


Abstract
With the rapid advances of various high-throughput technologies, generation of ‘-omics’ data is commonplace in almost every biomedical field. Effective data management and analytical approaches are essential to fully decipher the biological knowledge contained in the tremendous amount of experimental data. Meta-analysis, a set of statistical tools for combining multiple studies of a related hypothesis, has become popular in genomic research. Here, we perform a systematic search from PubMed and manual collection to obtain 620 genomic meta-analysis papers, of which 333 microarray meta-analysis papers are summarized as the basis of this paper and the other 249 GWAS meta-analysis papers are discussed in the next companion paper. The review in the present paper focuses on various biological purposes of microarray meta-analysis, databases and software and related statistical procedures. Statistical considerations of such an analysis are further scrutinized and illustrated by a case study. Finally, several open questions are listed and discussed.
INTRODUCTION
With the rapid advances in biological high-throughput technology, generation of various kinds of genomic data is commonplace in almost every biomedical field. Effective data management and analytical approaches are essential to fully decipher the biological knowledge contained in the tremendous amount of experimental data. In the past decade, the accumulation of transcriptomic data mainly from microarray experiments was particularly significant, and resulted in several large public data depositories (such as Gene Expression Omnibus and ArrayExpress). Similarly, genome-wide association studies (GWAS) are another example: thousands of GWAS have been performed world-wide and results and/or raw data for many are publicly available (see companion review paper for GWAS meta-analysis). It is common that multiple transcriptomic studies or GWAS are available for the same or related disease condition and each study has relatively small sample size with limited statistical power. Combining information from these studies to increase sensitivity and validate conclusions is a natural step. Such genomic information integration is akin to the classical meta-analysis in statistics where results of multiple studies of a similar research hypothesis are combined for a conclusive finding.
A major distinction in the genome-wide setting compared with the classical one is that we are typically analyzing data on thousands of genes. We term genomic information integration in which we combine results from multiple transcriptomic studies or GWAS as ‘horizontal genomic meta-analysis’ (Figure 1A). Figure 1B demonstrates another type of multi-dimensional integrative analysis that combines multiple sources of -omics information on a given cohort of patients. The multi-dimensional -omics data usually include, but are not limited to, transcriptome profile, genotypes, DNA copy number variation, methylation, microRNA, proteome and phenome. Examples of publicly available databases that include this type of information include the Cancer Genome Atlas (TCGA; cancergenome.nih.gov) and the Therapeutically Applicable Research to Generate Effective Treatments (TARGET; target.cancer.gov). Integration of this type of data is called ‘vertical genomic integrative analysis’. In this article, we will focus on horizontal genomic meta-analysis through extensive search of PubMed database and manual literature referencing. Of the 582 papers related to genomic meta-analysis, we will concentrate on 333 microarray meta-analysis papers in this article. The other 249 GWAS meta-analysis papers are discussed in the companion paper. The goal of this article is 3-fold. First, we aim to provide a summary of the methodologies used in the microarray meta-analysis papers. In this light, the article can be viewed as a ‘meta’–meta-analysis paper. The second goal of the article is to provide a critique of the methodologies used in the literature. Finally, we outline some further issues in the field that need more attention.
Figure 1.

Types of information integration of genomic studies. (A) Horizontal genomic meta-analysis that combines different sample cohorts for the same molecular event. (B) Vertical genomic integrative analysis that combines different molecular events usually in the same sample cohort.
The article is structured as follows. ‘Comprehensive review’ section summarizes details of the comprehensive literature review. In ‘Purposes of Microarray Meta-Analysis’ and ‘Databases and Software’ sections, we discuss various purposes of microarray meta-analysis and related software and database resources. In ‘Meta-Analysis for DE Gene Detection’ section, we discuss statistical considerations behind meta-analysis for differentially expressed (DE) gene detection, an analysis commonly encountered in microarray meta-analysis. ‘Open questions’ section describes a list of open questions and further discussions. ‘Conclusion and discussion’ section provides final conclusions.
COMPREHENSIVE REVIEW
Papers under review came from two sources: PubMed search and manual collection. 745 papers were obtained from searching the PubMed database by keywords on 29 December 2010 (see legend of Figure 2), and 102 papers were identified from cross-referencing accumulated in our research activities. After removing duplicates and irrelevant papers, a total of 620 distinct papers were formally reviewed and summarized. Among them, 22 papers belong to the vertical genomic integrative analysis category and 598 papers were horizontal genomic meta-analysis. Of the 598 papers, 333 papers were related to microarray meta-analysis, 256 papers were in the GWAS meta-analysis category and 9 papers were meta-analysis of other categories (e.g. copy-number variation or genome-wide linkage scan). The flow diagram is shown in Figure 2.
Figure 2.

Flow chart of paper collection and categorization. Papers were collected from PubMed search and manual collection. After removing duplicates and irrelevant papers, 620 papers were formally reviewed. Commands used in PubMed search: a(“meta-analysis”[Title/Abstract]) AND ((“microarray”[Title/Abstract]) OR (“expression profiles”[Title/Abstract]) OR (“expression profile”[Title/Abstract]) OR (“gene expression”[Title/Abstract]) OR (“Affymetrix”[Title/Abstract]) OR (“Illumina”[Title/Abstract])); b(“meta-analysis”[Title/Abstract]) AND (“genome-wide association”[Title/Abstract]); c(“meta-analysis”[Title/Abstract]) AND ((“CGH”[Title/Abstract]) OR (“CNV”[Title/Abstract]) OR (“copy number”[Title/Abstract])); d(“meta-analysis”[Title/Abstract]) AND ((“miRNAs”[Title/Abstract]) OR (“miRNA”[Title/Abstract]) OR (“microRNAs”[Title/Abstract])).
Figure 3 illustrates a summary of our microarray meta-analysis review. Detailed information of the paper list and categorization to generate Figure 3 is available in the Supplementary Data. Of the 333 microarray meta-analysis papers, 7 (2%) were descriptive review without quantitative information integration, 42 (13%) were meta-analysis on one or several targeted genes (not at genome-wide scale) and the remaining 284 (85%) represented genome-wide meta-analysis on a global basis (Figure 3A). In Figure 3B, the 333 papers were categorized into review papers (11 papers; 3%), biological applications (201 papers; 60%), novel methodologies (83 papers; 25%) and database/software (38 papers; 12%). For different purposes of meta-analysis shown in Figure 3C, the majority of papers targeted on DE gene or pathway detection (218 papers; 66%). Other purposes include ‘network or co-expression analysis’ (32 papers; 10%), ‘classification analysis’ (25 papers; 8%), ‘reproducibility or bias analysis’ (19 papers; 6%) and ‘others’ (34 papers; 10%). We will further survey these various meta-analysis purposes later in ‘Purposes of microarray meta-analysis’ section. Since two-thirds (218 papers; 66%) of the microarray meta-analysis papers were related to DE gene or pathway detection which conceptually were extensions from traditional meta-analysis, we scrutinized this category and summarized four types of statistical methodologies used (Figure 3D). Of the 191 papers that could be clearly categorized, 81 papers (42%) used meta-analysis methods that combine P-values from individual studies, while 41 papers (22%) combined effect sizes, 18 papers (9%) combined ranks and 51 papers (27%) directly merged data after proper normalization. ‘Types of meta-analysis methods’ section will go over these four types of statistical methodologies in more detail.
Figure 3.

Summary of microarray meta-analysis review. (A) Types of information integration; (B) Types of paper; (C) Purposes of meta-analysis; and (D) Types of statistical methods for DE gene detection.
PURPOSES OF MICROARRAY META-ANALYSIS
When the term ‘microarray meta-analysis’ is used, it usually means meta-analysis for DE gene (or marker) detection. Although two-thirds of identified publications (Figure 3C) were of this type, microarray studies have also been combined for many other biological purposes, as described below.
DE gene detection (218 papers)
DE gene detection is a commonly used downstream analysis in microarray that identifies genes differentially expressed across two or more conditions with statistical significance and/or biological significance (e.g. fold change). In the simple case that we are looking at one gene, this type of analysis is usually performed using a two-sample t-test or a Wilcoxon rank-sum test. However, when this analysis is performed genome-wide, a major issue becomes the fact that there can be many spurious associations that are expected by chance. To counteract this problem, some type of multiple comparisons adjustment is usually done; a popular one is to use the q-value (1). The task is usually a first step to identify gene targets for understanding genetic mechanisms under a disease or for guiding the search of treatment targets. From Figure 3C, detection of DE genes covers two-thirds of papers (218 papers) in the microarray meta-analysis literature. Most existing methods or applications are for two-class comparison (e.g. identify DE genes comparing cases versus controls). Other types of outcome variables (e.g. multi-class, continuous, censored survival or time series) have also been considered in microarray meta-analysis (2). Details of these methods will be further described in ‘Types of meta-analysis Methods’ section.
Pathway analysis
Pathway analysis (a.k.a. gene set analysis) is a statistical tool to infer correlation of differential expression evidence in the data with pathway knowledge from established databases (3,4). The idea behind pathway analysis is to determine if there is enrichment in the detected DE genes based on an a priori defined biological category. Such a category might come from one or multiple databases such as Gene Ontology (GO; www.geneontology.org), the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/), Biocarta Pathways (http://www.biocarta.com/) and the comprehensive Molecular Signatures Database (MSigDB; http://www.broadinstitute.org/gsea/msigdb/). For the majority of recent microarray meta-analysis applications, pathway analysis has been a standard follow-up to identify pathways associated with detected DE genes [e.g. (5) and many others]. The result provides more insightful biological interpretation and it has been reported that pathway analysis results are usually more consistent and reproducible across studies than DE gene detection (6). Shen and Tseng (7) developed a systematic framework of Meta-Analysis for Pathway Enrichment (MAPE) by combining information at gene level, at pathway level and a hybrid of the two.
Network and co-expression analysis (32 papers)
Co-expression analysis and network analysis of microarray data are used to investigate potential transcriptional co-regulation and gene interactions. Network analyses typically work with the gene–gene co-expression matrix, which represents the correlation between each pair of genes in the study. A crucial assumption is that the magnitude of the co-expression between any pair of genes is associated with a greater likelihood that the two genes interact. Thus, networks of interactions between genes are inferred from the co-expression matrix. Many papers have extended this analysis to the meta-analysis scenario. Of the 32 papers identified, some directly merge multiple studies to construct a network as if from a single study (8–15). Others combine pairwise gene interaction evidence across studies by vote counting (16–18) or Fisher's (19,20) method, similar to meta-analysis for DE gene detection. Segal et al. (21) was probably the first large-scale microarray meta-analysis for network or co-expression analysis. They developed a ‘module map’ by combining 1975 arrays in 26 cancer studies to characterize expression behavior of 2849 modules collected from various sources (e.g. Gene Ontology, KEGG pathways and gene expression clusters). Wang et al. (22) formulated a regularized approach to combine multiple time-course microarray studies for inferring gene regulatory networks. Zhou et al. (23) proposed a 2nd-order correlation analysis to construct network and functional annotation by combining 39 yeast data sets. Huttenhower et al. (24) used a scalable Bayesian framework to combine studies for pairwise meta-correlation and predicted functional relationship. Wang et al. (25) developed a semi-parametric meta-analysis approach for combining co-expression relationships from multiple expression profile data sets to evaluate similarity and dissimilarity of gene network across species. Steele et al. (26) proposed a weighted meta-analysis Bayesian network based on combining statistical confidences attached to network edges and a consensus Bayesian network to identify consistent network features across all studies.
Inter-study prediction analysis (25 papers)
Prediction analysis (a.k.a. classification analysis or supervised machine learning) is probably the most commonly applied microarray analysis that leads to clinical utility. In this type of analysis, the goal is to construct an improved discrimination between two or more study populations with accuracy beyond existing criteria in clinical practice (27). There now exists an extensive literature on classification methods for gene expression data; we refer the reader to Perez-Diaz et al. (28) for a recent review. In a single microarray study analysis, cross-validation has been routinely used by splitting the entire cohort into training and testing groups, constructing a prediction rule in the training group and finally validating in the test group. To demonstrate validity of microarray signatures or prediction models in other studies, two major strategies for developing prognostic signatures have been pursued. The first approach focuses on validity of biomarkers in external data. The prognostic signatures (a small number of genes) generated from training data are usually subsequently developed from a more traditional platform such as qRT–PCR. Reasons for failure of external validation in this regard have been widely surveyed and discussed in the literature (27,29–35). The second type of external validation focuses on inter-study prediction (i.e. construct a prediction model in one study and use the model to make predictions in another study). Although external validation of a gene expression-based prediction model has been shown valid in some publications (36,37), it has been found to be difficult in general. The failure of direct inter-study prediction is mainly due to discrepancy of probe design and experimental protocols across array platforms, plus possible heterogeneous patient cohorts across studies. Some reports avoided the major cross-platform obstacle by directly merging studies of the same platform (usually Affymetrix) to construct a prediction signature (38–42) and conventional cross-validation can be performed. Others developed sophisticated normalization techniques to solve or alleviate such a problem, including cross-platform normalization (XPN) (43), distance-weighted discrimination (DWD) (44), ratio-adjusted gene-wise normalization (rGN) (45) and module-based prediction (MBP) (46). In these approaches, data are normalized across studies so the prediction model can be applied across studies (47–50). Rank-based robust approaches have also been used (41,51).
Reproducibility and bias analysis (19 papers)
Evaluating reproducibility and bias across microarray studies was an important topic, especially when array technology and experimental protocols were in an early developmental stage. Simple Pearson correlation and Venn diagrams have been widely used (52–55). Other sophisticated statistical measures have been proposed to quantify similarity of any two microarray studies, including integrative correlation coefficient (56), similarities of ordered gene lists (SOGL) (57,58), BayesGen (59) and co-inertia analysis (CIA) (60).
Others (34 papers)
Additional purposes of microarray meta-analysis include: (i) discover or validate disease subtypes (61–65); (ii) predict unknown gene functions (66,67) or transcriptional regulations (13); (iii) dimension reduction (68); (iv) gene clustering (69). Targeted gene detections other than classical DE gene analysis have also been pursued. For example, phase-coupled models (70) or Bayesian approaches (71) have been used to combine multiple studies to detect periodic or cell cycle-related genes. Sequence information and gene expression have been combined for cyclic gene detection (72). Others have also combined large-scale microarray studies to identify house-keeping genes (defined as genes having consistent expression across various cellular or environmental changes) (73–75) or conversely highly variable genes (76,77).
DATABASES AND SOFTWARE
Databases
Many web databases are available for public storage and meta-analysis of microarray data sets. Gene Expression Omnibus (GEO) from NCBI and ArrayExpress from EBI are probably the two largest public repositories. On 3 April 2011, GEO contained 22 170 data series and 546 633 samples. Several other databases are housed in specific universities or groups, including Stanford Microarray Database (SMD), caArray at NCI, UPenn RAD Database, UNC Microarray Database, Yale Microarray Database, MUSC Database and UPSC-BASE. These websites are considered primary databases, where the main purpose is to provide downloadable and searchable microarray data sets. Other secondary databases import data sets from primary data archives, preprocess the data, perform in-depth analyses and deliver it through convenient interfaces for fast query, data mining and information integration. GEO Profiles and Gene Expression Atlas (78) are two secondary databases that accompany GEO and ArrayExpress. Other secondary databases include Genevestigator (79), ArrayTrack (80), Gemma, NextBio (81), LOLA (82), L2L (83), A-MADMAN (84), PrognoScan (85), MiMiR (86), Microarray retriever (87), TranscriptomeBrowser (88), M2DB (89), MAMA (90) and GeneSigDB (91). These tools contain various types of gene signature, regulatory network and differential expression information available for fast query, retrieval and evaluation.
In addition to the general-purpose microarray databases listed above, many databases are specialized to particular disease or species, including aging databases [AGEMAP (92) and Gene Age Nexus (93)], Pancreatic Expression database (94), COXPRESdb for gene networks in mammals (95), CYCLONET for cell cycle regulation (96), HCNet for heart and calcium functional network (14), and general cancer databases [Oncomine (97) and Cancer Genome Workbench (CGWB) (98)]. Of these, Oncomine has been used and cited widely in cancer research particularly when only a few targeted genes are scrutinized. While the statistical methods in these databases are relatively simple, a major advantage of these is the ease of use for biological scientists who are generating microarray data sets.
Software
Despite the availability of many web databases and many microarray meta-analysis methods (to be discussed in detail in the ‘Types of meta-analysis methods’ section), there exist surprisingly few user-friendly software packages for microarray meta-analysis implementation, in terms of their documentation and workflow. Compared with popular microarray packages (e.g. SAM, LIMMA or BRB array tool), existing meta-analysis packages are relatively primitive and difficult to use. In the R and Bioconductor environment, GeneMeta (implements fixed and random effects model; http://www.bioconductor.org/packages/release/bioc/html/GeneMeta.html; version 1.24.20), metaMA (implements random effects model and Stouffer's method; http://cran.r-project.org/web/packages/metaMA/; version 2.1), metaArray (implements meta-analysis of probability of expression, POE; http://www.bioconductor.org/packages/release/bioc/html/metaArray.html; version 1.28.20) (99), OrderedList (compares ordered gene lists; http://www.bioconductor.org/packages/release/bioc/html/OrderedList.html; version 1.24.20) (100), SequentialMA (for determining sensitivity and judge whether more samples are needed to assure firm conclusion) (101), RankProd (implement rank product method; http://www.bioconductor.org/packages/release/bioc/html/RankProd.html; version 2.24.20) (102) and RankAggreg (implements various rank aggregation methods; http://cran.r-project.org/web/packages/RankAggreg/; version 0.4-2) (103) are available. GODiff (104) (http://fishgenome.org/bioinfo/godiff/index.htm version 1.2) allows investigation of functional differentiation across studies using Gene Ontology annotation. Integrative Array Analyzer (105) (http://zhoulab.usc.edu/iArrayAnalyzer.htm; version 1.1.13) provides data mining and visualization tools to combine studies for simple co-expression analysis and differential expression analysis. For visualization, UCSC Genome Browser (106) and Genome Graphs provide flexible tools to compare and explore multiple genomic studies. Other commercial packages, including JMP Genomics from SAS (http://www.jmp.com/software/genomics/index.shtml; version 5.1) and Partek Genomic Suite (http://www.partek.com/software), also provide similar or more advanced visualization and graphical tools but with less statistical information integration capabilities.
In addition to scarcity of software packages in the field, quality of software packages should be enhanced. The concept of ‘literate programming’ (107) (e.g. the ‘sweave’ package in R) has been developed for reproducible research and should be promoted in future software development. For example, all packages available in Bioconductor now meet this requirement. Such a programing practice allows users to easily understand program design and rationale in the source code and to reproduce the results by other researchers.
META-ANALYSIS FOR DE GENE DETECTION
Ramasamy et al. (108) outlined a seven-step practical guidelines for conducting microarray meta-analysis: ‘(1) Identify suitable microarray studies; (2) Extract the data from studies; (3) Prepare the individual datasets; (4) Annotate the individual datasets; (5) Resolve the many-to-many relationship between probes and genes; (6) Combine the study-specific estimates; (7) Analyze, present, and interpret results’. In the section below, we will focus on steps 6 and 7 for DE gene detection of microarray meta-analysis. We will discuss four major types of statistical meta-analysis methods in the ‘Types of meta-analysis methods’ section. In the ‘Statistical considerations behind the methods’ and ‘A case study’ sections, related statistical considerations and a case study are discussed to illustrate the issue of choosing a suitable method.
Types of meta-analysis methods
As shown in Figure 3C, microarray meta-analysis for DE gene detection is a commonly encountered application. In this sub-section, we will discuss four categories of methods to combine information for DE gene detection: combine P-values, combine effect sizes, combine ranks and directly merge after normalization. In addition to these major categories, sophisticated latent variable approaches have also been developed.
Combining P-values (81 papers)
Combining P-values from multiple studies for information integration has a long history in statistical science. It has two major advantages (e.g. compared with another popular category of combining effects sizes below), including its simplicity and extensibility to different kinds of outcome variables. When the outcome variable is not binary (e.g. multi-class, continuous or censored survival), effects sizes may not be well defined, while association P-values can still be calculated. Below, we briefly introduce five P-value combination methods and use the examples in the ‘A case study’ section for illustration later. A major advantage of the P-value-based approaches is that they allow for standardization of the associations from genomic studies to a common scale.
Rhodes et al. (109) was among the earliest to demonstrate use of sophisticated statistical meta-analysis for DE gene detection. They applied the famous Fisher's method that summed up minus log-transformed P-values. For example, two-sided P-values of the PTTG1 gene were obtained from differential expression analysis in four prostate cancer studies separately in Table 1. The Fisher's statistics was calculated as SFisher = −2 × [log(1.6 × 10−3) + log(4.7 × 10−7) + log(1.7 × 10−4) + log(4.7 × 10−7)] = 88.52, where larger Fisher score reflects stronger aggregated differential expression evidence. Instead of log-transformation, Stouffer's method (110) adopted a different alternative by inverse normal transformation. In the PTTG1 example, 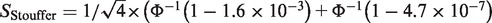
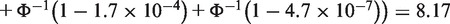 [where
[where 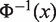 is the inverse cumulative distribution function of standard normal distribution]. Similar to Fisher score, smaller P-values result in larger
is the inverse cumulative distribution function of standard normal distribution]. Similar to Fisher score, smaller P-values result in larger 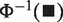 values and thus generate larger Stouffer score to reflect stronger aggregated statistical evidence. For the third and fourth methods, minimum or maximum P-values are taken as the test statistics: SminP = min(1.9E-5, 1E-20, 2E-5, 1E-20) = 1E-20 and SmaxP = max(1.9E-5, 1E-20, 2E-5, 1E-20) = 2E-5. Smaller minP or maxP statistics reflects stronger differential expression evidence. Conceptually, minP claims a DE gene if any study used to combine has a small P-value while maxP tends to be more conservative that detected DE genes should have small P-values in all studies combined. Differences of these two methods that correspond to the two hypothesis settings will be discussed in the ‘Statistical considerations behind the methods’ section. Recently, Li and Tseng (111) introduced an adaptively weighted Fisher's method (AW) that characterizes effective studies contributing to the meta-analysis so that the meta-analysis result has better biological interpretation. Take the ‘TPM2’ gene in Table 1 as an example. AW searched all possible 0-1 weights for the four studies (a total of 24 − 1 = 15 possibilities) and identified (1,0,1,1) as the best adaptive weight, meaning that combination of the three effective studies (Lapointe, Varambally and Yu) contributes the best to the DE evidence in the meta-analysis. For all the five methods, statistical inference can be performed parametrically under the assumption that P-values are uniformly distributed under the null hypothesis or can be done non-parametrically by permutation-based analysis (109,112).
values and thus generate larger Stouffer score to reflect stronger aggregated statistical evidence. For the third and fourth methods, minimum or maximum P-values are taken as the test statistics: SminP = min(1.9E-5, 1E-20, 2E-5, 1E-20) = 1E-20 and SmaxP = max(1.9E-5, 1E-20, 2E-5, 1E-20) = 2E-5. Smaller minP or maxP statistics reflects stronger differential expression evidence. Conceptually, minP claims a DE gene if any study used to combine has a small P-value while maxP tends to be more conservative that detected DE genes should have small P-values in all studies combined. Differences of these two methods that correspond to the two hypothesis settings will be discussed in the ‘Statistical considerations behind the methods’ section. Recently, Li and Tseng (111) introduced an adaptively weighted Fisher's method (AW) that characterizes effective studies contributing to the meta-analysis so that the meta-analysis result has better biological interpretation. Take the ‘TPM2’ gene in Table 1 as an example. AW searched all possible 0-1 weights for the four studies (a total of 24 − 1 = 15 possibilities) and identified (1,0,1,1) as the best adaptive weight, meaning that combination of the three effective studies (Lapointe, Varambally and Yu) contributes the best to the DE evidence in the meta-analysis. For all the five methods, statistical inference can be performed parametrically under the assumption that P-values are uniformly distributed under the null hypothesis or can be done non-parametrically by permutation-based analysis (109,112).
Table 1.
Results of the case study
| PT: primary tumor Met: metastasis | Types of hypothesis setting | Total number of detected DE genes (FDR = 1%) | PTTG1 | FOLR3 | TPM2 | BRAF |
|---|---|---|---|---|---|---|
| Study analysis | ||||||
| Lapointe (62 PT, 9 Met) | – | 364 | P = 1.6E-3; q = 1.5E-2; FC = 2.75 | P = 0.65; q = 0.80; FC = 0.92 | P = 9.4E-7; q = 9.3E-5; FC = 0.36 | P = 2.9E-4; q = 5E-3; FC = 1.65 |
| Tomlins (30 PT, 19 Met) | – | 598 | P = 4.7E-7; q = 3.4E-5; FC = 1.42 | P = 1E-20; q = 0; FC = 0.58 | P = 0.92; q = 0.95; FC = 0.99 | P = 3.4E-3; q = 1.9E-2; FC = 0.81 |
| Varambally (7 PT, 6 Met) | – | 587 | P = 1.7E-4; q = 3E-3; FC = 8.49 | P = 0.96; q = 0.97; FC = 1.02 | P = 1E-20; q = 0; FC = 0.04 | P = 1.4E-2; q = 4.8E-2; FC = 0.58 |
| Yu (65 PT, 25 Met) | – | 1073 | P = 4.7E-7; q = 8.1E-6; FC = 3.34 | P = 0.43; q = 0.56; FC = 1.13 | P = 1E-20; q = 0; FC = 0.16 | P = 8.5E-6; q = 9E-5; FC = 2.3 |
| Meta-analysis | ||||||
| Fisher | HSB | 2287 | P = 0; q = 0 | P = 0; q = 0 | P = 0; q = 0 | P = 4E-10; q = 3E-9 |
| Stouffer | HSB | 1472 | P = 0; q = 0 | P = 1.1E-5; q = 4.9E-3 | P = 0; q = 0 | P = 0.36; q = 0.97 |
| minP | HSB | 1740 | P = 4E-20 (q = 4E-19) | P = 4E-20 (q = 4E-19) | P = 4E-20 (q = 4E-19) | P = 1E-5 (q = 9E-5) |
| AW | HSB | 2312 | P = 0 (q = 0) (1,1,1,1) | P = 0 (q = 0) (0,1,0,0) | P = 0 (q = 0) (1,0,1,1) | P = 0 (q = 0) (1,1,1,1) |
| RankSum | ||||||
| Up | HSB | 672 | P = 0 (q = 0) | P = 0.93 (q = 1) | P = 1 (q = 1) | P = 2E-6 (q = 4E−5) |
| Down | HSB | 626 | P = 1 (q = 1) | P = 0.06 (q = 0.23) | P = 0 (q = 0) | P = 0.99 (q = 1) |
| RankProd | ||||||
| Up | HSB | 490 | P = 0 (q = 0) | P = 0.84 (q = 1) | P = 1 (q = 1) | P = 0 (q = 0) |
| Down | HSB | 462 | P = 1 (q = 1) | P = 0.02 (q = 0.02) | P = 0 (q = 0) | P = 0.99 (q = 1) |
| Vote counting | ||||||
| S ≥ 3, P = .01 | HSA or HSA− | 453 | Yes | No | Yes | Yes |
| S ≥ 3, P = .05 | HSA or HSA− | 1021 | Yes | No | Yes | Yes |
| S = 4, P = .01 | HSA or HSA− | 80 | Yes | No | No | Yes |
| S = 4, P = .05 | HSA or HSA− | 217 | Yes | No | No | Yes |
| Random effects model | HSA | 350 | P = 2E-14 (q = 1E-11) | P = 0.33 (q = .56) | P = 0.002 (q = 0.02) | P = 0.89 (q = 0.95) |
| maxP | HSA | 549 | P = 2E-19 (q = 2E-16) | P = 0.79 (q = 0.86) | P = 0.05 (q = 0.13) | P = 2E-8 (q = 1E-6) |
Results of DE gene detection from individual study analysis and meta-analysis (using nine different methods) are listed. Four representative genes are scrutinized for the P-value and q-value results.
Despite availability of powerful statistical tools described above, many biological applications we surveyed chose to apply naïve Venn diagram (used in 21 papers in our survey) or vote counting methods (used in 24 papers) for convenience. Venn diagram is a useful visualization tool, when combining few (usually 2–4) studies, to demonstrate the intersection and union distribution of DE gene lists detected by each individual study under a fixed threshold (e.g. FDR = 5%). The naïve diagram, however, does not perform real information integration but only displays a consistency summary. When many studies are combined, naïve vote counting is often chosen by biologists instead. For each gene, the method simply counts the number of studies with P-values under a given threshold (e.g. P < 0.05). In the statistical literature, it is well known that vote counting is statistically inefficient (113,114). On the other hand, vote counting is useful when raw data and complete P-value information of all genes are unavailable while only a list of DE genes under certain P-value threshold is available. This happened frequently in many early microarray studies, in which DE gene lists were summarized in supplemental tables of publications but raw data were not uploaded to public domain. Due to the significant loss of information and efficiency, the vote counting method should be avoided whenever possible in the applications.
Combining effect sizes (41 papers)
Many meta-analysis methods have been based on the assumption that the standardized effect sizes are combinable across studies. Fixed and random effects models (FEM & REM) are the two most popular approaches in this category. In FEM, the estimated effect size in each study is assumed to come from an underlying true effect size plus measurement error (that may come from experimental or population sampling error). In REM, each study further contains a random effect that can incorporate unknown cross-study heterogeneities in the model. Choi et al. (115) was among the first to apply these models to microarray meta-analysis. In a given application, a Q-statistic was used to determine the need for a random effects model and the underlying effect size was estimated under FEM or REM. Bayesian meta-analysis was also developed with Markov Chain Monte Carlo (MCMC) simulation to estimate the underlying effect size. Others have also developed different variations of effect size models (116–118).
Combining ranks (18 papers)
One apparent downside of methods combining P-values or effect sizes is that the results can often be dominated by outliers. This can be a significant problem when thousands of genes are analyzed simultaneously in the noisy nature of microarray experiments. Methods combining robust rank statistics are used to alleviate this problem. Instead of P-values or effect sizes, the ranks of DE evidence are calculated for each gene in each study. The product, mean (119) or equivalently sum (120) of ranks from all studies is then calculated as the test statistic. Permutation analysis can be performed to assess the statistical significance and to control FDR. Hong et al. (102) proposed a more advanced RankProd algorithm that calculates the product of the ranks of fold change in each inter-group pair of samples. In a follow-up comparative study, they showed its better performance as compared to Fisher's method and the random effects model (121). DeConde (122) applied various ‘rank aggregation’ methods, which were developed for the meta-search problem for combining top-k lists in the computer science literature. The methods effectively aggregate the rankings of, say the top 100 most upregulated or downregulated genes in each study.
Directly merging the raw data (51 papers)
Despite the concern of heterogeneity across studies, many microarray meta-analysis applications chose to normalize across studies and directly merge data sets for DE gene detection. This approach is often called ‘mega-analysis’, especially in GWAS meta-analysis. In microarray meta-analysis, such applications usually restrict selection of studies from the same or similar array platform, e.g. a single Affymetrix U133 or multiple Affymetrix platforms (38,123). The collection of only Affymetrix arrays allows pre-processing by model-based robust multi-array (RMA) normalization (124) on the CEL files of all samples simultaneously. Others have developed advanced normalization techniques to eliminate cross-study discrepancy and allow direct merge of studies [e.g. XPN (43), DWD (44) and rGN (45)]. Although direct merging can be attractive in applications for its convenience, cautions have to be taken that normalizations do not guarantee to remove all cross-study discrepancies. In fact, Goldstein et al. (125) demonstrated that RMA does not remove batch effects even when two studies are from the same lab and same Affymetrix platform but performed at different time.
Latent variable approaches
There are more sophisticated approaches in place that attempt to model the pre-processed microarray data sets using latent variable-based models and attendant inference using either expectation–maximization routines or Markov Chain Monte Carlo algorithms. For example, the probability of expression (POE) was a latent variable used in several papers that was not observable in the data but could be inferred from other observed variables. Papers of this category include metaArray (99) which employs two types of inferential strategies, frequentist and Bayesian (see the ‘Statistical considerations behind the methods’ section) for modeling data from multiple platforms, and XDE (126), which fits a joint parametric Bayesian model for multi-study meta-analysis. In particular, the latter paper shows some compelling simulation evidence for a joint modeling strategy using these latent variable models. For more specialized settings, Conlon et al. (127) and Fan et al. (71) have presented Bayesian modeling approaches for combining data from multiple microarray studies. While the hierarchical models used in these papers are statistically more sophisticated than the methods described in the previous section, they offer the potential of pooling information across genes to sharpen inferences about which genes are differentially expressed. However, due to their complexity, they have not been used much in practice. One notable exception is Shen et al. (128), which applied a precursor of the metaArray algorithm to identification of gene expression signatures for aggressive breast cancer.
Statistical considerations behind the methods
Null and alternative hypothesis assumptions behind the methods
Although the concept of combining studies for meta-analysis is seemingly straightforward, the targeted biomarker characteristics implicitly reflected by different statistical hypothesis settings behind the methods can be varied. Following the convention of Birnbaum (129), Li and Tseng (111) presented two major hypothesis settings behind microarray meta-analysis methods described in the ‘Types of meta-analysis methods’ section. Suppose K studies are combined and θk is the effect size of study k. The first hypothesis setting (HSA) detects candidate genes differentially expressed in ‘all’ studies (H0: θ1 = θk = 0 for one or more k versus Ha: θk ≠ 0, 1 ≤ k ≤ K) whereas, HSB identifies markers differentially expressed in ‘partial’ (one or more) studies (H0: θ1 = … = θk = 0 versus Ha: θk ≠ 0 for one or more k). For example, Fisher's method takes sum of log-transformed P-values as the statistics. If, for a given gene, a study has very significant P-value (e.g. P = 1E-20) but all other studies do not have significant P-values (e.g. the FOLR3 gene in the ‘A case study’ section), the Fisher's method still concludes a large Fisher's score and declares this gene as a DE gene. As a result, Fisher's method pursues the second hypothesis setting, HSB. Similarly, Stouffer, minP, maxP, AW, as well as rank sum and RankProd, all adopt similar hypothesis setting HSB. On the other hand, the maxP method takes the maximum P-value as the statistics. It requires that P-values from all studies are small and thus it pursues the first hypothesis setting, HSA. The random effects model has the same hypothesis setting that all studies have the same overall effect size while each study may contain an additional random effect component. One might somewhat relax HSA to detect genes differentially expressed in ‘majority’ of studies (denoted as HSA−). The vote counting method follows this relaxed hypothesis setting. The hypothesis setting of each method is presented in Table 1.
Frequentist versus Bayesian inference
Implicit in the discussion about inference has been the use of a frequentist framework. In particular, we assume that there is a test statistic, larger values which indicate stronger evidence against the null hypothesis. However, one could also perform Bayesian hypothesis testing using these hypotheses. This is done by consideration of posterior probabilities of the specific hypotheses (e.g. P(θ1 = … = θk = 0|data) versus P(θk ≠ 0 ∀k|data)). Computation of these posterior probabilities requires the use of a likelihood for the parameters of interest along with prior probabilities of the specific hypotheses being tested. The prior probabilities are typically selected based on the relative costs of a type I error (rejecting the null hypothesis when it is true) versus a type II error (accepting the null hypothesis when it is false). The larger the relative cost, the larger the prior probability for the null hypothesis should be. Bayesian hypothesis testing procedures are amenable with the latent variable models for meta-analysis described in the ‘Databases and software’ section. In the literature, another advantage of Bayesian approach is the use of Bayes factor that does not require a prior probability of the two hypotheses and can work as an alternative of classical hypothesis testing.
Consistent up or downregulation
Comparing the first three categories of meta-analysis methods in the ‘Types of meta-analysis methods’ section, combining effects sizes (e.g. random or fixed effects model) automatically identifies genes that have consistent up or downregulation in all studies. This may not be the case for methods combining P-values or ranks if the P-values and ranks are obtained from two-sided hypothesis testing. In this case, up- and down-regulation are treated as equally strong evidence and a gene may be detected from the meta-analysis with strong up-regulation evidence in one study but strong down-regulation evidence in another study, which leads to confusing conclusions. Theoretically, the discordance may reflect underlying biological truth due to population heterogeneity but it may as well be a result of technical artifacts such as gene annotation mistakes or cross-hybridization. Distinguishing the two is often a difficult, if not impossible, task. A convenient solution to avoid detecting genes with such discordances is by combining P-values or ranks from one-sided tests. For example, a modified Stouffer's method can apply a z-transformation that automatically utilizes one-sided tests and splits up- and downregulation evidences into positive and negative z-scores, respectively. Owen (130) applied a similar Pearson one-sided test adjustment for Fisher's method and the modification can be extended to minP, maxP and other methods. Note that the consistent up- or downregulation issue only exists in two-class comparison in DE gene detection and does not apply to other types of response variables (e.g. multi-class, continuous or survival).
A case study
To illustrate some properties of the methods described in the ‘Types of meta-analysis methods’ section, we performed a simple case study. The motivation of this small case study was to help understand how the algorithm of each method works and to explain pros and cons of each method. The result provides general insight for selecting an adequate method in applications. This case study is, however, neither comprehensive nor conclusive enough as a comparative study to judge performance of the methods. In this case study, four prostate cancer expression profiles (Lapointe, Tomlins, Varambally and Yu) containing metastasis versus primary tumor samples were combined for meta-analysis. After gene matching by official gene symbols, pre-processing and filtering, 4260 genes were analyzed in the meta-analysis. We used the R package ‘siggenes’ to perform DE gene analysis in each study. ‘siggenes’ allows implementation of the Significance Analysis of Microarray (SAM) method and the Empirical Bayes Analyses of Microarrays (EBAM) method. For simplicity, we applied the popular SAM method with B = 500 permutation. According to Phipson and Smyth (131), the P-values from permutation analysis should never be zero but the ‘siggenes’ package does occasionally generate zero P-values. If P = 0 is obtained for a certain gene in an individual study, we set it to P = 1E-20 to avoid failure of logarithmic or inverse normal transformation in the Fisher's and Stouffer's methods. After P-values are generated, Benjamini–Hochberg procedure is applied to calculate q-values and correct for multiple comparison (‘p.adjust’ function in R is used). The random effects model was implemented using the ‘GeneMeta’ package in R. RankSum and RankProd methods were performed in the R package ‘RankProd’. In the ‘RankProd’ package, the RankSum and RankProd methods could only be implemented with up- and downregulation analysis separately. Theoretically, it is easy to modify the algorithm to analyze up- and downregulation simultaneously. For the vote counting method, the method determines a DE gene if it has P-values smaller than a threshold P in greater or equal to S studies among the four studies combined. In Table 1, we list results for P = 0.01 or 0.05 and S = 3 or 4. Table 1 shows results of four single-study analyses and nine meta-analysis methods in four selected genes.
The first example gene, ‘PTTG1’, was up-regulated in the metastatic group with strong statistical significance in all four studies (P = 1.9E-5, 1E-20, 2E-5 and 1E-20). As expected, all nine meta-analysis methods concluded very strong statistical significance even after multiple comparison correction. As a comparison, the second selected gene ‘FOLR3’ was down-regulated with strong statistical significance in the Tomlins study (P = 1E-20; fold change FC = 0.58) but was not statistically significant in the other three studies (P = 0.65, 0.96 and 0.43). Such sporadic high statistical significance in a subset of studies might be a result of unknown experimental artifacts (e.g. non-specific probe design that causes cross-hybridization in the cDNA array design) but might instead be a biological truth in the specific cohort. Fisher, minP, AW, RankSum and RankProd all obtained strong to moderate statistical significance after meta-analysis for this gene (see FOLR3 column in Table 1). This reflected the underlying HSB hypothesis setting of these methods to detect a DE gene if the gene is differentially expressed in one or more studies (see ‘Statistical considerations behind the methods’ section). On the other hand, vote counting, the random effects model and maxP required a gene to be differentially expressed in all or ‘majority’ of the studies (i.e. hypothesis setting HSA) and thus did not generate significant q-values. The third gene, ‘TPM2’, was differentially expressed in three studies (P = 9.4E-7, 1E-20 and 1E-20 in Lapointe, Varambally and Yu) but not differentially expressed in Tomlins (P = 0.92). Among the nine methods, it was detected by seven methods, excepting only maxP (q = 0.13) and vote counting (S = 4). This result shows that methods to detect genes differentially expressed in ‘all’ studies might be too stringent and could ignore an important marker gene when many studies are combined. It was interesting that, in the random effects model, although it is aimed at HSA, the random effects assumption provided robustness so that TPM2 was statistically significant (q = 0.02). The fourth example gene, ‘BRAF’, was differentially expressed in all four studies but was surprisingly down-regulated in two studies but up-regulated in the other two studies. Among the nine methods, Fisher, minP, AW, vote counting and maxP detected BRAF as a DE gene because the methods combined two-sided P-values without distinguishing DE direction. RankSum and RankProd, although considered DE directions in the algorithm, still determined BRAF as an upregulated DE gene. Stouffer and random effects model were two methods that considered DE directions in the algorithm and generated non-significance q-values. Whether detecting a discordant gene like BRAF is favorable or not depends on the inferential goals of the experiment. It can be the case that BRAF is an important marker and the discordance is generated from an unknown meaningful confounding variable (e.g. race; say, BRAF is up-regulated in black but down-regulated in white). It is equally possible that the discordance comes from unknown technical artifacts.
Below, we further scrutinize the biological functions of the four genes using the NCBI database. PTTG1 has been related to DNA repair, cell division and mitosis cell cycle and has been correlated with tumor aggressiveness in multiple tumors. The strong statistical significance in all four studies is biologically verified. On the contrary, there is no direct evidence of cancer association found for FOLR3. The strong DE statistical significance in the Tomlins study might indeed be an artifact. For TPM2, a recent paper has identified a novel splice variant of TPM2 related to prostate cancer cell lines (132). The high statistical significance in three out of four studies might be strong enough evidence for its association with metastasis. The fourth gene, ‘BRAF’, plays a role in regulating the MAP kinase/ERKs signaling pathway, has been associated to multiple cancers and is in the KEGG prostate cancer pathway (05215). Indeed, the confusing discordant direction of fold changes might be the result of unknown confounding factors such as age or race. Further investigation of demographic or experimental information for the four studies might help elucidate the mystery. We also note that interpretation of detected DE genes also depend on other genes due to gene dependency.
OPEN QUESTIONS
Despite the popularity of microarray meta-analysis, many issues remain unresolved that can hamper the effectiveness of its application. In this section, we discuss a few open questions and related problems.
Quality assessment and inclusion/exclusion criteria
To date, the decision to include or exclude microarray studies in a meta-analysis has been mostly ad hoc and subjective in the literature. Researchers usually apply arbitrary criteria, such as number of samples or array platforms (e.g. (112,133,134) and many others), to make the decision. Inclusion of a low quality or outlying study into the meta-analysis, however, can greatly reduce the statistical power or even result in a false conclusion. As a first step, keyword searching in primary data repositories can provide a useful initial screening to identify studies to combine. Some biological terminology systems (e.g. Unified Medical Language System, UMLS) may help provide a refined and unbiased selection for more homogeneous studies. Ramaswamy et al. (108) has suggested to apply the integrative correlation technique by Parmagiani et al. (56) to select ‘reproducible’ genes for meta-analysis. This approach potentially can be extended for objective inclusion/exclusion decisions. In general, a data-driven quantitative evaluation for inclusion/exclusion criteria is still an open question in the field. This is tied to the classical question of between-study variation. In the case of a single gene, the issue of between-study variation has been carefully studied; a review of available methods can be found in (135). How to adapt this to the genomic, high-dimensional data setting is still an open question. This issue is also discussed in the companion paper for GWAS meta-analysis, under the terminology of ‘heterogeneity’.
Practical guidelines from large-scale comparative study and simulation
Among the papers we have surveyed, only two papers performed systematic comparative analysis on microarray meta-analysis methods: Hong et al. (121) and Campain and Yang (136). Although the two studies provided insightful conclusions, the number of methods compared (three and five methods, respectively) and the number of real examples examined (two and three examples, respectively with each example combining only 2–5 microarray studies) were very limited. Some key conclusions from the two papers were even contradictory. A large-scale comparative study and simulation study with adequate evaluation measures will help provide insights and practical guidelines for choosing the ‘best’ meta-analysis method(s) in practice.
Combining studies with censored information
As mentioned in ‘Types of meta-analysis methods’ section, vote counting has a natural advantage to combine information from studies with censored P-value information (i.e. raw data are not accessible but only a top ranked DE gene list under certain P-value threshold is available), though it suffers greatly from low statistical power. Although many grant agencies and journals now enforce data sharing policies, many old studies or new studies funded by private foundations are still not openly accessible. Studies with censored information can be an obstacle for meta-analysis. Researchers are forced to either drop studies with censored information or use inefficient vote counting methods in the meta-analysis. In the literature, Bushman and Wang (137) have transformed P-values to pseudo effect sizes to combine vote counting and effect size combination methods. Extension of other existing methods, such as Fisher, Stouffer and maxP, to analyze such censored P-value data in partial studies will provide a more powerful solution to this practical problem.
Meta-analysis to guide and design future studies
In modern evidence-based medicine, meta-analysis is often used (or required) to combine existing evidence in the literature when planning for a new study. Similarly, genomic meta-analysis should be used more frequently to narrow down gene targets or scope of study when designing new studies (e.g. targeted sequencing).
Meta-analysis on a pathway basis
While the work of authors such as Shen et al. (37) and Shen and Tseng (7) has led the way in the area of combining information from multiple studies at the pathway level, there are several issues that remain to be addressed. Adjusting for inference due to pathway dependence remains an important open problem, as the dependence in pathway data might render many of the statistical methods available for multiple testing (e.g. q-values/false discovery rate control) invalid.
Development of user-friendly software
In our review, only a few microarray meta-analysis methods are developed with R packages. When we tested the packages, most of them either did not have clear manuals or had functions that were not easy to apply (especially compared with mature and popular microarray packages such as SAM, PAM, LIMMA, BRB Array Tool or GSEA). Convenient R packages or packages in a programmable environment will allow researchers to test and compare methods and motivate further methodological development. Software with friendly graphical user interfaces (GUI) will further assist biologists in daily applications.
Adjust for potential confounding variables
Heterogeneities caused by demographic, clinical and technical variables often exist within and across studies. Failure to consider these variables in the statistical models and meta-analysis can result in reduced statistical power or false positives. In a microarray meta-analysis, these systematic variabilities should be considered and incorporated in the analysis whenever possible. Leek and Storey (138) proposed surrogate variable analysis (SVA) to further account for unmeasured and unmodeled factors in a genome-wide expression analysis. The result has shown improved sensitivity and accuracy. Similar techniques can be extended to microarray meta-analysis.
CONCLUSION AND DISCUSSION
In this article, we performed a comprehensive review of microarray meta-analysis and discussed the related statistical issues. Although many methods have been proposed and used in published applications, the detailed meta-analysis workflow and the hypothesis behind the analysis needs more attention. Selection of a suitable method depends on the type of analysis desired (various purposes described in ‘Purposes of microarray meta-analysis’ section) and the hypothesis setting behind each method (‘Statistical considerations behind the methods’ section). In our review, we noticed that easy to use software packages are rare in the field. We have also addressed several important open questions (‘Open questions’ section), including developing a quantitative inclusion/exclusion evaluation, performing comparative study for a practical guideline and adjusting for confounding variables. As many high-throughput experimental technologies are rapidly developed and widely applied nowadays, data management and effective integrative analysis will become more and more essential to fully utilize the rich information contained in the tremendous amount of data. The analytical techniques and concepts may also extend to information integration of other types of genomic data.
One limitation of this review article is the restricted scope of literature search by PubMed. We have attempted to include 102 manually collected references. The inclusion, however, cannot be exhaustive. For example, many related approaches are termed ‘integrative analysis’ in the literature and thus cannot be included in the review. This is especially true in categories other than DE gene analysis (e.g. pathway analysis, prediction analysis or network analysis). We attempted to include ‘integrative analysis’ in the keyword search but failed because it generated thousands of publications with most of them irrelevant to the purpose of this article.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table.
FUNDING
National Institutes of Health (NIH) (R01MH077159 and RC2HL101715, to G.C.T.); (R01HD38979 and R01DE14899, to E.F. and F.B.); NIH (R01GM72007, to D.B.); Huck Institute for Life Sciences (to D.B.). Funding for open access charge: University of Pittsburgh.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank C. Song, X. Wang and G. Liao for collecting and printing papers.
REFERENCES
- 1.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu S, Li J, Song C, Shen K, Tseng GC. Biomarker detection in the integration of multiple multi-class genomic studies. Bioinformatics. 2010;26:333–340. doi: 10.1093/bioinformatics/btp669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Setlur SR, Royce TE, Sboner A, Mosquera JM, Demichelis F, Hofer MD, Mertz KD, Gerstein M, Rubin MA. Integrative microarray analysis of pathways dysregulated in metastatic prostate cancer. Cancer Res. 2007;67:10296–10303. doi: 10.1158/0008-5472.CAN-07-2173. [DOI] [PubMed] [Google Scholar]
- 6.Manoli T, Gretz N, Grone HJ, Kenzelmann M, Eils R, Brors B. Group testing for pathway analysis improves comparability of different microarray datasets. Bioinformatics. 2006;22:2500–2506. doi: 10.1093/bioinformatics/btl424. [DOI] [PubMed] [Google Scholar]
- 7.Shen K, Tseng GC. Meta-analysis for pathway enrichment analysis when combining multiple genomic studies. Bioinformatics. 2010;26:1316–1323. doi: 10.1093/bioinformatics/btq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mabbott NA, Kenneth Baillie J, Hume DA, Freeman TC. Meta-analysis of lineage-specific gene expression signatures in mouse leukocyte populations. Immunobiology. 215:724–736. doi: 10.1016/j.imbio.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Carrera J, Rodrigo G, Jaramillo A, Elena SF. Reverse-engineering the Arabidopsis thaliana transcriptional network under changing environmental conditions. Genome Biol. 2009;10:R96. doi: 10.1186/gb-2009-10-9-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jupiter DC, VanBuren V. A visual data mining tool that facilitates reconstruction of transcription regulatory networks. PLoS One. 2008;3:e1717. doi: 10.1371/journal.pone.0001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frericks M, Meissner M, Esser C. Microarray analysis of the AHR system: tissue-specific flexibility in signal and target genes. Toxicol. Appl. Pharmacol. 2007;220:320–332. doi: 10.1016/j.taap.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 12.Ucar D, Neuhaus I, Ross-MacDonald P, Tilford C, Parthasarathy S, Siemers N, Ji RR. Construction of a reference gene association network from multiple profiling data: application to data analysis. Bioinformatics. 2007;23:2716–2724. doi: 10.1093/bioinformatics/btm423. [DOI] [PubMed] [Google Scholar]
- 13.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong SE, Rho SH, Yeom YI, Kim do H. HCNet: a database of heart and calcium functional network. Bioinformatics. 2006;22:2053–2054. doi: 10.1093/bioinformatics/btl331. [DOI] [PubMed] [Google Scholar]
- 15.Mehan MR, Nunez-Iglesias J, Kalakrishnan M, Waterman MS, Zhou XJ. An integrative network approach to map the transcriptome to the phenome. J. Comput. Biol. 2009;16:1023–1034. doi: 10.1089/cmb.2009.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niida A, Imoto S, Nagasaki M, Yamaguchi R, Miyano S. A novel meta-analysis approach of cancer transcriptomes reveals prevailing transcriptional networks in cancer cells. Genome Inform. 2009;22:121–131. [PubMed] [Google Scholar]
- 17.Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell. 2006;11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 18.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava GP, Li P, Liu J, Xu D. Identification of transcription factor's targets using tissue-specific transcriptomic data in Arabidopsis thaliana. BMC Syst. Biol. 2010;4(Suppl. 2):S2. doi: 10.1186/1752-0509-4-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez-Zas SL, Ko Y, Adams HA, Southey BR. Advancing the understanding of the embryo transcriptome co-regulation using meta-, functional, and gene network analysis tools. Reproduction. 2008;135:213–224. doi: 10.1530/REP-07-0391. [DOI] [PubMed] [Google Scholar]
- 21.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat. Genet. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Joshi T, Zhang XS, Xu D, Chen L. Inferring gene regulatory networks from multiple microarray datasets. Bioinformatics. 2006;22:2413–2420. doi: 10.1093/bioinformatics/btl396. [DOI] [PubMed] [Google Scholar]
- 23.Zhou XJ, Kao MC, Huang H, Wong A, Nunez-Iglesias J, Primig M, Aparicio OM, Finch CE, Morgan TE, Wong WH. Functional annotation and network reconstruction through cross-platform integration of microarray data. Nat. Biotechnol. 2005;23:238–243. doi: 10.1038/nbt1058. [DOI] [PubMed] [Google Scholar]
- 24.Huttenhower C, Hibbs M, Myers C, Troyanskaya OG. A scalable method for integration and functional analysis of multiple microarray datasets. Bioinformatics. 2006;22:2890–2897. doi: 10.1093/bioinformatics/btl492. [DOI] [PubMed] [Google Scholar]
- 25.Wang K, Narayanan M, Zhong H, Tompa M, Schadt EE, Zhu J. Meta-analysis of inter-species liver co-expression networks elucidates traits associated with common human diseases. PLoS Comput. Biol. 2009;5:e1000616. doi: 10.1371/journal.pcbi.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steele E, Tucker A. Consensus and Meta-analysis regulatory networks for combining multiple microarray gene expression datasets. J. Biomed Inform. 2008;41:914–926. doi: 10.1016/j.jbi.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J. Natl Cancer Inst. 2010;102:464–474. doi: 10.1093/jnci/djq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Diez A, Morgun A, Shulzhenko N. Microarrays for cancer diagnosis and classification. Adv. Exp. Med. Biol. 2007;593:74–85. doi: 10.1007/978-0-387-39978-2_8. [DOI] [PubMed] [Google Scholar]
- 29.Baker SG. Improving the biomarker pipeline to develop and evaluate cancer screening tests. J. Natl Cancer Inst. 2009;101:1116–1119. doi: 10.1093/jnci/djp186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diamandis EP. Cancer biomarkers: can we turn recent failures into success? J. Natl Cancer Inst. 2010;102:1462–1467. doi: 10.1093/jnci/djq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J. Natl Cancer Inst. 2007;99:147–157. doi: 10.1093/jnci/djk018. [DOI] [PubMed] [Google Scholar]
- 32.Ransohoff DF. Bias as a threat to the validity of cancer molecular-marker research. Nat. Rev. Cancer. 2005;5:142–149. doi: 10.1038/nrc1550. [DOI] [PubMed] [Google Scholar]
- 33.Ransohoff DF. How to improve reliability and efficiency of research about molecular markers: roles of phases, guidelines, and study design. J. Clinical Epidemiol. 2007;60:1205–1219. doi: 10.1016/j.jclinepi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 34.Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J. Clin. Oncol. 2005;23:7332–7341. doi: 10.1200/JCO.2005.02.8712. [DOI] [PubMed] [Google Scholar]
- 35.Simon R. Genomic biomarkers in predictive medicine: an interim analysis. EMBO Mol. Med. 2011;3:429–435. doi: 10.1002/emmm.201100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 37.Shen R, Chinnaiyan AM, Ghosh D. Pathway analysis reveals functional convergence of gene expression profiles in breast cancer. BMC Med. Genomics. 2008;1:28. doi: 10.1186/1755-8794-1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee Y, Scheck AC, Cloughesy TF, Lai A, Dong J, Farooqi HK, Liau LM, Horvath S, Mischel PS, Nelson SF. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genomics. 2008;1:52. doi: 10.1186/1755-8794-1-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandberg R, Ernberg I. The molecular portrait of in vitro growth by meta-analysis of gene-expression profiles. Genome Biol. 2005;6:R65. doi: 10.1186/gb-2005-6-8-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang H, Deng Y, Chen HS, Tao L, Sha Q, Chen J, Tsai CJ, Zhang S. Joint analysis of two microarray gene-expression data sets to select lung adenocarcinoma marker genes. BMC Bioinformatics. 2004;5:81. doi: 10.1186/1471-2105-5-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu CC, Hu J, Kalakrishnan M, Huang H, Zhou XJ. Integrative disease classification based on cross-platform microarray data. BMC Bioinformatics. 2009;10(Suppl. 1):S25. doi: 10.1186/1471-2105-10-S1-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu L, Tan AC, Winslow RL, Geman D. Merging microarray data from separate breast cancer studies provides a robust prognostic test. BMC Bioinformatics. 2008;9:125. doi: 10.1186/1471-2105-9-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shabalin AA, Tjelmeland H, Fan C, Perou CM, Nobel AB. Merging two gene-expression studies via cross-platform normalization. Bioinformatics. 2008;24:1154–1160. doi: 10.1093/bioinformatics/btn083. [DOI] [PubMed] [Google Scholar]
- 44.Qiao X, Zhang HH, Liu Y, Todd MJ, Marron JS. Weighted distance weighted discrimination and its asymptotic properties. J. Am. Statist. Assoc. 2010;105:401–414. doi: 10.1198/jasa.2010.tm08487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng C, Shen K, Song C, Luo J, Tseng GC. Ratio adjustment and calibration scheme for gene-wise normalization to enhance microarray inter-study prediction. Bioinformatics. 2009;25:1655–1661. doi: 10.1093/bioinformatics/btp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mi Z, Shen K, Song N, Cheng C, Song C, Tseng GC. Module-based prediction approach for robust inter-study prediction in microarray data. Bioinformatics. 2010;26:2586–2593. doi: 10.1093/bioinformatics/btq472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fielden MR, Nie A, McMillian M, Elangbam CS, Trela BA, Yang Y, Dunn RT, 2nd, Dragan Y, Fransson-Stehen R, Bogdanffy M, et al. Interlaboratory evaluation of genomic signatures for predicting carcinogenicity in the rat. Toxicol. Sci. 2008;103:28–34. doi: 10.1093/toxsci/kfn022. [DOI] [PubMed] [Google Scholar]
- 48.Lu Y, Lemon W, Liu PY, Yi Y, Morrison C, Yang P, Sun Z, Szoke J, Gerald WL, Watson M, et al. A gene expression signature predicts survival of patients with stage I non-small cell lung cancer. PLoS Med. 2006;3:e467. doi: 10.1371/journal.pmed.0030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warnat P, Eils R, Brors B. Cross-platform analysis of cancer microarray data improves gene expression based classification of phenotypes. BMC Bioinformatics. 2005;6:265. doi: 10.1186/1471-2105-6-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bloom G, Yang IV, Boulware D, Kwong KY, Coppola D, Eschrich S, Quackenbush J, Yeatman TJ. Multi-platform, multi-site, microarray-based human tumor classification. Am. J. Pathol. 2004;164:9–16. doi: 10.1016/S0002-9440(10)63090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu L, Tan AC, Naiman DQ, Geman D, Winslow RL. Robust prostate cancer marker genes emerge from direct integration of inter-study microarray data. Bioinformatics. 2005;21:3905–3911. doi: 10.1093/bioinformatics/bti647. [DOI] [PubMed] [Google Scholar]
- 52.Kuo WP, Jenssen TK, Butte AJ, Ohno-Machado L, Kohane IS. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics. 2002;18:405–412. doi: 10.1093/bioinformatics/18.3.405. [DOI] [PubMed] [Google Scholar]
- 53.Jarvinen AK, Hautaniemi S, Edgren H, Auvinen P, Saarela J, Kallioniemi OP, Monni O. Are data from different gene expression microarray platforms comparable? Genomics. 2004;83:1164–1168. doi: 10.1016/j.ygeno.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Mah N, Thelin A, Lu T, Nikolaus S, Kuhbacher T, Gurbuz Y, Eickhoff H, Kloppel G, Lehrach H, Mellgard B, et al. A comparison of oligonucleotide and cDNA-based microarray systems. Physiol. Genomics. 2004;16:361–370. doi: 10.1152/physiolgenomics.00080.2003. [DOI] [PubMed] [Google Scholar]
- 55.Lee JK, Bussey KJ, Gwadry FG, Reinhold W, Riddick G, Pelletier SL, Nishizuka S, Szakacs G, Annereau JP, Shankavaram U, et al. Comparing cDNA and oligonucleotide array data: concordance of gene expression across platforms for the NCI-60 cancer cells. Genome Biol. 2003;4:R82. doi: 10.1186/gb-2003-4-12-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parmigiani G, Garrett-Mayer ES, Anbazhagan R, Gabrielson E. A cross-study comparison of gene expression studies for the molecular classification of lung cancer. Clin. Cancer Res. 2004;10:2922–2927. doi: 10.1158/1078-0432.ccr-03-0490. [DOI] [PubMed] [Google Scholar]
- 57.Yang X, Bentink S, Scheid S, Spang R. Similarities of ordered gene lists. J. Bioinform. Comput. Biol. 2006;4:693–708. doi: 10.1142/s0219720006002120. [DOI] [PubMed] [Google Scholar]
- 58.Yang X, Sun X. Meta-analysis of several gene lists for distinct types of cancer: a simple way to reveal common prognostic markers. BMC Bioinformatics. 2007;8:118. doi: 10.1186/1471-2105-8-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen VA, Lio P. Measuring similarity between gene expression profiles: a Bayesian approach. BMC Genomics. 2009;10(Suppl. 3):S14. doi: 10.1186/1471-2164-10-S3-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Culhane AC, Perriere G, Higgins DG. Cross-platform comparison and visualisation of gene expression data using co-inertia analysis. BMC Bioinformatics. 2003;4:59. doi: 10.1186/1471-2105-4-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanga S, Broom BM, Cristini V, Edgerton ME. Gene expression meta-analysis supports existence of molecular apocrine breast cancer with a role for androgen receptor and implies interactions with ErbB family. BMC Med. Genomics. 2009;2:59. doi: 10.1186/1755-8794-2-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoshida Y, Brunet JP, Tamayo P, Golub TR, Mesirov JP. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS One. 2007;2:e1195. doi: 10.1371/journal.pone.0001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li A, Walling J, Ahn S, Kotliarov Y, Su Q, Quezado M, Oberholtzer JC, Park J, Zenklusen JC, Fine HA. Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Res. 2009;69:2091–2099. doi: 10.1158/0008-5472.CAN-08-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wren JD. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics. 2009;25:1694–1701. doi: 10.1093/bioinformatics/btp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Srivastava GP, Qiu J, Xu D. Genome-wide functional annotation by integrating multiple microarray datasets using meta-analysis. Int. J. Data Min. Bioinform. 2010;4:357–376. doi: 10.1504/ijdmb.2010.034194. [DOI] [PubMed] [Google Scholar]
- 68.Tamayo P, Scanfeld D, Ebert BL, Gillette MA, Roberts CW, Mesirov JP. Metagene projection for cross-platform, cross-species characterization of global transcriptional states. Proc. Natl Acad. Sci. USA. 2007;104:5959–5964. doi: 10.1073/pnas.0701068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pennings JL, Kimman TG, Janssen R. Identification of a common gene expression response in different lung inflammatory diseases in rodents and macaques. PLoS One. 2008;3:e2596. doi: 10.1371/journal.pone.0002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pyne S, Gutman R, Kim CS, Futcher B. Phase Coupled Meta-analysis: sensitive detection of oscillations in cell cycle gene expression, as applied to fission yeast. BMC Genomics. 2009;10:440. doi: 10.1186/1471-2164-10-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fan X, Pyne S, Liu JS. Bayesian meta-analysis for identifying periodically expressed genes in fission yeast cell cycle. Ann. Appl. Stat. 2010;4:988–1013. [Google Scholar]
- 72.Lu Y, Rosenfeld R, Bar-Joseph Z. Identifying cycling genes by combining sequence homology and expression data. Bioinformatics. 2006;22:e314–322. doi: 10.1093/bioinformatics/btl229. [DOI] [PubMed] [Google Scholar]
- 73.Saviozzi S, Cordero F, Lo Iacono M, Novello S, Scagliotti GV, Calogero RA. Selection of suitable reference genes for accurate normalization of gene expression profile studies in non-small cell lung cancer. BMC Cancer. 2006;6:200. doi: 10.1186/1471-2407-6-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Frericks M, Esser C. A toolbox of novel murine house-keeping genes identified by meta-analysis of large scale gene expression profiles. Biochim. Biophys. Acta. 2008;1779:830–837. doi: 10.1016/j.bbagrm.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 75.Byun J, Logothetis CJ, Gorlov IP. Housekeeping genes in prostate tumorigenesis. Int. J. Cancer. 2009;125:2603–2608. doi: 10.1002/ijc.24680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hao P, Zheng S, Ping J, Tu K, Gieger C, Wang-Sattler R, Zhong Y, Li Y. Human gene expression sensitivity according to large scale meta-analysis. BMC Bioinformatics. 2009;10(Suppl. 1):S56. doi: 10.1186/1471-2105-10-S1-S56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morgan AA, Dudley JT, Deshpande T, Butte AJ. Dynamism in gene expression across multiple studies. Physiol. Genomics. 2010;40:128–140. doi: 10.1152/physiolgenomics.90403.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kapushesky M, Emam I, Holloway E, Kurnosov P, Zorin A, Malone J, Rustici G, Williams E, Parkinson H, Brazma A. Gene expression atlas at the European bioinformatics institute. Nucleic Acids Res. 2010;38:D690–698. doi: 10.1093/nar/gkp936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zimmermann P, Laule O, Schmitz J, Hruz T, Bleuler S, Gruissem W. Genevestigator transcriptome meta-analysis and biomarker search using rice and barley gene expression databases. Mol. Plant. 2008;1:851–857. doi: 10.1093/mp/ssn048. [DOI] [PubMed] [Google Scholar]
- 80.Fang H, Harris SC, Su Z, Chen M, Qian F, Shi L, Perkins R, Tong W. ArrayTrack: an FDA and public genomic tool. Methods Mol. Biol. 2009;563:379–398. doi: 10.1007/978-1-60761-175-2_20. [DOI] [PubMed] [Google Scholar]
- 81.Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cahan P, Ahmad AM, Burke H, Fu S, Lai Y, Florea L, Dharker N, Kobrinski T, Kale P, McCaffrey TA. List of lists-annotated (LOLA): a database for annotation and comparison of published microarray gene lists. Gene. 2005;360:78–82. doi: 10.1016/j.gene.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 83.Newman JC, Weiner AM. L2L: a simple tool for discovering the hidden significance in microarray expression data. Genome Biol. 2005;6:R81. doi: 10.1186/gb-2005-6-9-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bisognin A, Coppe A, Ferrari F, Risso D, Romualdi C, Bicciato S, Bortoluzzi S. A-MADMAN: annotation-based microarray data meta-analysis tool. BMC Bioinformatics. 2009;10:201. doi: 10.1186/1471-2105-10-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med. Genomics. 2009;2:18. doi: 10.1186/1755-8794-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tomlinson C, Thimma M, Alexandrakis S, Castillo T, Dennis JL, Brooks A, Bradley T, Turnbull C, Blaveri E, Barton G, et al. MiMiR—an integrated platform for microarray data sharing, mining and analysis. BMC Bioinformatics. 2008;9:379. doi: 10.1186/1471-2105-9-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ivliev AE, t Hoen PA, Villerius MP, den Dunnen JT, Brandt BW. Microarray retriever: a web-based tool for searching and large scale retrieval of public microarray data. Nucleic Acids Res. 2008;36:W327–W331. doi: 10.1093/nar/gkn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lopez F, Textoris J, Bergon A, Didier G, Remy E, Granjeaud S, Imbert J, Nguyen C, Puthier D. TranscriptomeBrowser: a powerful and flexible toolbox to explore productively the transcriptional landscape of the Gene Expression Omnibus database. PLoS One. 2008;3:e4001. doi: 10.1371/journal.pone.0004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng WC, Tsai ML, Chang CW, Huang CL, Chen CR, Shu WY, Lee YS, Wang TH, Hong JH, Li CY, et al. Microarray meta-analysis database (M(2)DB): a uniformly pre-processed, quality controlled, and manually curated human clinical microarray database. BMC Bioinformatics. 2010;11:421. doi: 10.1186/1471-2105-11-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Z, Fenstermacher D. An Introduction to MAMA (Meta-Analysis of MicroArray data) System. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2005;7:7730–7733. doi: 10.1109/IEMBS.2005.1616304. [DOI] [PubMed] [Google Scholar]
- 91.Culhane AC, Schwarzl T, Sultana R, Picard KC, Picard SC, Lu TH, Franklin KR, French SJ, Papenhausen G, Correll M, et al. GeneSigDB—a curated database of gene expression signatures. Nucleic Acids Res. 2009;38:D716–D725. doi: 10.1093/nar/gkp1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, et al. AGEMAP: a gene expression database for aging in mice. PLoS Genet. 2007;3:e201. doi: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pan F, Chiu CH, Pulapura S, Mehan MR, Nunez-Iglesias J, Zhang K, Kamath K, Waterman MS, Finch CE, Zhou XJ. Gene Aging Nexus: a web database and data mining platform for microarray data on aging. Nucleic Acids Res. 2007;35:D756–D759. doi: 10.1093/nar/gkl798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chelala C, Hahn SA, Whiteman HJ, Barry S, Hariharan D, Radon TP, Lemoine NR, Crnogorac-Jurcevic T. Pancreatic Expression database: a generic model for the organization, integration and mining of complex cancer datasets. BMC Genomics. 2007;8:439. doi: 10.1186/1471-2164-8-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Obayashi T, Hayashi S, Shibaoka M, Saeki M, Ohta H, Kinoshita K. COXPRESdb: a database of coexpressed gene networks in mammals. Nucleic Acids Res. 2008;36:D77–D82. doi: 10.1093/nar/gkm840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kolpakov F, Poroikov V, Sharipov R, Kondrakhin Y, Zakharov A, Lagunin A, Milanesi L, Kel A. CYCLONET—an integrated database on cell cycle regulation and carcinogenesis. Nucleic Acids Res. 2007;35:D550–D556. doi: 10.1093/nar/gkl912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang J, Finney RP, Rowe W, Edmonson M, Yang SH, Dracheva T, Jen J, Struewing JP, Buetow KH. Systematic analysis of genetic alterations in tumors using Cancer Genome WorkBench (CGWB) Genome Res. 2007;17:1111–1117. doi: 10.1101/gr.5963407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Choi H, Shen R, Chinnaiyan AM, Ghosh D. A latent variable approach for meta-analysis of gene expression data from multiple microarray experiments. BMC Bioinformatics. 2007;8:364. doi: 10.1186/1471-2105-8-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lottaz C, Yang X, Scheid S, Spang R. OrderedList—a bioconductor package for detecting similarity in ordered gene lists. Bioinformatics. 2006;22:2315–2316. doi: 10.1093/bioinformatics/btl385. [DOI] [PubMed] [Google Scholar]
- 101.Marot G, Mayer CD. Sequential analysis for microarray data based on sensitivity and meta-analysis. Stat. Appl. Genet. Mol. Biol. 2009;8 doi: 10.2202/1544-6115.1368. Article 3. [DOI] [PubMed] [Google Scholar]
- 102.Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22:2825–2827. doi: 10.1093/bioinformatics/btl476. [DOI] [PubMed] [Google Scholar]
- 103.Pihur V, Datta S. RankAggreg, an R package for weighted rank aggregation. BMC Bioinformatics. 2009;10:62. doi: 10.1186/1471-2105-10-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen Z, Wang W, Ling XB, Liu JJ, Chen L. GO-Diff: mining functional differentiation between EST-based transcriptomes. BMC Bioinformatics. 2006;7:72. doi: 10.1186/1471-2105-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pan F, Kamath K, Zhang K, Pulapura S, Achar A, Nunez-Iglesias J, Huang Y, Yan X, Han J, Hu H, et al. Integrative Array Analyzer: a software package for analysis of cross-platform and cross-species microarray data. Bioinformatics. 2006;22:1665–1667. doi: 10.1093/bioinformatics/btl163. [DOI] [PubMed] [Google Scholar]
- 106.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 39:D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Knuth DE. Literate Programming. Comput. J. 1984;27:97–111. [Google Scholar]
- 108.Ramasamy A, Mondry A, Holmes CC, Altman DG. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008;5:e184. doi: 10.1371/journal.pmed.0050184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62:4427–4433. [PubMed] [Google Scholar]
- 110.Stouffer SA, Suchman EA, DeVinnery L, Star S, Williams RM., Jr . The American Soldier, Volume I: Adjustement during Army Life. Princeton, NJ: Princeton University Press; 1949. [Google Scholar]
- 111.Li J, Tseng GC. An adaptively weighted statistic for detecting differential gene expression when combining multiple transcriptomic studies. Ann. App. Stat. 2011;5:994–1019. [Google Scholar]
- 112.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl Acad. Sci. USA. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Friedman L. Why vote-count reviews don't count. Biol. Psychiatry. 2001;49:161–162. [Google Scholar]
- 114.Hedges L, Olkin I. Vote-counting methods in research synthesis. Psychol. Bull. 1980;88:359–369. [Google Scholar]
- 115.Choi JK, Yu U, Kim S, Yoo OJ. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19(Suppl. 1):i84–i90. doi: 10.1093/bioinformatics/btg1010. [DOI] [PubMed] [Google Scholar]
- 116.Marot G, Foulley JL, Mayer CD, Jaffrezic F. Moderated effect size and P-value combinations for microarray meta-analyses. Bioinformatics. 2009;25:2692–2699. doi: 10.1093/bioinformatics/btp444. [DOI] [PubMed] [Google Scholar]
- 117.Hu P, Greenwood CM, Beyene J. Integrative analysis of multiple gene expression profiles with quality-adjusted effect size models. BMC Bioinformatics. 2005;6:128. doi: 10.1186/1471-2105-6-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Conlon EM, Song JJ, Liu A. Bayesian meta-analysis models for microarray data: a comparative study. BMC Bioinformatics. 2007;8:80. doi: 10.1186/1471-2105-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zintzaras E, Ioannidis JP. Meta-analysis for ranked discovery datasets: theoretical framework and empirical demonstration for microarrays. Comput. Biol. Chem. 2008;32:38–46. doi: 10.1016/j.compbiolchem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 120.Dreyfuss JM, Johnson MD, Park PJ. Meta-analysis of glioblastoma multiforme versus anaplastic astrocytoma identifies robust gene markers. Mol. Cancer. 2009;8:71. doi: 10.1186/1476-4598-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hong F, Breitling R. A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics. 2008;24:374–382. doi: 10.1093/bioinformatics/btm620. [DOI] [PubMed] [Google Scholar]
- 122.DeConde RP, Hawley S, Falcon S, Clegg N, Knudsen B, Etzioni R. Combining results of microarray experiments: a rank aggregation approach. Stat. Appl. Genet. Mol. Biol. 2006;5:Article15. doi: 10.2202/1544-6115.1204. [DOI] [PubMed] [Google Scholar]
- 123.Sims AH, Smethurst GJ, Hey Y, Okoniewski MJ, Pepper SD, Howell A, Miller CJ, Clarke RB. The removal of multiplicative, systematic bias allows integration of breast cancer gene expression datasets—improving meta-analysis and prediction of prognosis. BMC Med. Genomics. 2008;1:42. doi: 10.1186/1755-8794-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Goldstein DR, Delorenzi M, Luthi-Carter R, Sengstag T. Meta-Analysis and Combining Informationi in Genetics and Genomics. In: Guerra R, Goldstein DR, editors. Florence, KY: Chapman & Hall/CRC; 2010. pp. 135–156. [Google Scholar]
- 126.Scharpf RB, Tjelmeland H, Parmigiani G, Nobel AB. A Bayesian model for cross-study differential gene expression. J. Am. Stat. Assoc. 2009;104:1295–1310. doi: 10.1198/jasa.2009.ap07611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Conlon EM, Song JJ, Liu JS. Bayesian models for pooling microarray studies with multiple sources of replications. BMC Bioinformatics. 2006;7:247. doi: 10.1186/1471-2105-7-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shen R, Ghosh D, Chinnaiyan AM. Prognostic meta-signature of breast cancer developed by two-stage mixture modeling of microarray data. BMC Genomics. 2004;5:94. doi: 10.1186/1471-2164-5-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Birnbaum A. Combining independent tests of significance. J. Am. Stat. Assoc. 1954;49:559–574. [Google Scholar]
- 130.Owen AB. Karl Pearson's meta-analysis revisited. Ann. Stat. 2009;37:3867–3892. [Google Scholar]
- 131.Phipson B, Smyth GK. Permutation P-values should never be zero: calculating exact P-values when permutations are randomly drawn. Stat. Appl. Genet. Mol. Biol. 2010;9 doi: 10.2202/1544-6115.1585. Article 39. [DOI] [PubMed] [Google Scholar]
- 132.Assinder SJ, Au E, Dong Q, Winnick C. A novel splice variant of the beta-tropomyosin (TPM2) gene in prostate cancer. Mol. Carcinog. 2010;49:525–531. doi: 10.1002/mc.20626. [DOI] [PubMed] [Google Scholar]
- 133.Grutzmann R, Boriss H, Ammerpohl O, Luttges J, Kalthoff H, Schackert HK, Kloppel G, Saeger HD, Pilarsky C. Meta-analysis of microarray data on pancreatic cancer defines a set of commonly dysregulated genes. Oncogene. 2005;24:5079–5088. doi: 10.1038/sj.onc.1208696. [DOI] [PubMed] [Google Scholar]
- 134.Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, Desmedt C, Ignatiadis M, Sengstag T, Schutz F, et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10:R65. doi: 10.1186/bcr2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Normand SL. Meta-analysis: formulating, evaluating, combining, and reporting. Stat. Med. 1999;18:321–359. doi: 10.1002/(sici)1097-0258(19990215)18:3<321::aid-sim28>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 136.Campain A, Yang YH. Comparison study of microarray meta-analysis methods. BMC Bioinformatics. 11:408. doi: 10.1186/1471-2105-11-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bushman BJ, Wang MC. The Handbook of Research Synthesis and Meta-analysis. 2nd edn. New York: Russell Sage Foundation; 2009. [Google Scholar]
- 138.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–1735. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.