafilomycin (Fig. 6H). Other cathepsin-containing vesicular organelles such as LysoTracker-positive lysosomes and Rab7-positive late endosomes were also sequestered in swellings after leupeptin treatment (Fig. 7A,B), whereas DsRed-positive mitochondria (Fig. 7C) or early endosomes detected by immunocytochemistry for early endosome membrane protein Rab5 (Fig. 7D) were only occasionally observed in the swellings. In live-imaging analyses of GFP-LC3 and DsRed-Mito in relation to axonal swellings, we observed that mitochondria enter and exit the swollen region, whereas LC3 vesicles accumulate (Fig. 7J), establishing that cathepsin inhibition or accumulated AVs in axonal swellings did not block the trafficking of mitochondria. In further support of our observation that global axonal transport is not affected by proteolysis inhibition, additional immunocytochemical analyses showed that nonvesicular axonal constituents such as β-tubulin and NF-L were also not enriched in swellings (Fig. 7E,F). These organelles have been previously observed to accumulate above and below a global block in axonal transport (Griffin et al., 1977; Nagatsu et al., 1978).
Figure 6.

Lysosomal proteolysis inhibition accumulates LC3 vesicles. A, Endogenous LC3-immunoreactive vesicles (arrows) are abundant and accumulated (arrowhead) after treatment with bafilomycin (10 nm, 4 h). The cell body contains relatively few LC3 puncta compared with the axon. B, C, Representative live image of GFP-LC3 neuron before (B) or after (C) treatment with leupeptin (20 μm, 24 h). LC3 vesicles accumulate in swellings (arrows). The enlarged area of axonal swellings is shown below. D, E, Live image of axonal GFP-LC3 vesicles after treatment with leupeptin (20–40 μm, 24 h; D) or bafilomycin A (50 nm, 4 h; E) where vesicles are both dispersed (arrows) and accumulated in a focal swelling (arrowhead). F, Quantification of the percentage of GFP-LC3 neurons with neuritic swellings containing GFP-LC3. G, The number of GFP-LC3 vesicle-enriched swellings per millimeter length of neurite after treatment with various protease inhibitors (see Materials and Methods). H, Quantification of the number of GFP-LC3 vesicles per 100 μm in control or after leupeptin treatment (n = 50). The frequency of GFP-LC3 vesicles increases ∼2.5-fold after leupeptin. Values represents means ± SEM. All scale bars are as indicated. **p < 0.01; ***p < 0.0001. No Tx, No treatment; BafA, bafilomycin A.
Figure 7.
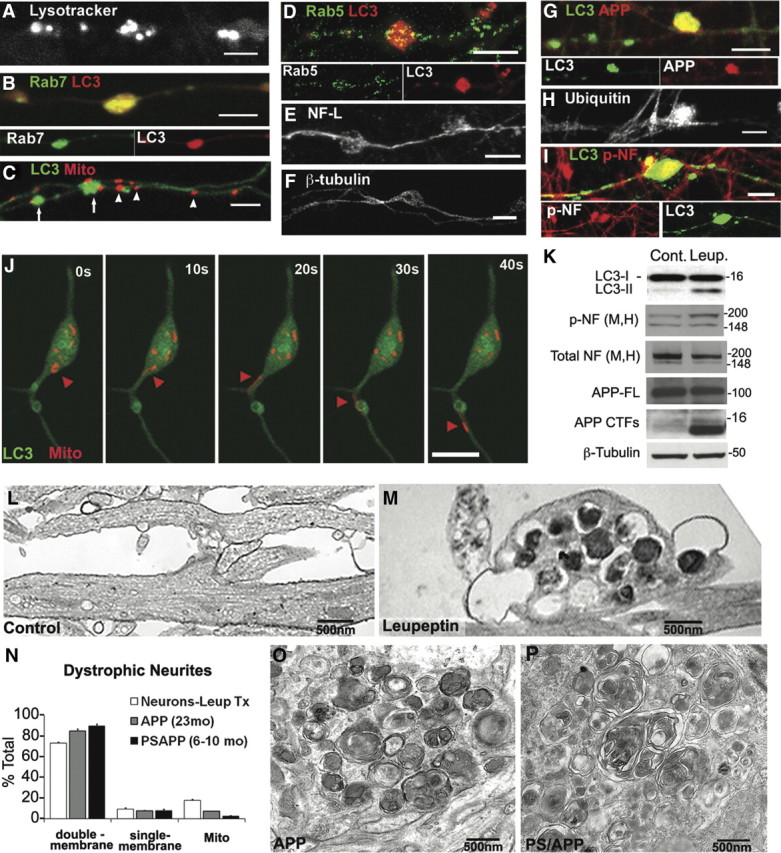
Biochemical and ultrastructural profiles of neurites after lysosomal clearance inhibition resemble AD dystrophic neurites. A–F, Swellings preferentially accumulate with lysosomal (proteolytic) vesicles after treatment with leupeptin (20 μm, 24 h). LysoTracker Red vesicles (A), Rab7 vesicles (B), and LC3 vesicles (C, arrows) are preferentially accumulated, whereas mitochondria (C, arrowheads), Rab5-positive early endosomes (D), neurofilament-light chain (E), or β-tubulin (F) are relatively evenly distributed along the axon. G–I, Swellings accumulate proteins that identify AD-dystrophic neurites including APP-containing autophagic vesicles (G; C1/6.1 antibody for C terminus of APP double labeled with LC3), ubiquitinated proteins (H), and phospho-neurofilaments (I; NF-M/H; double labeled with LC3). Phosphorylated neurofilament accumulation occurs in swollen regions containing accumulated LC3 vesicles. Scale bars, 5 μm. J, Time lapse of swelling with GFP-LC3 and DsRed-Mito cotransfection and treatment with leupeptin (20 μm, 5 h). The cell body is at the top. Although both GFP-LC3 vesicles and DsRed-mitochondria are accumulated, mitochondria occasionally resume transport (red arrowheads), whereas LC3 movement out from the swelling is not observed. K, Western blots with indicated antibodies in leupeptin-treated neurons compared with controls. Leupeptin increases the ratio of LC3-II/I, phospho-neurofilaments (SMI-31), and APP-CTFs, without increasing overall levels of neurofilaments or APP holoprotein (full length). Molecular mass is shown in kilodaltons. L, M, Representative electron micrographs of neurites after leupeptin (20 μm, 24 h; M) compared with controls (L). Leupeptin-induced accumulation of double-membrane, amorphous electron-dense AVs in neurites is shown. N, Quantification of morphometric analysis of organelles in dystrophic neurites from leupeptin-treated neurons (Leup Tx; n = 20) or AD mouse models (APP, n = 25; PSAPP, n = 25). O, P, Representative electron micrographs of dystrophic neurites from APP (O) and PSAPP (P) mouse brain. Organelles within dystrophic neurites are mostly double-membrane AVs. Values represents means ± SEM. Scale bars: A–J, 5 μm.
We also investigated the presence of other antigens related to neuritic dystrophy that are found to be associated with AVs. Vesicles immunolabeled with antibodies to APP and ubiquitin, which are commonly seen in dystrophic neurites in senile plaques in the AD brain (Dickson et al., 1990), were enriched within LC3-positive swellings (Fig. 7G,H). Notably, endogenous APP C-terminal fragments were accumulated in the lysates in leupeptin-treated neurons (Fig. 7K). Phosphorylation of neurofilaments, another marker of dystrophic neurites, was also increased in swellings with LC3 accumulation (Fig. 7I), although the total levels of these proteins (the sum of phosphorylated and nonphosphorylated NF) were not elevated (Fig. 7K), suggesting that neurofilament kinases are locally activated within axonal swellings.
Consistent with the transport analyses, neuritic swellings in leupeptin-treated neurons viewed ultrastructurally were filled predominantly with autophagy-related structures (Boland et al., 2008) (Fig. 7M), including double-membrane AVs, amphisomes, and autolysosomes, and were similar in composition of profiles to the dystrophic neurites seen in PSAPP and APP at ages after plaques have developed [6- to 10-month-old PSAPP (Fig. 7P), 23-month-old APP (Fig. 7O)]. Morphometric analysis of over 50 dystrophic neurites in these mouse models showed that AVs constitute nearly 90% of the organelles in dystrophic neurites, whereas mitochondria and clear lumen vesicles each accounted for fewer than 5% of accumulated organelles (Fig. 7N). Dystrophic neurites from leupeptin-treated neurons exhibited a similar organelle composition to dystrophic neurites in vivo (Fig. 7N) whereas autophagosomes are absent in normal neurites (Fig. 7L).
Reversibility of proteolysis inhibition induced axonal transport deficits and axonal dystrophy
We replaced leupeptin-containing medium with normal medium after 24 h to investigate the possibility of recovery. During 3 d in normal medium, cathepsin B activity was restored to normal levels (Fig. 8A,B). To determine whether axonal transport of protease-containing vesicles could also be recovered, we measured the movements of GFP-Rab7 vesicles after replacing from leupeptin-containing medium with normal medium. At 24 h of recovery, the motility and percentage of net retrograde movements of GFP-Rab7 vesicles were nearly identical to the levels of control-recovery neurons (Fig. 8C,D). The significant increase in stationary behavior and reduction in retrograde movement of GFP-Rab7 vesicles caused by leupeptin (Figs. 5A, 8C,D, independent experiments) was completely reversed, although the mean velocity of retrograde vesicles remained slower after recovery (Table 2).
Figure 8.

Recovery of lysosomal proteolysis restores transport of GFP-Rab7-positive late endosomes and autolysosomes. A, Active cathepsin B (Magic Red) is severely reduced by leupeptin treatment (20 μm, 24 h) but restored after recovery for 3 d in normal medium. B, Quantification of Magic Red cathepsin B loading in A. Values represent mean intensity. C, Quantification of GFP-Rab7 movements after leupeptin (n = 55 vesicles), or 24 h recovery after leupeptin (leupeptin recovery; n = 119) compared with untreated controls (n = 101 vesicles) and recovery controls (normal medium replacement without leupeptin treatment; n = 119 vesicles). D, Leupeptin recovery restores the percentage of moving vesicles and the retrograde transport of GFP-Rab7 vesicles (C, D). Values represents means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Table 2.
Time-lapse summary of GFP-Rab7 vesicles after recovery from leupeptin
| GFP-Rab7 | Control | Control recovery | Leupeptin | Leupeptin recovery |
|---|---|---|---|---|
| Total vesicles (n) | 101 | 87 | 55 | 119 |
| Total movements | 4515 | 3975 | 4166 | 6658 |
| Mean retrograde velocity (μm/s) | 0.44 ± 0.04 | 0.41 ± 0.04 | 0.36 ± 0.05 | 0.38 ± 0.04 |
| Net anterograde vesicles | 16 | 14 | 11 | 21 |
| Net retrograde vesicles | 76 | 61 | 23 | 85 |
| Stationary vesicles | 10 | 12 | 21 | 13 |
Axonal transport of GFP-Rab7 vesicles under normal conditions (control) or after leupeptin treatment (20 μm, 24 h), leupeptin, and 24 h recovery (leupeptin recovery) or control recovery (no leupeptin) data are shown. Leupeptin recovery significantly improves motility and net retrograde movement direction, although the mean retrograde velocity is not yet significantly recovered.
Regarding biochemical markers of axonal dystrophies and autophagic contents, recovery of lysosomal proteases by replacing leupeptin-containing medium with normal medium resulted in a decline in the phosphorylation of neurofilaments (SMI-31) (Fig. 9C) and in levels of p62 to the level seen in untreated neurons (Fig. 9C,D). Using SMI-31, an antibody used to label axonal swellings (defined as focal regions of 7×-fold increased diameter), the threefold increase in the number of SMI-31-positive axon swellings after leupeptin (Fig. 9B) was nearly completely reversed during the recovery period, although LC3-II/I ratios remained elevated (Fig. 9C). Recovery was also evident ultrastructurally. By 4 d of recovery, AVs in both the cell bodies and neurites compared with those in leupeptin-treated neurons (large and compact with electron-dense material) (Fig. 9E,F) had now become clear single-membrane vesicles [i.e., autolysosomes (Boland et al., 2008)] (Fig. 9G,H), indicative of maturation caused by restoration of intralumenal proteolysis. The reversibility of this pathology indicates that dystrophies that are produced after lysosomal failure are not committed to irreversible neurodegeneration.
Discussion
Growing genetic and biochemical evidence indicates that lysosomal proteolysis is markedly impaired in neurons in AD (Boland et al., 2008; Lee et al., 2010; Yang et al., 2011). Here we show that lysosomal proteolysis inhibition, but not strong autophagy induction, selectively disrupts the transport of cathepsin-containing AVs, late endosomes, and lysosomes. The impaired transport of these cargoes resulted in the formation of axonal swellings selectively enriched in this same class of autophagic and endosomal–lysosomal vesicles and are also immunopositive for APP, ubiquitin, and hyperphosphorylated neurofilaments, three well known markers of the hallmark dystrophic neurites seen in AD brain. The strong resemblance of these axonal swellings to the dystrophic neurites in AD brain suggests that lysosomal proteolysis impairment is a driving force behind the highly characteristic axonopathy that develops in AD.
Origin and fate of autophagosomes in axons
In agreement with previous evidence that neuronal autophagy is constitutive (Komatsu et al., 2005; Boland et al., 2008), autophagosome formation of GFP-LC3-positive vesicles was active in both the somatodendritic and axonal regions. The higher frequency of GFP-LC3 vesicles in axons than cell bodies was expected given the longer residence time of AVs in an axon than in the cell body where degradation is facilitated by the higher density of lysosomes. It is also possible that autophagosomes form at higher rates at axon terminals, as proposed previously (Kaasinen et al., 2008).
The extreme polar shapes of neurons, which prolong the maturation of LC3 vesicles, facilitated our investigations on the fates of AVs. Based on quantitative data on nearly 300 GFP-LC3 vesicles, we observed that the overwhelming majority of LC3-positive vesicles in axons move in the retrograde direction (Fig. 2). After they appear, LC3 vesicles rapidly fuse with late endosomes (Rab7/LAMP2 vesicles) abundant all along the axons to form amphisomes or with a lysosome to form an autolysosome: very few LC3 vesicles were seen that were not fused with Rab7 or LAMP. Interestingly, the extensive colocalization of LC3 with Rab7 supports previous findings that fusion between AVs and endosomes is active (Morvan et al., 2009) and may be necessary for autophagy completion (Filimonenko et al., 2007; Fader et al., 2008; Razi et al., 2009).
Although lysosomes are often assumed to be restricted to the perikarya, lysosomes have been identified in axons of cells in pituitary axons in human brain (Cataldo and Broadwell, 1984), hippocampal pyramidal neurons (McGuinness et al., 2007), and dorsal root ganglia (Perrot and Julien, 2009). We observed vesicles containing LAMP1, active cathepsins (B and D), and LysoTracker in axons and established that AVs fuse with proteolytically active lysosomes in axons, although fusion is more active near the cell body where the lysosomes are most concentrated. This mechanism of progressive maturation of AVs as they approach the cell body was proposed in previous studies that provided strong support for this proposal (Hollenbeck, 1993; Overly and Hollenbeck, 1996). Although LC3 has been shown to bind to the anterograde motor kinesin via FYCO (FYVE and coiled-coil domain-containing protein) (Pankiv et al., 2010), the extensive acquisition of Rab7/LAMP on LC3-positive vesicles likely converts them into mainly retrogradely moving AVs, thus favoring transport to the cell body for complete degradation.
Cathepsin inhibition selectively disrupts the axonal transport of cathepsin-containing organelles leading to AD-like axonal dystrophy
Proteolysis inhibition, by either of two independent mechanisms, selectively slowed the anterograde and retrograde transport of LAMP- or LysoTracker-positive vesicles and rendered a greater number of them stationary for relatively long periods. Lysosomal protease inhibition also slowed or halted the axonal transport of LC3-positive AVs, nearly all of which had fused with cathepsin-containing LAMP1/Rab7-positive late endosomes. Significantly, the negative effects of protease inhibition on Rab7 vesicles (which overlap extensively with LC3 and LAMP vesicles) (Fig. 3) were completely reversible after removal of the proteolysis block. In contrast, the movement of RhoB vesicles or mitochondria that lack cathepsins was unaffected. The similar effects on transport of cathepsin inhibitors or bafilomycin A, which inhibits lysosome/autolysosome acidification, suggest that altered motility of AVs/endo-lysosomes involves a change in the intralumenal environment of lysosomes that, in turn, alters the transport properties of these vesicles.
The selective transport failure of AV/endo-lysosomal compartments and axonal accumulation within dystrophic axonal swellings in response to proteolysis inhibition provides a possible molecular basis for axonal dystrophy in AD. This selective organelle accumulation is highly characteristic of AD and distinct from dystrophies that result from a global disruption of microtubule-based axonal transport, where all varieties of vesicular and nonvesicular cargoes are impeded and accumulate above or below the site of blockade (Griffin et al., 1977). Moreover, global transport failure is often seen in the context of neurodegeneration, and distinguishing primary effects on transport from those secondary to neurodegenerative events is usually difficult. In contrast, we showed that transport impairments and axonal dystrophy are reversible after withdrawal of the proteolytic block, consistent with evidence that a given dystrophic neurite may persist in AD brain for relatively long periods of time (Meyer-Luehmann et al., 2008; Adalbert et al., 2009) and is potentially reversible (Garcia-Alloza et al., 2007). Dystrophic axons with a more selective content of fibrillar and vesicular components have been seen in diseases associated with early axonal transport deficits and axonal dystrophy (Williamson and Cleveland, 1999; Morfini et al., 2009; Hadano et al., 2010); however, the accumulated organelles in these conditions are distinguishable from those in the dystrophic neurites in AD, which are predominantly composed of autophagy-related structures (Nixon et al., 2005) (Fig. 7).
Several of our observations may be relevant to the molecular mechanism by which lysosomal proteolysis inhibition impedes transport of specific organelles. Cathepsin inhibition significantly lowered the proportion of LC3 vesicles containing dynein, which roughly matched the proportion of LC3-positive vesicles that became stationary after protease inhibition, suggesting that detachment from dynein motors from these compartments may contribute to their defective transport, although we cannot exclude the possibility that protease inhibition stabilizes a population of AVs that are normally detached from dynein. Dynein malfunction disrupts axonal transport ultimately causing neurodegeneration (LaMonte et al., 2002; Hafezparast et al., 2003), although structures accumulating in axonal swellings are different from those observed in AD models. However, organelle-specific deficits in axonal transport, as we propose in AD, have been seen in several other diseases where, interestingly, the pathogenic protein is a constituent of the affected organelle (Kasher et al., 2009; Lim and Kraut, 2009). Notably, the ablation of snapin, a Rab7-binding protein that interacts with dynein, was shown to selectively impede Rab7-positive vesicle transport and disrupt autophagic and lysosomal turnover (Cai et al., 2010). The dynein mechanism suggested by our preliminary observations represents only one of various possible mechanisms, which may even include alterations of a different motor than dynein or involve more than one motor since, for example, protease inhibition impaired the anterograde movement of LAMP vesicles. An excess of APP or specific APP metabolites may also alter vesicular transport (Stokin et al., 2008; Muresan et al., 2009). APP metabolites including, notably, the trans-membrane C-terminal fragments, which accumulate after leupeptin treatment (Siman et al., 1993; Boland et al., 2010), are known to disturb endosomal trafficking and size (Jiang et al. 2010). Consistent with these findings, the increase in APP-CTF levels after leupeptin in our experiments was robust and may represent an additional pathogenic consequence of lysosomal dysfunction relevant to AD (Jiang et al., 2010).
We did not observe significant changes in AV/lysosome transport or axonal morphology after strong autophagy induction by rapamycin. Nerve growth factor deprivation in PC12 cells (Yang et al., 2008) and excitotoxicity induce axonal swellings and activate autophagy, but these changes are associated with neurodegeneration (Batistatou and Greene, 1991; Yue et al., 2002; Wang et al., 2006). In our study, however, stimulating autophagy by TOR (target of rapamycin) inhibition (rapamycin) did not induce axonal swellings or alter the dynamics of AV or lysosome transport, consistent with similar observations in non-neuronal cell types using similar treatments (Jahreiss et al., 2008; Katsumata et al., 2010). We cannot, however, exclude the possibility that autophagy overactivation via non-TOR-mediated pathways as occurs in some neuronal injuries differentially affects axonal transport.
Although the detailed mechanism by which changes in the proteolytic activity within a vesicle regulates its transport requires further investigation, our data strongly support a novel hypothesis linking deficient endo-lysosomal proteolysis to the development of a neuritic dystrophy resembling the highly characteristic pattern seen in AD. Our additional studies indicate that the mechanism involves a selective disruption of axonal transport of the same types of vesicular organelles that accumulate in dystrophic neurites in AD. The results provide insight into the mechanism by which mutations of Presenilin 1, which cause lysosomal proteolysis inhibition, dramatically accelerate neuritic dystrophy and associated pathologies in familial AD (Cataldo et al., 2004; Lee et al., 2010). Finally, our observation that the dystrophy may be reversed by relieving the inhibition of lysosomal proteolysis is consistent with the significant therapeutic effects of restoring a more normal level of lysosomal proteolysis in mouse models of AD (Yang et al., 2011) and underscores the promise of this approach for developing effective AD therapies.
Footnotes
This work was supported by National Institutes of Health Grant P01AG017617 (R.A.N.). We thank Asok Kumar for expert technical advice and assistance with electron microscopy and Takahiro Sasaki for assistance with live imaging. We also thank Ana-Maria Cuervo (Albert Einstein College of Medicine, Bronx, NY) and members of the Nixon laboratory for helpful discussions.
The authors declare no competing financial interests.
References
- Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M, Misgeld T, Coleman MP. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132:402–416. doi: 10.1093/brain/awn312. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Bendiske J. The neuropathogenic contributions of lysosomal dysfunction. J Neurochem. 2002;83:481–489. doi: 10.1046/j.1471-4159.2002.01192.x. [DOI] [PubMed] [Google Scholar]
- Bains M, Heidenreich KA. Live-cell imaging of autophagy induction and autophagosome-lysosome fusion in primary cultured neurons. Methods Enzymol. 2009;453:145–158. doi: 10.1016/S0076-6879(08)04007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Bampton ETW, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dymanics of autophagy visualizes in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Batistatou A, Greene L. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol. 1991;15:461–471. doi: 10.1083/jcb.115.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Smith DA, Mooney D, Jung SS, Walsh DM, Platt FM. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem. 2010;285:37415–37426. doi: 10.1074/jbc.M110.186411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Lu L, Tian JH, Zhu Y, Qiao H, Seng ZH. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010;68:73–86. doi: 10.1016/j.neuron.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Broadwell RD. Cytochemical staining of the endoplasmic reticulum and glycogen in mouse anterior pituitary cells. J Histochem Cytochem. 1984;32:1285–1294. doi: 10.1177/32.12.6094657. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M, Mathews PM, Nixon RA. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol. 2004;63:821–830. doi: 10.1093/jnen/63.8.821. [DOI] [PubMed] [Google Scholar]
- Ciechomska IA, Tolkovsky AM. Non-autophagic GFP-LC3 puncta induced by saponin and other detergents. Autophagy. 2007;3:586–590. doi: 10.4161/auto.4843. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Wertkin A, Mattiace LA, Fier E, Kress Y, Davies P, Yen SH. Ubiquitin immunoelectron microscopy of dystrophic neurites in cerebellar senile plaques of Alzheimer's disease. Acta Neuropathol. 1990;79:486–493. doi: 10.1007/BF00296107. [DOI] [PubMed] [Google Scholar]
- Dunn WA., Jr Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994;4:139–143. doi: 10.1016/0962-8924(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem. 2007;102:1095–1104. doi: 10.1111/j.1471-4159.2007.04613.x. [DOI] [PubMed] [Google Scholar]
- Griffin JW, Price DL, Engel WK, Drachman DB. The pathogenesis of reactive axonal swellings: role of axonal transport. J Neuropathol Exp Neurol. 1977;36:214–227. doi: 10.1097/00005072-197703000-00002. [DOI] [PubMed] [Google Scholar]
- Hadano S, Otomo A, Kunita R, Suzuki-Utsunomiya K, Akatsuka A, Koike M, Aoki M, Uchiyama Y, Itoyama Y, Ikeda JE. Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS One. 2010;5:e9805. doi: 10.1371/journal.pone.0009805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Hollenbeck P. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:305–315. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Klionsky DJ. Regulation of macroautophagy in Saccharomyces cerevisiae. Semin Cell Dev Biol. 2010;21:664–670. doi: 10.1016/j.semcdb.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer's-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasinen SK, Harvey L, Reynolds AJ, Hendry IA. Autophagy generates retrogradely transported organelles: a hypothesis. Int J Dev Neurosci. 2008;26:625–634. doi: 10.1016/j.ijdevneu.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasher PR, De Vos KJ, Wharton SB, Manser C, Bennett EJ, Bingley M, Wood JD, Milner R, McDermott CJ, Miller CCJ, Shaw PJ, Grierson AJ. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J Neurochem. 2009;110:34–44. doi: 10.1111/j.1471-4159.2009.06104.x. [DOI] [PubMed] [Google Scholar]
- Katsumata K, Nishiyama J, Inoue T, Mizushima N, Takeda J, Yuzaki M. Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy. 2010;6:378–385. doi: 10.4161/auto.6.3.11262. [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli G, Schiavo G. Analysis of retrograde transport in motor neurons reveals common endocytic carriers for tetanus toxin and neurotrophin receptor p75NTR. J Cell Biol. 2002;156:233–239. doi: 10.1083/jcb.200106142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur ELF. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require Presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim A, Kraut R. The Drosophila BEACH family protein, Blue Cheese, links lysosomal axon transport with motor neuron degeneration. J Neurosci. 2009;29:951–963. doi: 10.1523/JNEUROSCI.2582-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews PM, Jiang Y, Schmidt SD, Grbovic OM, Mercken M, Nixon RA. Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of calpains enhances endosomal generation of beta-cleaved C-terminal APP fragments. J Biol Chem. 2002;277:36415–36424. doi: 10.1074/jbc.M205208200. [DOI] [PubMed] [Google Scholar]
- McGuinness L, Bardo SJ, Emptage NJ. The lysosome or lysosome-related organelle may serve as a Ca2+ store in the boutons of hippocampal pyramidal cells. Neuropharmacology. 2007;52:126–135. doi: 10.1016/j.neuropharm.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-[bgr] plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvan J, Kochl R, Watson R, Collinson LM, Jefferies HB, Tooze SA. In vitro reconstitution of fusion between immature autophagosomes and endosomes. Autophagy. 2009;5:676–689. doi: 10.4161/auto.5.5.8378. [DOI] [PubMed] [Google Scholar]
- Muresan V, Varvel NH, Lamb BT, Muresan Z. The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J Neurosci. 2009;29:3565–3578. doi: 10.1523/JNEUROSCI.2558-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu I, Karasawa N, Shimizu N. Early changes in the structure of rat sciatic nerves by ligation. Arch Histol Jpn. 1978;41:439–451. doi: 10.1679/aohc1950.41.439. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- Nishiyama J, Matsuda K, Kakegawa W, Yamada N, Motohashi J, Mizushima N, Yuzaki M. Reevaluation of neurodegeneration in lurcher mice: constitutive ion fluxes cause cell death with, not by, autophagy. J Neurosci. 2010;30:2177–2187. doi: 10.1523/JNEUROSCI.6030-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon R, Cataldo A. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimer's Disease. 2006;9:277–289. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM, Mathews PM. The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res. 2000;25:1161–1172. doi: 10.1023/a:1007675508413. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Ohm TG, Treiber-Held S, Distl R, Glöckner F, Schönheit B, Tamanai M, Meske V. Cholesterol and tau protein–findings in Alzheimer's and Niemann Pick C's disease. Pharmacopsychiatry. 2003;36(Suppl 2):S120–S126. doi: 10.1055/s-2003-43060. [DOI] [PubMed] [Google Scholar]
- Overly CC, Hollenbeck PJ. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J Neurosci. 1996;16:6056–6064. doi: 10.1523/JNEUROSCI.16-19-06056.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot R, Julien JP. Real-time imaging reveals defects of fast axonal transport induced by disorganization of intermediate filaments. FASEB J. 2009;23:3213–3225. doi: 10.1096/fj.09-129585. [DOI] [PubMed] [Google Scholar]
- Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009;185:305–321. doi: 10.1083/jcb.200810098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondanino C, Rojas R, Ruiz WG, Wang E, Hughey RP, Dunn KW, Apodaca G. RhoB-dependent modulation of postendocytic traffic in polarized Madin-Darby canine kidney cells. Traffic. 2007;8:932–949. doi: 10.1111/j.1600-0854.2007.00575.x. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Mistretta S, Durkin JT, Savage MJ, Loh T, Trusko S, Scott RW. Processing of the beta-amyloid precursor. Multiple proteases generate and degrade potentially amyloidogenic fragments. J Biol Chem. 1993;268:16602–16609. [PubMed] [Google Scholar]
- Stokin GB, Almenar-Queralt A, Gunawardena S, Rodrigues EM, Falzone T, Kim J, Lillo C, Mount SL, Roberts EA, McGowan E, Williams DS, Goldstein LS. Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum Mol Genet. 2008;17:3474–3486. doi: 10.1093/hmg/ddn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller-Steiner S, Devidze N, Wang X, Grubb A, Gan L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun-Wada GH, Wada Y, Futai M. Lysosome and lysosome-related organelles responsible for specialized functions in higher organisms, with special emphasis on vacuolar-type proton ATPase. Cell Struct Funct. 2003;28:455–463. doi: 10.1247/csf.28.455. [DOI] [PubMed] [Google Scholar]
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- Walkley SU. Cellular pathology of lysosomal storage disorders. Brain pathology. 1998;8:175–193. doi: 10.1111/j.1750-3639.1998.tb00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Xu K, Koike T, Zheng X. Transport of autophagosomes in neurites of PC12 cells during serum deprivation. Autophagy. 2008;4:243–245. doi: 10.4161/auto.5431. [DOI] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–933. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]