


Abstract
Special AT-rich sequence-binding protein 1 (SATB1) is a global chromatin organizer and gene expression regulator essential for T-cell development and breast cancer tumor growth and metastasis. The oligomerization of the N-terminal domain of SATB1 is critical for its biological function. We determined the crystal structure of the N-terminal domain of SATB1. Surprisingly, this domain resembles a ubiquitin domain instead of the previously proposed PDZ domain. Our results also reveal that SATB1 can form a tetramer through its N-terminal domain. The tetramerization of SATB1 plays an essential role in its binding to highly specialized DNA sequences. Furthermore, isothermal titration calorimetry results indicate that the SATB1 tetramer can bind simultaneously to two DNA targets. Based on these results, we propose a molecular model whereby SATB1 regulates the expression of multiple genes both locally and at a distance.
INTRODUCTION
Global gene regulation is essential for cell differentiation and maturation. Local and long-range coordinated gene regulations provide flexibility for the coregulation and modulation of related genes to meet various biological requirements during development or tumors growth and metastasis (1). The chromatin architecture in the nucleus plays an important role in regulating gene expression (2). The sequences of the nuclear matrix attachment regions (MARs), also referred to as base-unpairing regions (BURs), are generally 70% AT rich (3). The unwinding property of BURs has been shown to be essential for binding to the nuclear matrix and enhancing promoter activity (4). Special AT-rich sequence-binding protein 1 (SATB1) binds to the core unwinding elements of BURs and recruits various chromatin remodeling/modifying enzymes to regulate gene expression by directly influencing various gene promoter activities (5–8). Strikingly, SATB1 is also able to regulate gene expression at long distances, up to several hundred kb (9–11). A recent report showed that SATB1 is aberrantly expressed in human metastatic breast cancer and coordinately regulates the expression of sets of genes that promote breast cancer tumor growth and metastasis (12).
SATB1 was initially identified as a cell type-specific MAR DNA-binding protein, predominantly expressed in the thymus (3). SATB1 consists of an N-terminal domain, a C-terminal homeodomain (HD) and tandem CUT domains in the center (Figure 1A). The sequence-specific binding of SATB1 to its DNA targets is mediated by the HD and CUT tandem domains (13,14) and requires oligomerization of the N-terminal domain (15,16). In addition, chemical interference assays suggested a rapid association and dissociation kinetics of DNA binding by SATB1, and the dissociation rate (Koff) for the multiple AT-rich-containing DNA fragments (such as seven repeats) is slower than the less AT-rich-containing DNA fragments (such as two repeats) (3). In addition to its DNA-binding ability, SATB1 also acts as a ‘docking site’ for various chromatin remodeling/modifying enzymes and transcription factors (5,7,9,11,17). The transcriptional activity of SATB1 is regulated by several post-translational modifications such as phosphorylation (18), acetylation (7,18) and sumoylation (19). Taken together, these studies demonstrate that SATB1 acts as a linker between DNA loop organization, chromatin modification/remodeling and the association of transcription factors with MARs, and thus it functions as a ‘genome organizer’ that is essential for T-cell development (17,20).
Figure 1.

Structure of the SATB1 ULD. (A) Schematic representation of the domain organization of mouse SATB1. The ULD domain boundary identified in this work is located from Gly71 to Ser172, and a novel CUTL domain is located from His186 to Lys244. The two mutants used in this study, KWN–AAA and EFH–AAA, were created by substituting the 136K137W138N and the 97E98F162H motifs with ‘AAA’ cassettes. (B) Cartoon representation of the overall structure of ULD. The N- and C-termini of the protein are labeled. (C) Stereo view showing the superimposed structures of ULD and ubiquitin (PDB code: 1UBI).
A structural study of the SATB1 CUT1 domain bound to DNA showed that the CUT1 domain binds to the major groove of DNA (21). However, the molecular mechanism of how SATB1 oligomerization regulates DNA binding has remained elusive. In this study, we present the crystal structure of the N-terminus of SATB1. Surprisingly, it resembles a ubiquitin domain instead of the previously identified PDZ domain. We also found that SATB1 can assemble into a tetramer, which provides insight into molecular basis of its ability to regulate gene expression at long distances.
MATERIALS AND METHODS
Expression and purification of various N-terminal fragments of SATB1
Various fragments (Supplementary Data and Supplementary Figure S1) of the mouse SATB1 gene were PCR amplified from a mouse thymus cDNA library and cloned into an in-house modified version of the pET32a (Novagen) vector and confirmed by DNA sequencing. The resulting protein contained a His6-tag in its N-terminus. All point mutations of SATB1 described here were created using the standard PCR-based mutagenesis method and confirmed by DNA sequencing. The recombined protein was expressed in BL21 (DE3) Escherichia coli cells at 16°C for 16–18 h. The His6-tagged protein was purified by Ni-NTA (QIAGEN) affinity chromatography followed by size exclusion chromatography on a HiLoad 26/60 Superdex 200 (GE Healthcare). After digestion with PreScission Protease to cleave the N-terminal His6-tag, the target protein was purified on a Mono Q 10/100 GL (GE Healthcare) anion-exchange column. The final purification step was size exclusion chromatography on a HiLoad 26/60 Superdex 200 column in 50 mM Tris pH 8.0, 50 mM NaCl, 1 mM EDTA and 1 mM DTT.
Se–Met-recombined protein was expressed in B834 (DE3) E. coli cells at 20°C for 20 h. The B834 (DE3) cells were cultured in LeMaster medium in which methionine was replaced by selenomethionine. The Se–Met substituted protein was purified as wild-type protein as described above.
Expression and purification of full-length protein
The outline for the expression and purification of full-length protein is summarized in Supplementary Figure S7. Briefly, SATB1 and its mutants were expressed in E. coli as a fusion protein with an N-terminal Trx-His6-tag and C-terminal His6-tag. The Trx-His6-tagged protein was purified by Ni-NTA affinity chromatography followed by size exclusion chromatography on a HiLoad 26/60 Superdex 200 column. After digestion with PreScission Protease to cleave the N-terminal Trx-His6-tag, the target protein was purified on a Mono Q 10/100 GL anion-exchange column. The protein was then denatured with 6 M guanidine hydrochloride and purified by Ni-NTA affinity chromatography under denaturing conditions by its C-terminal His6-tag. The eluted protein was refolded by extensively dialyzing out the denaturant in 50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA and 1 mM DTT at 4°C for 12 h at least three times. The refolded protein was further purified on a Mono Q 10/100 GL anion-exchange column followed by size exclusion chromatography on a HiLoad 26/60 Superdex 200 column in 50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA and 1 mM DTT.
Crystallization and data collection
The wild-type protein was crystallized using the sitting drop vapor diffusion method equilibrated against a reservoir solution of 15% polyethylene glycol 3350, 0.15 M ammonium citrate dibasic and 4% polypropylene glycol P400. Selenium-substituted protein was crystallized under similar conditions to the native protein, except that 4% polypropylene glycol P400 was replaced by 3% dextran sulfate. Crystals grew at 20°C and were frozen in a cryoprotectant solution consisting of the reservoir solution supplemented with 15% glycerol. All crystals belonged to the space group of P212121. The wild-type crystals diffracted to 1.7 Å with unit cell dimensions of a = 35.90 Å, b = 71.03 Å and c = 153.76 Å and Se–Met substituted crystals diffracted to 2.1 Å with unit cell dimensions of a = 36.14 Å, b = 71.23 Å and c = 154.23 Å. A native data set and a single anomalous dispersion (SAD) data set were collected at the peak wavelength for Se on station BL17U1 of the Shanghai Synchrotron Radiation Facility (SSRF). Both data sets were processed using HKL2000 software (22).
Structure determination and refinement
The program HKL2MAP (23) was used to search for 16 Se sites and the initial SAD phases were then calculated using PHENIX program (24). A model covering 60% of the protein molecule was automatically built into the SAD map using the PHENIX program (24), and additional residues were manually built into the electron density with the Coot program (25). Then, molecular replacement by the Phaser program (26) was performed to locate the exact position of four protein molecules in one asymmetric unit of the wild-type data. The final tetramer model was refined iteratively by the CNS (27) and the Coot programs (25). The orientations of the amino acid side chains and bound water molecules were modeled based on sigmaA weighted 2Fobs − Fcalc and Fobs − Fcalc Fourier electron density maps. The final structure had an Rcrystal value of 15.8% and an Rfree value of 21.8%. The statistics on the structure refinement are summarized in Table 1.
Table 1.
Data collection and refinement statistics for SATB1structures
| Crystal name | Wild-type | Se–Met–crystal |
|---|---|---|
| Space group | P212121 | P212121 |
| Unit cell (Å) | a = 35.90, b = 71.03, c = 153.76 | a = 36.14, b = 71.23, c = 154.23 |
| Wavelength (Å) | 0.9792 | 0.9795 (peak) |
| Resolution range (Å) | 26–1.70 (1.76–1.70)b | 30–2.14 (2.18–2.14)b |
| No. of unique reflections | 36 665 | 24 131 |
| Redundancy | 6.2 (2.4)b | 6.3 (3.6)b |
| Rsym (%)a | 6.6 (19.0)b | 11.9 (32.0)b |
| I/σ | 24.7 (3.0)b | 22.5 (2.6)b |
| Completeness (%) | 95.4 (68.9)b | 98.6 (93.5)b |
| Figure of merit | – | 0.653 |
| Refinement | ||
| Rcrystal (%)c | 15.8 | – |
| Rfree (%)d | 21.8 | – |
| RMSDbond (Å) | 0.005 | – |
| RMSDangle (°) | 0.9 | – |
| Number of | ||
| Protein atoms | 2983 | – |
| Ligand atoms | 0 | – |
| Solvent atoms | 296 | – |
| Residues in (%) | ||
| Most favored | 95.3 | – |
| Additional allowed | 4.4 | – |
| Generously allowed | 0.3 | – |
| Disallowed | 0 | – |
| Average B factor (Å2) of | ||
| chain A | 31.3 | – |
| chain B | 31.8 | – |
| chain C | 31.7 | – |
| chain D | 33.8 | – |
| solvent | 47.2 | – |
aRsym = 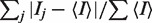 where
where 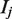 is the intensity of the jth reflection and
is the intensity of the jth reflection and 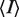 is the average intensity.
is the average intensity.
bthe highest resolution shell.
cRcrystal = 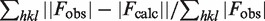 .
.
dRfree, calculated the same as Rcrystal, but from a test set containing 5% of data excluded from the refinement calculation.
Analytical ultracentrifugation
Sedimentation velocity (SV) and sedimentation equilibrium (SE) experiments were performed in a Beckman/Coulter XL-I analytical ultracentrifuge using double-sector or six-channel centerpieces and sapphirine windows. An additional protein purification step on a HiLoad 26/60 Superdex 200 size exclusion column in 50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA and 1 mM TCEP was performed before the experiments. SV experiments were conducted at 50 000 rpm and 20°C using absorbance detection and double-sector cells loaded with ∼80 μM for ubiquitin-like domain (ULD). For the full-length protein, SV experiments were conducted at 40 000 rpm and 4°C with 7.5 and 15 μM protein. For the SE experiment, data were collected at 20°C and 18 000 rpm with ∼16, 24 and 40 μM ULD, at 4°C and 4300, 6400 and 8000 rpm with 2.9, 4.4 and 7.3 µM wild-type full-length SATB1 and at 4°C and 6000, 8000, 10 000 and 12 000 rpm with 3.3, 4.9 and 8.2 μM KWN–AAA mutant, respectively. The buffer composition (density and viscosity) and protein partial specific volume (V-bar) were obtained using the program SEDNTERP (http://www.rasmb.bbri.org/). The SV and SE data were analyzed using the programs SEDFIT and SEDPHAT (28,29).
Electrophoretic mobility shift assay
The forward and reverse oligonucleotides for a particular set were mixed together. To anneal the labeled oligonucleotides, the mixtures were heated to 95°C for 10 min and allowed to cool slowly at room temperature. Annealed oligonucleotides were end labeled with γ-32P ATP using T4 polynucleotide kinase (New England Biolabs). Binding reactions were performed in a 10 μl total volume containing 10 mM Tris pH 7.5, 1 mM DTT, 50 mM KCl, 5 mM MgCl2, 2.5% glycerol, 0.05% NP-40 and the appropriate amount of annealed oligonucleotides and recombinant proteins. Samples were incubated on ice for 1 h and separated by electrophoresis on a 5% native polyacrylamide gel (PAGE). The gels were dried under vacuum and exposed to X-ray film. The radioactive intensities of protein-bound DNA bands and free DNA probe reduction due to protein binding were calculated with Adobe Photoshop CS4 and normalized to that of the free DNA probe.
Analytical gel filtration
Size exclusion chromatography was performed on an AKTA FPLC system using a Superose 12 10/300 column (GE Healthcare) for ULD or Superdex 200 10/300 column (GE Healthcare) for full-length SATB1. Protein samples were dissolved in buffer containing 50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA and 1 mM DTT. The column was calibrated with a gel filtration standard from Bio-Rad.
Circular dichroism
Circular dichroism (CD) spectra of various proteins were collected on a MOS450 spectropolarimeter (BioLogic) at room temperature. The protein samples (∼6 μM) were dissolved in 50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA and 1 mM DTT.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) measurements were performed on a MicroCal™ Isothermal Titration Calorimeter iTC200 (GE Healthcare) in 20 mM PBS pH 7.2 and 50 mM NaCl. For wild-type SATB1, ∼232 μM of DNA probe was titrated into 11.7 μM of protein. For the KWN-AAA mutant, 239 μM of DNA probe was titrated into 16 μM of protein. The titration consisted of an initial injection of 0.4 μl followed by 26 injections of 1.5 μl every 120 s at 15°C. To determine the baseline, the DNA probe was titrated into the same buffer without protein under the same conditions. The titration data and binding plot after baseline subtraction were analyzed with the MicroCal Origin software with the two-site and one-site-binding models for wild-type SATB1 and the KWN–AAA mutant, respectively.
Chemical cross-linking assay
Chemical cross-linking assay was carried out by incubating (1–263)AA or SATB1 proteins with Lys-specific cross-linker disuccinimidyl glutarate (DSG, Thermo Pierce). The reaction was carried out in 20 mM Hepes buffer (pH 7.5) containing various concentrations of NaCl and glycerol with a protein concentration of 0.5 mg/ml for 15 min on ice. The concentration of DSG was adjusted to be 5 M equivalent of total Lys concentration of each protein. The cross-linking reactions were quenched by addition of 50 mM Tris to the reaction solution. GST and MBP were served as positive and negative control, respectively.
RESULTS
Overall structure of the ULD domain of SATB1
To identify a stable region of the N-terminus of SATB1 for crystallization, several constructs were designed and tested for protein expression and purification. Among them, seven purified proteins formed tetramers, as determined by analytical ultracentrifugation SV experiments (Supplementary Figure S1). After extensive crystal screening, high-quality crystals were obtained only with the construct containing residues 71–172. The crystal structure of residues 71–172 of SATB1 was solved by SAD at a resolution of 1.8 Å (Table 1). Strikingly, the overall structure of this region contains four antiparallel β-sheets (β1–β4) flanked by four α-helices (α1–α4) (Figure 1B), and does not show any similarity to the previously defined PDZ domain (15). Instead, it resembles a typical ubiquitin domain. The superposition of the N-terminal domain of SATB1 onto the classic ubiquitin domain results in a root-mean-square deviation of 2.7 Å for the 67 Cα-atoms (Figure 1C), even though the two proteins share no sequence homology. Therefore, we renamed this domain a ULD (Figure 1A).
Prior studies have proposed that residues 90–204 of SATB1 form a PDZ-like domain based on primary sequence alignment (15). So far, two structures of residues 179–250 of SATB1 have been deposited in the Protein Data Bank with accession codes 3NZL and 2L1P. Interestingly, the structure of residues 179–250 resembles the CUT1 domain of SATB1, and this region was designated a CUT1-like (CUTL) domain (Supplementary Figure S2 and Figure 1A). Therefore, according to current structural studies, it appears that the N-terminus of SATB1 does not contain a PDZ domain. A sequence alignment of SATB family proteins (SATB1 and SATB2) across species shows that the residues of the ULD domain are all highly conserved (Supplementary Figure S3), indicating that the ULD domain we identified in this study may exist in all SATB family members and have similar biological functions.
ULD-mediated SATB1 tetramerization
Our crystal structure contains four ULD molecules, which seem to form a tightly packed tetramer, in one asymmetric unit (Figure 2A). Consistent with this observation, ULD was eluted as a single peak from a size exclusion column with a molecular mass corresponding to a tetramer (Supplementary Figure S4A). Analytical ultracentrifugation further confirmed that ULD assembles into a tetramer with a molecular mass of ∼43.5 kDa (Supplementary Figure S4B). These results demonstrate that ULD forms a homotetramer in solution.
Figure 2.

Interface of the SATB1 ULD homotetramer. (A) Ribbon diagram of a representative SATB1 ULD homotetramer. The four individual ULDs are colored red, cyan, green and yellow. Schematic diagrams showing the detailed interactions within the cyan–yellow dimer (B) and the green–yellow dimer (C). The color of the residues within each dimer interface is same as that of the individual ULDs in (A). Hydrogen bonds and hydrophobic interactions are shown as red and gray dotted lines, respectively.
In the structure of the ULD tetramer, two dimers (cyan–yellow and green–yellow) are formed with multiple hydrogen bonds and hydrophobic interactions within their interfaces (Figure 2B and C, Supplementary Figure S5). It has been noted that high concentrations of NaCl and glycerol disrupt neither the ULD tetramer nor ULD-mediated full-length SATB1 tetramer (Supplementary Figure S6). The buried surface area calculated by the program AREAIMOL (30) is 1343.6 and 1647.5 Å2 within the interface of the cyan–yellow and green–yellow dimers, respectively, indicating that both dimers could play a role in the oligomerization of full-length SATB1. The interactions within the interface of both dimers were analyzed in detail. These interactions display non-crystallographic 2-fold symmetry. Within the interface of the cyan–yellow dimer (Figure 2B and Supplementary Figure S5A), Glu97 from the cyan monomer makes several hydrogen bonds with Thr72 from the yellow monomer. Additionally, Phe98 and His162 from the cyan monomer form stacking interactions with Pro75 and His162 from the yellow monomer, respectively. Within the green–yellow interface (Figure 2C and Supplementary Figure S5B), three residues (Ile132, Val134 and Val145) from the green monomer form a hydrophobic pocket to accommodate the bulky side chain of Trp137 from the yellow monomer. Atom NE1 of the side chain of Trp137 from the green monomer forms a hydrogen bond with the main chain oxygen atom of Met156 from the yellow monomer. Asn138 and Asp159 from the green monomer make hydrogen bonds with Asp147 and Lys136 from the yellow monomer, respectively. All of the above interactions are also reciprocal. Therefore, the ‘97E98F162H’ motif (residue Glu97, Phe98 and His162) is presumably important for the formation of the cyan–yellow ULD dimer, and the ‘136K137W138N’ motif (residues 136–138) is important for the formation of the green–yellow ULD dimer. For the convenience of further discussion, the cyan–yellow ULD dimer interface (Figure 2B) and the green–yellow ULD dimer interface (Figure 2C) are referred to as the EFH and KWN interfaces, respectively.
To investigate the oligomerization of SATB1 in solution, we mutated residues within the two interfaces to test the ULD domain-mediated oligomerization state of full-length SATB1. The first mutant was designed to disrupt the EFH interface-mediated ULD dimer by replacing the 97E98F162H motif with an ‘AAA’ cassette, and the second mutant was designed to disrupt the KWN interface-mediated ULD dimer by substituting the 136K137W138N motif with an ‘AAA’ cassette (referred to as EFH–AAA and KWN–AAA, respectively) (Figure 1A). Wild-type SATB1 and the two mutants were purified to homogeneity by the protocol summarized in Supplementary Figure S7 (Figure 3A, inset) and analyzed by SV analysis. Wild-type SATB1 exhibited a narrow sedimentation coefficient distribution in continuous size distribution analysis (black lines in Figure 3A) (28), suggesting that it is mono-disperse, and it is estimated to be a stable tetramer. SE analysis of analytical ultracentrifugation further confirmed that wide-type full-length SATB1 assembled into a tetramer with a molecular mass of ∼344.4 kDa (Figure 3B and Supplementary Figures S8A–S8C). The KWN–AAA mutant also exhibited a mono-disperse sedimentation coefficient distribution (red lines in Figure 3A), suggesting that it is a dimer. The dimeric state of the KWN–AAA mutant in solution was confirmed by SE analysis, yielding a calculated molecular weight of ∼174.5 kDa (Figure 3B and Supplementary Figures S8D–S8F). In sharp contrast, the EFH–AAA mutant exhibited a wide range of sedimentation coefficients in the c(S) distribution (cyan lines in Figure 3A) indicating that it exists as multiple forms of oligomers including dimmers, tetramers and even high-order form. These data suggest that this mutation drastically interferes with the oligomerization of SATB1 and that the EFH–AAA mutant does not have a unique form of oligomers, which is consistent with its elution profile by size exclusion chromatography (Supplementary Figure S9). Taken together, we conclude that both dimers in the ULD tetramer contribute to the oligomerization of full-length SATB1.
Figure 3.

Oligomeric assembly of wild-type and SATB1 mutants measured by analytical ultracentrifugation. (A) c(S) distributions from SV runs for SATB1 (7.5 μM, dotted black line; 15 μM, solid black line), EFH–AAA (7.5 µM, dotted cyan line; 15 μM, solid cyan line) and KWN–AAA (7.5 μM, dotted red line; 15 μM, solid red line). Representative SE profiles for SATB1 (B) and KWN–AAA (C) derived from a global fit MW ∼344.4 kDa (1σ confidence interval 342.6–345.8) and ∼174.5 kDa (1σ confidence interval 174.1–174.9), indicating that wild-type SATB1 and the KWN–AAA mutant assemble into a tetramer and dimer in solution, respectively.
SATB1 oligomerization is required for DNA binding
SATB1 has been shown to bind to AT-rich DNA of BURs through its C-terminal tandem CUT domains and HD (13,14) and lost its DNA binding activity after deletion the N-terminal 248 amino acids of SATB1 (15). We therefore investigated the role of ULD domain-mediated oligomerization in SATB1's DNA-binding activity. To accomplish this, we assessed the ability of wild-type SATB1 and the KWN–AAA and EFH–AAA mutants to bind a 37-bp DNA fragment known to bind wild-type SATB1 (Figure 4A) (6) via an electrophoretic mobility shift assay (EMSA). The DNA-binding affinity of wild-type SATB1 to the 37-bp DNA was stronger than that of the KWN–AAA and EFH–AAA mutants in dose-dependent EMSAs varying either the protein or the DNA probe concentration (Figure 4B and C). Thus, our data indicate that ULD-mediated oligomerization of SATB1 is required for binding to target DNA.
Figure 4.

DNA binding affinity of wild-type and SATB1 mutants measured by EMSA and ITC. (A) Nucleotide sequence of the interleukin-2 (IL-2) promoter region spanning base pairs −447 to −441 from the translation site. (B and C) EMSA assay. The binding of SATB1 and its mutants to a target DNA probe was tested by EMSA using the radiolabeled synthetic duplex oligonucleotides shown in (A). The EMSA was carried out with various protein (B, top) or probe concentrations (C, top). Bar graph of DNA binding affinity from the dose-dependent EMSA of protein (B, bottom) or DNA probe (C, bottom). The radioactive intensities of protein-bound DNA bands and free DNA probe reduction due to protein binding were calculated with Adobe Photoshop CS4 and normalized to that of the free DNA probe. The error bars indicate the standard error mean (n = 3 separate experiments). *P < 0.05; **P-value < 0.01; ***P < 0.005. The dissociation constants (Kd's) of SATB1 (D) and the KWN–AAA mutant (E) with IL-2 37-bp DNA shown in (A) were measured by ITC. The Kd’s were calculated by a two-site binding model for wild-type SATB1 to give Kd1 of ∼0.36 µM and Kd2 of ∼1.99 µM. For the KWN–AAA mutant, the data were fit to a one-site binding model to give a Kd of ∼1.95 µM.
To quantitatively study the binding of wild-type and mutant SATB1 to target DNA, we used ITC. The binding of wild-type SATB1 to DNA fit well to a two-site binding model with calculated Kd values of ∼0.36 and ∼1.99 µM, indicating that the SATB1 tetramer is capable of binding to two DNA fragments simultaneously (Figure 4D). However, the binding of the KWN–AAA mutant to target DNA fit well to a one-site binding model with a Kd of ∼1.95 µM (Figure 4E). The EFH–AAA mutant was not amenable to analysis by ITC because it exists as multiple forms of oligomers (Figure 3A and Supplementary Figure S9). These ITC results indicate that the wild-type SATB1 can possibly bind to two DNA segments in an allosteric interaction with one comparable and another 5-fold increase binding affinity comparing to the KWN–AAA mutant binding to DNA, which is consistent with the observation in EMSA assays that the wild-type SATB1 binds to the target DNA stronger than the KWN–AAA mutant does.
DISCUSSION
Oligomerization of SATB1 plays a very important role in DNA binding and has been implicated in gene regulation by SATB1. The structure of the SATB1 ULD domain reported here provides the molecular basis for how the ULD domain mediated the oligomerization state of SATB1. SATB1 assembles into a tetramer in vitro (Figure 5A), and the tetramerization of SATB1 is essential for recognizing specific DNA sequences (such as multiple AT-rich DNA fragments). Thus, SATB1 may regulate gene expression directly by binding to various promoters and upstream regions and thereby influencing promoter activity (Figure 5B). This local gene regulation model is consistent with experimental observations that SATB1 directly regulates the expression of a number of genes, including globin, interleukin-2, interleukin-2 receptor α and interleukin-5, by recruiting either coactivators or corepressors (5–8). Furthermore, we showed that the SATB1 tetramer can simultaneously bind to two DNA segments, and thus the tetramerization of SATB1 may organize high-order chromatin architecture by anchoring specialized DNA sequences in close proximity and recruiting various chromatin remodeling factors to coordinately regulate gene expression over long distances (Figure 5C). This long-range gene regulation model is also consistent with the observations that SATB1 regulates the coordinated expression of genes located both at the 200-kb T-helper 2 cytokine locus (10) and at the 300-kb major histocompatibility Class I locus (11), and that it reprograms chromatin organization and the transcriptional profiles of breast tumors to promote growth and metastasis (12,31,32).
Figure 5.

Model of SATB1-mediated transcriptional regulation. (A) Schematic showing SATB1 assembles into a tetramer by oligomerization of its N-terminal ULD domain. (B and C) Schematic representation of a possible model for SATB1 oligomer-mediated transcriptional regulation. The SATB1 dimer or tetramer may regulate gene expression by recognizing specific DNA sequences in the promoter regions of various genes (B), and the SATB1 tetramer may organize higher-order chromatin architecture to coordinately regulate gene expression over long distances by anchoring specialized DNA sequences in close proximity and recruiting various chromatin remodeling factors (bottom).
Long-range gene regulation plays a role in many biological activities such as regulation of the β-globin locus (33), cytokine gene cluster (34), estrogen-induced gene expression (35) and mating-type switching in yeast (36). Moreover, it has been found that the CI protein of bacteriophage λ regulates gene expression over a long distance via cooperative binding of its oligomers to specific target DNA (37). Our proposed model for SATB1 oligomerization-mediated long-range gene regulation is consistent with their finding and likely represents a general mechanism for spatiotemporal and quantitative regulation of gene expression.
Any assembly of a functional protein complex in a living cell must be dynamically regulated. The oligomeric assembly of SATB1 is not an exception. The mechanism that regulates the dynamic assembly of SATB1 tetramers remains unclear. Understanding the dynamic regulatory assembly mechanism of SATB1 (or SATB2) is an important area of future research. SATB1 is known to be acetylated by P300/CBP-associated factor at residue Lys136, located just within the 136K137W138N motif, which is important for SATB1 tetramerization and to mediate gene regulation in coordination with C-terminal-binding protein 1 during Wnt signaling in T cells (7). The identification of key residues (i.e. 97E98F162H and 136K137W138N motifs) in the assembly of SATB1 oligomers (and likely SATB2) should be helpful in designing mutants of SATB family proteins to evaluate their functional roles in living cells and/or animal models.
The results of EMSA and ITC indicate that the ULD-mediated SATB1 oligomerization can affect the DNA-binding affinity and stoichiometry for SATB1 (Figure 4). It is possible that, similar to DNA binding, the ULD-mediated SATB1 oligomerization may also affect the binding affinity and stoichiometry for its various protein-binding partners. Furthermore, the ITC data showed that SATB1 may allosterically bind to two DNA fragments, indicating that SATB1 binds to multiple AT-rich-containing DNA fragments with higher affinity by its tetramerization. In addition, a previous study suggested a rapid association and dissociation kinetics of DNA binding by SATB1 (3). Whether the oligomerization of SATB1 influencing the kinetic constants of DNA-binding by SATB1 needs to be investigated in the future study by other technique such as surface plasmon resonance.
Although the N-terminus of SATB1 (residues 90–204) was previously identified as a PDZ domain (15), structural studies have shown that this region is made up of ULD and CUTL domains. Detailed sequence alignments show that CUTL has the evolutionarily conserved amino acids involved in CUT1 DNA binding (Supplementary Figures S2B and S3). It would be interesting to investigate the functional role of the CUTL domain of SATB1 in future studies.
ACCESSION NUMBER
The atomic coordinates and structure factors for the structure of SATB1 ULD have been deposited in the Protein Data Bank with accession code 3TUO.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–9.
FUNDING
973 Program (grant 2009CB825504); National Natural Science Foundation of China (grants 31100527, 31140029 and 31170684); Tianjin Basic Research program (grants 08QTPTJC28200 and 08SYSYTC00200); Fundamental Research Funds for the Central Universities (grants 65011621 and 65020241). Funding for open access charge: grant 2009CB825504.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dr Lingyi Chen and Miss Lipin Ma for help with the EMSA experiments and Mr Wentao Diao for his technical help in CD experiments. We are also grateful to the staff at the beamline BL17U1 of the SSRF and at the beamline 3W1A of the Beijing Synchrotron Radiation Facility for excellent technical assistance during data collection.
REFERENCES
- 1.Kleinjan DA, van Heyningen V. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spector DL. The dynamics of chromosome organization and gene regulation. Annu. Rev. Biochem. 2003;72:573–608. doi: 10.1146/annurev.biochem.72.121801.161724. [DOI] [PubMed] [Google Scholar]
- 3.Dickinson LA, Joh T, Kohwi Y, Kohwi-Shigematsu T. A tissue-specific MAR/SAR DNA-binding protein with unusual binding site recognition. Cell. 1992;70:631–645. doi: 10.1016/0092-8674(92)90432-c. [DOI] [PubMed] [Google Scholar]
- 4.Bode J, Kohwi Y, Dickinson L, Joh T, Klehr D, Mielke C, Kohwi-Shigematsu T. Biological significance of unwinding capability of nuclear matrix-associating DNAs. Science. 1992;255:195–197. doi: 10.1126/science.1553545. [DOI] [PubMed] [Google Scholar]
- 5.Wen J, Huang SM, Rogers H, Dickinson LA, Kohwi-Shigematsu T, Noguchi CT. SATB1 family protein expressed during early erythroid differentiation modifies globin gene expression. Blood. 2005;105:3330–3339. doi: 10.1182/blood-2004-08-2988. [DOI] [PubMed] [Google Scholar]
- 6.Kumar PP, Purbey PK, Ravi DS, Mitra D, Galande S. Displacement of SATB1-bound histone deacetylase 1 corepressor by the human immunodeficiency virus type 1 transactivator induces expression of interleukin-2 and its receptor in T cells. Mol. Cell. Biol. 2005;25:1620–1633. doi: 10.1128/MCB.25.5.1620-1633.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purbey PK, Singh S, Notani D, Kumar PP, Limaye AS, Galande S. Acetylation-dependent interaction of SATB1 and CtBP1 mediates transcriptional repression by SATB1. Mol. Cell. Biol. 2009;29:1321–1337. doi: 10.1128/MCB.00822-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahlfors H, Limaye A, Elo LL, Tuomela S, Burute M, Gottimukkala K, Notani D, Rasool O, Galande S, Lahesmaa R. SATB1 dictates expression of multiple genes including IL-5 involved in human T helper cell differentiation. Blood. 2010;116:1443–1453. doi: 10.1182/blood-2009-11-252205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasui D, Miyano M, Cai ST, Varga-WEisz P, Kohwi-Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature. 2002;419:641–645. doi: 10.1038/nature01084. [DOI] [PubMed] [Google Scholar]
- 10.Cai ST, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat. Genet. 2006;38:1278–1288. doi: 10.1038/ng1913. [DOI] [PubMed] [Google Scholar]
- 11.Kumar P, Bischof O, Purbey PK, Notani D, Urlaub H, Dejean A, Galande S. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat. Cell. Biol. 2007;9:45–56. doi: 10.1038/ncb1516. [DOI] [PubMed] [Google Scholar]
- 12.Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature. 2008;452:187–193. doi: 10.1038/nature06781. [DOI] [PubMed] [Google Scholar]
- 13.Dickinson LA, Dickinson CD, KohwiShigematsu T. An atypical homeodomain in SATB1 promotes specific recognition of the key structural element in a matrix attachment region. J. Biol. Chem. 1997;272:11463–11470. doi: 10.1074/jbc.272.17.11463. [DOI] [PubMed] [Google Scholar]
- 14.Nakagomi K, Kohwi Y, Dickinson LA, Kohwi-Shigematsu T. A novel DNA-binding motif in the nuclear matrix attachment DNA-binding protein SATB1. Mol. Cell. Biol. 1994;14:1852–1860. doi: 10.1128/mcb.14.3.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galande S, Dickinson LA, Mian IS, Sikorska M, Kohwi-Shigematsu T. SATB1 cleavage by caspase 6 disrupts PDZ domain-mediated dimerization, causing detachment from chromatin early in T-cell apoptosis. Mol. Cell. Biol. 2001;21:5591–5604. doi: 10.1128/MCB.21.16.5591-5604.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purbey PK, Singh S, Kumar PP, Mehta S, Ganesh KN, Mitra D, Galande S. PDZ domain-mediated dimerization and homeodomain-directed specificity are required for high-affinity DNA binding by SATB1. Nucleic Acids Res. 2008;36:2107–2122. doi: 10.1093/nar/gkm1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, Mehta S, Purbey PK, Joseph J, Galande S. Global regulator SATB1 recruits beta-Catenin and regulates T(H)2 differentiation in Wnt-dependent manner. Plos Biol. 2010;8:e1000296. doi: 10.1371/journal.pbio.1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavan KP, Purbey PK, Sinha CK, Notani D, Limaye A, Jayani RS, Galande S. Phosphorylation of SATB1, a global gene regulator, acts as a molecular switch regulating its transcriptional activity in vivo. Mol. Cell. 2006;22:231–243. doi: 10.1016/j.molcel.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Tan JAT, Sun YJ, Song J, Chen Y, Krontiris TG, Durrin LK. SUMO conjugation to the matrix attachment region-binding protein, special AT-rich sequence-binding protein-1 (SATB1), targets SATB1 to promyelocytic nuclear bodies where it undergoes caspase cleavage. J. Biol. Chem. 2008;283:18124–18134. doi: 10.1074/jbc.M800512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez JD, Yasui DH, Niida H, Joh T, Loh DY, Kohwi-Shigematsu T. The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Genes Dev. 2000;14:521–535. [PMC free article] [PubMed] [Google Scholar]
- 21.Yamasaki K, Akiba T, Yamasaki T, Harata K. Structural basis for recognition of the matrix attachment region of DNA by transcription factor SATB1. Nucleic Acids Res. 2007;35:5073–5084. doi: 10.1093/nar/gkm504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Pape T, Schneider TR. HKL2MAP: a graphical user interface for phasing with SHELX programs. J. Appl. Cryst. 2004;37:843–844. [Google Scholar]
- 24.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.McCoy A. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macro-molecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 28.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 30.Lee B, Richards FM. Interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 31.Iorns E, Hnatyszyn HJ, Seo P, Clarke J, Ward T, Lippman M. The role of SATB1 in breast cancer pathogenesis. J. Natl. Cancer Inst. 2010;102:1284–1296. doi: 10.1093/jnci/djq243. [DOI] [PubMed] [Google Scholar]
- 32.Kohwi-Shigematsu T, Han HJ, Russo J, Kohwi Y. Re: the role of SATB1 in breast cancer pathogenesis. J. Natl. Cancer Inst. 2010;102:1879–1880. doi: 10.1093/jnci/djq440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nat. Genet. 2002;32:623–626. doi: 10.1038/ng1051. [DOI] [PubMed] [Google Scholar]
- 34.Spilianakis CG, Flavell RA. Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat. Immunol. 2004;5:1017–1027. doi: 10.1038/ni1115. [DOI] [PubMed] [Google Scholar]
- 35.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao WL, Hestermann EV, Geistlinger TR, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Jia ST, Yamada T, Grewal SI. Heterochromatin regulates cell type-specific long-range chromatin interactions essential for directed recombination. Cell. 2004;119:469–480. doi: 10.1016/j.cell.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 37.Dodd IB, Shearwin KE, Perkins AJ, Burr T, Hochschild A, Egan JB. Cooperativity in long-range gene regulation by the lambda CI repressor. Genes Dev. 2004;18:344–354.36. doi: 10.1101/gad.1167904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.