


Abstract
Quantitative analyses of next-generation sequencing (NGS) data, such as the detection of copy number variations (CNVs), remain challenging. Current methods detect CNVs as changes in the depth of coverage along chromosomes. Technological or genomic variations in the depth of coverage thus lead to a high false discovery rate (FDR), even upon correction for GC content. In the context of association studies between CNVs and disease, a high FDR means many false CNVs, thereby decreasing the discovery power of the study after correction for multiple testing. We propose ‘Copy Number estimation by a Mixture Of PoissonS’ (cn.MOPS), a data processing pipeline for CNV detection in NGS data. In contrast to previous approaches, cn.MOPS incorporates modeling of depths of coverage across samples at each genomic position. Therefore, cn.MOPS is not affected by read count variations along chromosomes. Using a Bayesian approach, cn.MOPS decomposes variations in the depth of coverage across samples into integer copy numbers and noise by means of its mixture components and Poisson distributions, respectively. The noise estimate allows for reducing the FDR by filtering out detections having high noise that are likely to be false detections. We compared cn.MOPS with the five most popular methods for CNV detection in NGS data using four benchmark datasets: (i) simulated data, (ii) NGS data from a male HapMap individual with implanted CNVs from the X chromosome, (iii) data from HapMap individuals with known CNVs, (iv) high coverage data from the 1000 Genomes Project. cn.MOPS outperformed its five competitors in terms of precision (1–FDR) and recall for both gains and losses in all benchmark data sets. The software cn.MOPS is publicly available as an R package at http://www.bioinf.jku.at/software/cnmops/ and at Bioconductor.
INTRODUCTION
Next-generation sequencing (NGS) has evolved into an important technology for genotyping (1) and genome assembly (2). NGS has also been applied to transcriptomics (mRNA-Seq), where it revealed new splice variants and new transcripts (3). Despite these successes, quantitative analyses of NGS data, for instance, determination of the expression levels of genes, are still challenging (4,5). Estimation of DNA copy numbers is another important kind of quantitative analysis, in which local depths of coverage must be mapped to integer copy numbers. Copy number analysis by NGS has the following potential advantages compared with array-based techniques: (i) estimation of integer copy numbers from NGS data is more accurate for large copy numbers, since depths of coverage scale linearly with copy numbers (6). (ii) Breakpoints of copy number regions can be determined more precisely (7) because they do not rely on predefined probes. (iii) Allele-specific copy numbers may be estimated for observed alleles, while array-based techniques are restricted to predefined alleles. Allele-specific copy numbers are of interest because they allow for determining whether an allele is fully functional, which is important, for example, for the identification of mutations leading to cancer development (8).
In the following, we review existing methods for estimating DNA copy numbers in NGS data. These methods represent the depth of coverage either as read counts or as log read counts in an interval and can be classified into (a) approaches detecting read count deviations and (b) reference-based approaches. Class (a) methods detect CNVs as deviations of (log) read counts from an average (log) read count of a chromosome. Class (b) methods detect CNVs as intervals for which (log) read count ratios between a sample and a reference deviate from 1 (0). This reference can either be a designated control sample or a constructed average sample.
MOFDOC (‘MOdel Free Depth Of Coverage’) belongs to class (a) and has been used by Alkan et al. (6), Campbell et al. (9), Wang et al. (10), Bentley et al. (11) and Wheeler et al. (12) for NGS copy number detection and earlier by Bailey et al. (13) for whole-genome shotgun sequencing. The variant of MOFDOC, as introduced by Alkan et al. (6), first divides the genome into non-overlapping segments of equal length in which reads are counted. Note that some variants of MOFDOC are based on log read counts. Using the overall mean and standard deviation of those segment read counts, each segment is characterized by the multiple of the standard deviation by which its read count differs from the overall mean. A segmentation algorithm combines consecutive segments into a gain segment if they have read counts larger than three times the standard deviation above the mean; analogously, it combines segments into a loss segment if they have read counts smaller than two times the standard deviation below the mean (see blue boxes in Figure 2). GC correction is crucial for proper performance of MOFDOC because of the GC content bias of NGS (14). However, MOFDOC, like most class (a) methods, has a high false discovery rate (FDR), even upon GC correction. The reason is that mean segment read counts for copy number two may vary along the chromosome due to technological biases or local genomic characteristics. These read count variations along the chromosome appear consistently across samples, for example, all samples tend to have either larger or smaller read counts (see Supplementary Figure S11 and the third bar in Figure 2). Class (a) methods confound these variations with copy number changes leading to false discoveries.
Figure 2.

Illustration of the basic concept of cn.MOPS: a CNV call incorporates the detection of variation across samples (I/NI call) and the detection of variation along a chromosome (segmentation). Curves show read counts along one chromosome for five samples. I/NI calls (green) detect variation across samples (green vertical boxes). A CNV (red box) is called if consecutive segments have high I/NI calls. Blue boxes mark segments that segmentation algorithm of class (a) methods (see the ‘Introduction’ section) would combine into a CNV. First vertical bar (from the left) and first sample: the I/NI call indicates variation across samples (‘I/NI call +’). However, too few adjacent segments show high I/NI calls. Second bar and third sample: the I/NI call indicates variation across samples (‘I/NI call +’) and sufficiently many adjacent segments show high I/NI calls, which leads to a CNV call (red box). Third bar: the read counts drop consistently and would thus be detected by a segmentation algorithm of class (a) methods (blue boxes). However, the read counts of the samples do not vary, which does not lead to an I/NI call (‘I/NI call −’). A CNV is not detected, which is correct as the copy number does not vary across samples. Fourth bar and samples numbers 2 and 4: I/NI call indicates variation across samples (‘I/NI call +’). As in the first bar, too few adjacent segments show high I/NI calls. Fifth bar and second sample: a segmentation algorithm of class (a) methods would combine adjacent read counts that are consistently small (blue box) into a CNV. However, the read counts are within the variation of the constant copy number at this location. Therefore, the I/NI call does not indicate variation across samples (‘I/NI call −’).
EWT (‘Event-Wise Testing’), introduced by Yoon et al. (15), is identical to MOFDOC except for the final segmentation algorithm. EWT uses a probabilistic approach to join consecutive segments that, under a Gaussian assumption, show either significantly larger or significantly smaller read counts than the overall mean (see blue boxes in Figure 2). As for MOFDOC, read count variations along the chromosome lead to false CNVs (see Supplementary Figure S11).
JointSLM (16) also belongs to class (a), and extends the idea of EWT to a simultaneous segmentation of multiple samples. Again, the genome is divided into equally sized, non-overlapping segments for which the logarithm of GC-corrected and normalized (divided by the median per sample) read counts is computed. A hidden Markov model (HMM) slides along the chromosome and simultaneously scans the log-normalized read counts of all samples. The more samples show large or small read counts, the more likely a segment is detected (see blue boxes in Figure 2). JointSLM hardly detects CNVs that occur only in a few samples, because its HMM uses a single state variable for simultaneously explaining the copy numbers of all individuals (see Supplementary Figure S9). Furthermore, CNV regions may contain both gains and losses (17), which impedes JointSLM in detecting such regions, since samples have propensities to transit to different HMM hidden states. Like other class (a) methods, JointSLM detects spurious regions if they contain read counts that, due to genomic and technical biases, are smaller or larger than the chromosome average (see Supplementary Figure S11). Note that we consider JointSLM as a class (a) method because it detects simultaneous deviations of log read counts from an average log read count.
SeqSeg (7) is a class (b) method that was designed to identify copy number aberrations (CNAs) in tumor samples by comparing them to references, that is, their matched controls. SeqSeg evaluates the likelihood of each tumor read being a CNA breakpoint and keeps the most likely ones, thereby segmenting the chromosome. For each segment, the ratio between sample read counts and reference read counts is computed. Segments are called gains if their ratios are above 1.5, which corresponds to a copy number of at least 3, or losses if their ratios are below 0.5, which corresponds to a copy number of at most 1. Read count variations along the chromosome stemming, for instance, from the GC bias are implicitly corrected because these variations affect both the tumor sample and the reference in a similar way. SeqSeg relies on a single reference and does not consider local read count variations across replicates or multiple samples. Consequently, SeqSeg falsely detects CNVs in genomic regions where read counts of replicates are highly variable (see Supplementary Figure S12).
rSW-seq (18) improves SeqSeg with respect to breakpoint identification, but the local read count variability remains disregarded. Note that both methods, SeqSeg and rSW-seq, were designed for CNA detection in tumor samples, especially at the breakpoint identification step.
CNAseg (19) was also designed to detect CNAs in tumor samples, using an approach similar to that of SeqSeg. CNAseg, like JointSLM for a single sample, employs an HMM for joining equally sized, non-overlapping segments using the difference in segment read counts between tumor and reference. The resulting segments are joined on the basis of a χ2 statistic.
CNV-Seq (20) is another class (b) method. It also divides the genome into equally sized, non-overlapping segments for which read count ratios are computed using a reference sample. The read counts are assumed to follow a Poisson distribution, which is approximated by a Gaussian distribution. Subsequently, the Geary-Hinkley transformation is applied to the ratios of Gaussians to produce an approximately Gaussian output. In a final step, a segmentation algorithm joins consecutive segments with log ratios above or below a certain threshold. Like all other methods, CNV-Seq is prone to falsely detecting CNVs since it does not take local read count variability into account (see Supplementary Figure S12).
FREEC (‘control-FREE Copy number calling’) is a class (b) method suggested by Boeva et al. (21). FREEC also counts reads in equally sized, non-overlapping segments and computes read count ratios per segment using a reference sample. Hypothetical read counts estimated by a polynomial function of the segment's GC content can be used instead of a reference. In the segmentation step, the breakpoints are determined by LASSO (‘Least Absolute Shrinkage eStimatOr’) regression (22). FREEC does not consider local read count variability either, which makes it susceptible to falsely discovered CNVs (see Supplementary Figure S12).
In summary, existing methods suffer from a high FDR that results (i) from read count variations along the chromosome, especially if no references are used, and (ii) from variations in read counts (noisy counts) across samples that may occur even for constant copy numbers. The high FDR can be moderated for paired-end reads by confirming CNVs by means of clusters of discordant read pairs—incorrect orientation, order or distance (23). However, this approach may considerably decrease the discovery power since clusters may be missed, especially in cases of low coverage.
Below we introduce cn.MOPS, a CNV detection method together with a data processing pipeline which, in contrast to most previous methods, (i) provides integer copy numbers, (ii) estimates variations in read counts across samples and (iii) uses these estimates for CNV calling, thereby keeping the FDR low. A high FDR is particularly critical in association studies between CNVs and diseases: a high FDR implies many false CNVs. Correction for multiple testing must then consider these false discoveries, which increases the corrected P-values and reduces the discovery power of a study. The novelty of cn.MOPS is modeling across samples, which improves the performance considerably, as technical and biological variations are estimated and taken into account. By ‘modeling across samples’ we mean the construction of a generative model that explains how the data have been produced. This model decomposes the read count of each sample into signal and noise. The idea of modeling across samples has already improved CNV detection in microarray data by reducing the FDR, as the recent cn.FARMS method (24) demonstrates.
METHODS
The cn.MOPS processing pipeline is depicted in Figure 1. The left column shows modeling across samples and integer copy number estimation that are unique to the cn.MOPS pipeline. On the right-hand side, GC correction is unique to some previous analysis pipelines; however, this step is not necessary for cn.MOPS, as the local model automatically captures GC content effects. The steps of the cn.MOPS processing pipeline and the central cn.MOPS model are described in the following subsections.
Figure 1.

The processing pipelines for CNV detection in NGS data. Left column: modeling across samples and integer copy number estimation are unique to cn.MOPS. Right column: either GC correction [class (a) methods] or read count ratios [class (b) methods] are required for previous pipelines.
Read mapping and segment read counts
After quality control, read mapping is the first step in analyzing NGS data with respect to CNVs (Figure 1). Depending on the technology, the read length and whether the reads are single or paired, the parameters of the mapping method should be adjusted to minimize the number of false positives without generating too many false negatives. The most important mapping parameters are the number of mismatches allowed and the gap parameters of the employed alignment algorithm. These parameters depend on the expected sequencing errors [see (14) for a statistical analysis of sequencing errors]. If multiple best mapping positions are found for a read, then the read can be randomly assigned to one of them, to all of them, or can be discarded. In our experiments, we mapped the reads by Bowtie (25) for paired reads, allowed two mismatches and mapped to one random best mapping position.
After read mapping, segments must be defined in which the reads are counted (Figure 1). For cn.MOPS, as for all other approaches except SeqSeg and rSW-seq, the genome is first divided into non-overlapping segments in which reads are counted. Previous methods compare read counts along the chromosome (depth of coverage) and must therefore have equally sized segments in which reads are counted. Although cn.MOPS also uses equally sized segments by default, equal size is not strictly required, since a separate model is generated for each segment. The later segmentation along the chromosome is based on the expected copy number, which is independent of the segment length. If the sizes of segments in which reads are counted are variable, then the resolution can be traded off against the confidence in the estimated copy numbers.
Sample normalization and GC correction
GC correction is a crucial first step for proper performance of class (a) methods, such as MOFDOC (Figure 1). These methods subsequently apply a segmentation algorithm to (log) read counts along the chromosome. Therefore, the (log) read counts must be normalized to be comparable between different genomic loci. Since the numbers of reads within segments depend on their GC content (14), the segments' (log) read counts must be normalized for their GC content.
Sample normalization is important for modeling across samples because the reads of all samples are assumed to be caused by the same model (Figure 1). Sample normalization corrects the read counts of one sample by the number of mappable reads of the sample to make read counts comparable across samples. Using data of HapMap individuals from the 1000 Genomes Project (26), we tested read counts of 25 kbp segments for being Poisson distributed with and without sample normalization. Without sample normalization, the Poisson assumption was rejected by a Poisson test (27) for 92% of the segments. Using normalization, however, the Poisson assumption was rejected for only 2% of the segments. Segments that were rejected coincide significantly with known CNV regions according to Fisher's exact test with p < 2.2e-16 (for details see Supplementary Table S1). Thus, our model assumption that, for a constant copy number, the read counts are Poisson distributed is justified with sample normalization. This assumption is also in concordance with the findings of Sathirapongsasuti et al. (28).
The mixture of Poissons model
Our main contribution and novelty is modeling of read count variations across samples in order to separate variations caused by copy numbers from local variations caused by technical or biological noise (Figure 1). For CNV detection, we use a mixture of Poissons model that is not affected by read count variations along the chromosome, because a separate model is constructed at each DNA locus. The model incorporates the linear dependency between average read counts in segments and copy numbers (6,7). In contrast to existing methods, cn.MOPS provides integer copy numbers together with their confidence intervals. Model selection in a Bayesian framework is based on maximizing the posterior by an expectation maximization (EM) algorithm. Most importantly, a Dirichlet prior on the mixture components prefers a constant copy number of 2 for all samples. Only if the data drive the posterior away from this prior, the segment receives a high informative/non-informative call (I/NI call), that is, the part of the CNV call which detects variation across samples.
The Model
cn.MOPS is a generative probabilistic model that explains the observed read counts by copy numbers and by measurement variations. Consequently, the model assumes that the read counts x in a segment are distributed across samples according to a mixture of Poissons, in which each mixture component corresponds to specific copy number and the Poisson parameter reflects the noise:
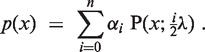 |
(1) |
In this model, αi is the percentage of samples with copy number i for 0 ≤ i ≤ n, and λ is the mean read count for copy number 2. P is the Poisson distribution:
| (2) |
The model integrates the assumption that the read counts are linearly related to the number of copies. For copy number i ≥ 1, the mean read count is thus 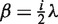 . For copy number i = 0, we assume a Poisson distribution with parameter
. For copy number i = 0, we assume a Poisson distribution with parameter 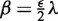 , which accounts for background noise stemming from wrongly or ambiguously mapped reads and for sample contamination by other DNA. See Supplementary Section S2.5, for a justification of the noise model. Note, that the results of cn.MOPS are robust against the choice of the hyperparameter ε (see Supplementary Section S3.8). The robustness is due to the fact that copy number zero can be detected with a broad range of ε values.
, which accounts for background noise stemming from wrongly or ambiguously mapped reads and for sample contamination by other DNA. See Supplementary Section S2.5, for a justification of the noise model. Note, that the results of cn.MOPS are robust against the choice of the hyperparameter ε (see Supplementary Section S3.8). The robustness is due to the fact that copy number zero can be detected with a broad range of ε values.
The model in Equation (1) allows estimation of integer copy numbers with fixed model parameters αi and λ. The prior probability that a read count stems from copy number i is p(i) = αi. The likelihood that a read count x is produced by the i-th mixture component is 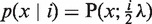 . Then Bayes' formula can be used to compute the posterior p(i ∣ x), that is, the probability that read count x is caused by the i-th component corresponding to copy number i. Consequently, a read count is assigned the integer copy number with the largest posterior probability.
. Then Bayes' formula can be used to compute the posterior p(i ∣ x), that is, the probability that read count x is caused by the i-th component corresponding to copy number i. Consequently, a read count is assigned the integer copy number with the largest posterior probability.
Model Selection by an EM Algorithm and Dirichlet Prior
Suppose that read counts {x1, … , xN} have been observed for N samples in a given segment. Model selection is concerned with fitting a model that best explains the training data {x1, … , xN}. In a Bayesian framework, α and λ are considered as random variables; thus, p(x) in Equation (1) becomes a conditional probability p(x ∣ α, λ), i.e. the likelihood that read count x has been produced by the model with parameters α and λ. If we assume that, for the prior distribution, the parameters α and λ are independent [p(α,λ) = p(α)p(λ)], then the parameter posterior is
| (3) |
We introduce a Dirichlet prior p(α) on α = (α0, α1, … , α n) to include the prior knowledge that almost all locations have copy number 2 for all samples, which is the null hypothesis of constant copy number 2. The Dirichlet prior
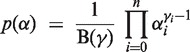 |
(4) |
with parameter vector γ = (γ0, γ1, … , γn) is well suited to express our prior assumptions about α. By setting γ2 ≫ γi ≥ 1 (for i ≠ 2), we ensure that vectors α with a large value for α2—the percentage of samples having copy number 2—are the most likely to be drawn. Each component i of the Dirichlet distribution p(α) is distributed according to a beta distribution with the mode (γi − 1)/(γs − n), where 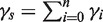 .
.
For the prior on λ, we simply choose a uniform distribution on a sufficiently large interval with left endpoint 0.
An EM algorithm minimizes an upper bound on the negative log-posterior of the parameters α and λ by following update rules (for details see Supplementary Section S2.2):
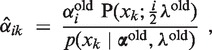 |
(5) |
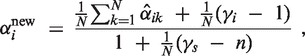 |
(6) |
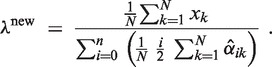 |
(7) |
Here, 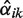 is an estimate (the E-step of the EM algorithm) of the posterior αik = p(i ∣ xk, α, λ) using current estimations αold and λold of the parameters. We simplify the hyperparameter vector γ to one intuitively interpretable hyperparameter G by setting γi = 1 for i ≠ 2 and γ2 = 1 + G. This setting ensures that α2 > G/(G + N) in the αi update Equation (6). Thus, a minimum percentage of individuals that have copy number 2 can be ensured by the hyperparameter G (for details see Supplementary Section S2.2).
is an estimate (the E-step of the EM algorithm) of the posterior αik = p(i ∣ xk, α, λ) using current estimations αold and λold of the parameters. We simplify the hyperparameter vector γ to one intuitively interpretable hyperparameter G by setting γi = 1 for i ≠ 2 and γ2 = 1 + G. This setting ensures that α2 > G/(G + N) in the αi update Equation (6). Thus, a minimum percentage of individuals that have copy number 2 can be ensured by the hyperparameter G (for details see Supplementary Section S2.2).
I/NI Call: Information Gain of Posterior over Prior
Based on the Bayesian framework, we define an informative/non-informative (I/NI) call analogous to that of the FARMS algorithm, which excelled at summarization and gene filtering of microarray data (29–31). In contrast to λ, which captures noise variation, α captures variation arising from CNVs; therefore, its posterior indicates CNVs in the data. The I/NI call measures the information gain of the posterior compared to its prior distribution p(α), which represents the null hypothesis that all samples have copy number 2. Therefore, the I/NI call measures the tendency to reject the null hypothesis based on the observed data.
We define the I/NI call as a weighted distance between the posterior's mode and the prior's mode (0, 0, 1, 0, … , 0), which results in the expected absolute log fold change relative to copy number 2 (for details see Supplementary Section S2.3):
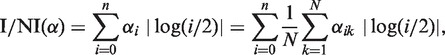 |
(8) |
with 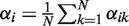 [this equation is derived in Supplementary Equation (S25)]. For notational convenience, we did not distinguish between i = 0 and i ≥ 1 in the above formula. ‘log(0/2)’ must be understood as log(ε/2)—in accordance with the fact that read counts for copy number 0 are Poisson distributed with parameter ελ/2 (see ‘The Model’ section). For α = (0, 0, 1, 0, … , 0), the I/NI call is zero, whereas any other α gives a positive I/NI call. The more copy numbers differ from 2, the higher is the I/NI call, where gains and losses are treated on the same level by the absolute value of the logarithm. The rightmost term in Equation (8) makes clear that the I/NI call can be understood as the sum of individual I/NI calls, I/NI(αk), that is, the contribution of the k-th sample to the I/NI call:
[this equation is derived in Supplementary Equation (S25)]. For notational convenience, we did not distinguish between i = 0 and i ≥ 1 in the above formula. ‘log(0/2)’ must be understood as log(ε/2)—in accordance with the fact that read counts for copy number 0 are Poisson distributed with parameter ελ/2 (see ‘The Model’ section). For α = (0, 0, 1, 0, … , 0), the I/NI call is zero, whereas any other α gives a positive I/NI call. The more copy numbers differ from 2, the higher is the I/NI call, where gains and losses are treated on the same level by the absolute value of the logarithm. The rightmost term in Equation (8) makes clear that the I/NI call can be understood as the sum of individual I/NI calls, I/NI(αk), that is, the contribution of the k-th sample to the I/NI call:
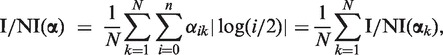 |
(9) |
where αk = (α0k, α1k, … , α nk) is the vector of posteriors for the read count xk.
Segmentation and CNV call
Segmentation is an important step in CNV detection as it determines the length and position of a CNV (Figure 1). Class (a) methods perform segmentation on the GC-corrected (log) read counts, while ratio-based methods perform segmentation on the (log) ratios. Some methods, such as JointSLM, apply an HMM for segmentation and CNV detection.
In the cn.MOPS pipeline, segmentation is based on the results of the modeling step. More specifically, cn.MOPS detects CNVs by segmenting the chromosomes of individuals based on their individual I/NI calls, joining genomically adjacent I/NI calls that show the same copy numbers. Note, however, that the I/NI call defined in Equation (9) does not distinguish between losses and gains with the same fold change. To avoid joining losses and gains, we define the signed individual I/NI call as the expected log fold change:
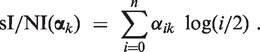 |
(10) |
The absolute value of the signed I/NI call |sI/NI(αk)| is not exactly the I/NI call I/NI(αk), but the two values are always very close (see Supplementary Section S2.4, for detailed mathematical analysis and experimental evaluations).
cn.MOPS applies either its own algorithm ‘fastseg’ or, alternatively, the circular binary segmentation algorithm [DNAcopy (32)] to sI/NI(αk) along the chromosome. The segmentation algorithm joins consecutive segments with large or small expected fold changes to make a candidate segment. It then supplies candidate segments that show variations along the chromosome and also across samples indicated by the signed individual I/NI calls.
All CNV detection methods except those based on HMMs decide on the basis of a threshold on the average/median (log) read count or (log) ratio over the segments whether the candidate segments are CNVs. In the cn.MOPS pipeline, a candidate segment is called a CNV segment if the median of the signed individual I/NI call sI/NI(αk) over the segment is at least 0.6 ≈ log2(3/2) for gains or at most −1 = log2(1/2) for losses. This CNV call incorporates two calls: (i) an I/NI call across samples and (ii) a segment call along the chromosome. Only if consecutive segments obtain an I/NI call, they are joined by the segmentation algorithm (see second bar and third sample in Figure 2). This idea of calling a CNV by detecting both variation across samples and variation along a chromosome has already led to improvements in CNV detection based on DNA microarray data using the cn.FARMS method (24).
Integer copy number estimation
The final step is concerned with estimating the integer copy numbers of the CNVs (Figure 1). Methods based on HMMs, such as JointSLM, automatically obtain integer copy numbers by means of their hidden states. However, most existing methods do not estimate the integer copy numbers of the CNVs.
cn.MOPS automatically supplies posterior estimates of the integer copy numbers for each segment (Figure 1). The estimated copy number of a CNV is the most probable posterior copy number, where the segment posteriors within the CNV are assumed to be independent.
RESULTS
In order to compare methods that detect copy number variations in NGS data, we first specify the evaluation procedure. Subsequently, we provide an overview of the methods compared and finally we present results on four benchmark data sets.
Evaluation of CNV detection results
We assume that the true CNVs are known and to be rediscovered. Each chromosome is split into equally large evaluation segments the size of which is chosen to accommodate the shortest known CNV. An evaluation segment is called a true positive (TP) if it is entirely contained both in a true CNV and in a detected CNV segment. It is called a false negative (FN) if it is entirely contained in a true CNV but does not overlap with any predicted CNV segment. An evaluation segment is called a false positive (FP) if it is entirely detected as a CNV segment but does not overlap with any true CNV. Finally, it is called a true negative (TN) if it overlaps neither with a true CNV nor with a detected CNV segment. These definitions imply that all evaluation segments that partly overlap with true CNVs or detected CNV segments remain ignored, as the copy numbers in these segments are ambiguous. Figure 3 illustrates the definitions of the four categories of evaluation segments. The two measures we employ hereafter are recall [#TP/(#TP + #FN)] and precision [#TP/(#TP + #FP)]. Note that precision is 1-FDR, in which we are especially interested. A CNV calling threshold governs the trade-off between recall and precision or, in other words, the trade-off between FNs and FPs, because more detected CNVs lead to more FPs but fewer FNs, and vice versa. To assess the performance of methods at different CNV calling thresholds, we use precision-recall curves. Precision-recall curves are independent of the number of TNs, which makes them an ideal tool for our evaluation, as the majority of samples are negatives (non-CNVs).
Figure 3.

Definitions for the evaluation of copy number detection methods. A genome is split into equally sized evaluation segments of a length shorter than the shortest CNV. Top panel: Knowing the true CNV regions (green), the evaluation segments are labeled as class 1 (CNV segment) or class −1 (non-CNV segment). Middle panel: A CNV detection method classifies each evaluation segment into CNV segments (blue, class 1) and non-CNV segments (class −1). Bottom panel: In the first line, positives (known CNV regions) are divided into true positives (TP, green) and false negatives (FN, red). In the second line, negatives (no overlap with known CNV regions) are divided into true negatives (TN, green) and false positives (FP, red). Segments partly overlapping with known or predicted CNV regions are not considered (‘na’).
Methods compared
We compared the following methods (see the ‘Introduction’ section for an overview):
cn.MOPS: our new model and pipeline,
MOFDOC: according to the variant of Alkan et al. (6),
EWT: Yoon et al. (15),
JointSLM: Magi et al. (16),
CNV-Seq: Xie and Tammi (20),
FREEC: Boeva et al. (21).
In this subsection, we provide an overview of the parameter settings, initializations and how these methods were employed.
cn.MOPS: we initialized the parameter λ with the median read count 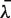 . We set ε = 0.05 and assumed nine possible copy numbers 0 ≤ i ≤ n = 8, which covers previously observed copy numbers in the HapMap individuals (17). The parameter α should be initialized close to the location of the prior's mode (0, 0, 1, 0, … , 0), which are the optimal parameters if all samples have copy number 2. However, initializing α with this vector would clamp all
. We set ε = 0.05 and assumed nine possible copy numbers 0 ≤ i ≤ n = 8, which covers previously observed copy numbers in the HapMap individuals (17). The parameter α should be initialized close to the location of the prior's mode (0, 0, 1, 0, … , 0), which are the optimal parameters if all samples have copy number 2. However, initializing α with this vector would clamp all 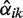 and
and 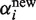 to zero according to Equation (6) and Equation (7). Therefore, we initialized α with (0.05, 0.05, 0.6, 0.05, … , 0.05).
to zero according to Equation (6) and Equation (7). Therefore, we initialized α with (0.05, 0.05, 0.6, 0.05, … , 0.05).
MOFDOC: we implemented MOFDOC using the CNV calling criterion of Alkan et al. (6). A CNV region is called if a out of b consecutive segments show a read count with a z-score below or above thresholds specified to call a loss or gain segment (default values a = 6 and b = 7). We generalized this ‘a-b-smoother’ to a smoothing algorithm that is not only able to smooth logical, but also real values arising from CNV calls, which improved MOFDOC's results.
EWT: we reimplemented event-wise testing as described by Yoon et al. (15), but improved the GC correction by using all samples to estimate the GC effect. Further, we restricted the minimum ‘event size’, a parameter that prevents EWT from testing for CNVs that are too short. We generalized EWT to a variety of segment sizes that are also apt for low coverage CNV detection. Our modifications to EWT improved its results.
JointSLM: we applied version 0.1 of the R package JointSLM adjusting the package to GC content correction for a variety of segment sizes.
CNV-Seq: we used the authors' implementation (http://tiger.dbs.nus.edu.sg/cnv-seq/), taking the median of the samples' read counts as reference read count.
FREEC: we used version 3.2 (http://bioinfo-out.curie.fr/projects/freec/), taking, analogously to CNV-Seq, the median of the samples' read counts as reference. Although FREEC can perform the analysis without a reference, we used it in ‘reference mode’ because of the improved performance.
We did not include SeqSeg (7) because we were not able to find suitable parameters, not even after an extensive search. The problem was that SeqSeg either did not detect any breakpoints or the thresholds for the P-values were not determined. However, the performance of SeqSeg can be estimated via CNV-Seq that is very similar. We also omitted CNAseg (19) from the comparison as its developers state that this method is specifically tailored to CNA detection in tumor samples.
To ensure a fair comparison, the parameters of the methods were optimized on simulated data sets similar to the one we used in our first experiment. More details, for instance, on the parameters that were used and computation time, can be found in the ‘Discussion’ section.
Simulated data with constructed CNVs
We constructed 100 artificial benchmark data sets, assuming an artificial genome to consist of a single chromosome of 125 Mb length, divided into 5000 segments of length 25 kb. We created 40 samples by sampling read counts for all segments and samples according to a Poisson process. The overall number of evaluation segments was therefore 40 × 5000 = 200 000.
The Poisson parameters λ of the Poisson process for simulating the read counts in the evaluation segments were drawn from a distribution of λ values that was estimated using median GC-corrected read counts of HapMap individuals from the 1000 Genomes Project. Therefore, the simulated Poisson distributions are similar to those found in real sequencing experiments. We scaled these λ values by a random number between 0.3 and 1 in order to simulate different coverages. This read count simulation yielded 290 000–770 000 reads, corresponding to a coverage of 0.18–0.46 and 0.08–0.22 for 75 and 36 bp reads, respectively.
Note that we consider low-coverage sequencing data here, because methods for analyzing SNPs and CNVs in low-coverage data will continue to be relevant in the future. Le and Durbin (33) showed that low-coverage data will remain important in the context of single nucleotide polymorphism (SNP) data analysis: in terms of a study's discovery power, where a fixed number of reads should rather be used for sequencing more samples with lower coverage than for sequencing fewer samples with higher coverage. The relationship between discovery power and coverage is similar for CNV data. In the ‘High Coverage Data Sets’ section below, we also simulate high coverage data sets.
On the basis of HapMap data (17), we determined CNV region characteristics and how copy numbers are distributed. We implanted 20 CNV regions into each of the benchmark chromosomes. The lengths of the CNV regions were chosen randomly from the interval 75–200 kb, which, according to Xie and Tammi (20), is the range of accurate detection for the given coverage. The 20 starting points of the CNV regions were chosen randomly along the chromosome. After having determined the 20 CNV regions, we had to decide how CNVs are implanted into the single samples. To this end, we first had to take into account that CNVs of different individuals cluster at specific regions of the DNA called ‘CNV regions’, of which many contain only losses or only gains. Based on characteristics of HapMap individuals, we assigned CNV region types such that 80% are of type ‘loss region’ (containing only losses), 15% of type ‘gain region’ (containing only gains), and 5% of type ‘mixed region’ (containing both losses and gains). Then the actual copy number for each sample was drawn according to the copy numbers observed for HapMap individuals (17): For a CNV region of type ‘loss region’, a sample has probabilities of 0.8, 0.15 and 0.05 of having copy numbers 2, 1 and 0, respectively. For a CNV region of type ‘gain region’, a sample has probabilities of 0.85, 0.08, 0.06 and 0.01 of having copy numbers 2, 3, 4 and 5, respectively. For a CNV region of type ‘mixed region’, a sample has probabilities 0.04, 0.16, 0.67, 0.11 and 0.02 of having copy numbers 0, 1, 2, 3 and 4, respectively. Of the 200 000 evaluation segments, on average 101 (±56) are gains and 612 (±104) are losses. The CNV lengths range from 75 006 bp to 199 848 bp with an average of 136 921 bp.
Table 1 reports the performance of the compared copy number detection methods separately for gains and losses. As evaluation measures, we use the area under the precision-recall curve and the recall for a fixed FDR of 0.05. All methods perform better at detecting losses because it is more likely for gains than for losses to have read counts in the range of copy number 2. This is due to the fact that an average copy number 2 read count is more likely to be produced by copy number 3 than by copy number 1 (see Supplementary Section S3.3). JointSLM performs worse than other methods because of the low percentage of samples showing an abnormal copy number (the rare events). cn.MOPS yielded the largest average area under the precision-recall curve. The improvement over the other methods is highly significant, as shown by a Wilcoxon signed-rank test. cn.MOPS achieved the highest recall (for FDR fixed at 0.05), which, according to a Wilcoxon test, was also significantly higher than those of the other methods. In summary, cn.MOPS significantly outperformed its competitors on the 100 simulated data sets.
Table 1.
Performance of the compared copy number detection methods on the artificial benchmark data set
| PR AUC | P-value | Recall | P-value | |
|---|---|---|---|---|
| Gains | ||||
| cn.MOPS | 0.94 | – | 0.88 | – |
| MOFDOC | 0.81 | 1.14e-13 | 0.76 | 9.75e-12 |
| EWT | 0.79 | 5.95e-14 | 0.74 | 1.34e-12 |
| JointSLM | 0.25 | 4.23e-18 | 0.22 | 2.80e-17 |
| CNV-Seq | 0.35 | 4.23e-18 | 0.35 | 3.98e-17 |
| FREEC | 0.65 | 1.95e-17 | 0.53 | 3.42e-14 |
| Losses | ||||
| cn.MOPS | 0.96 | – | 0.96 | – |
| MOFDOC | 0.92 | 3.50e-17 | 0.90 | 9.22e-17 |
| EWT | 0.91 | 3.20e-18 | 0.90 | 8.44e-17 |
| JointSLM | 0.34 | 1.98e-18 | 0.28 | 1.98e-18 |
| CNV-Seq | 0.81 | 1.98e-18 | 0.81 | 3.84e-17 |
| FREEC | 0.73 | 1.98e-18 | 0.72 | 3.32e-17 |
‘PR AUC’ gives the average area under the precision-recall curve of 100 experiments. The second column, ‘P-value’, reports the P-value of a Wilcoxon signed-rank test (over the 100 experiments) with the null hypothesis that cn.MOPS (in bold) and another method have the same area under the curve. ‘Recall’ reports the recall at a precision of 0.95, that is, an FDR of 0.05. The last column, ‘P-value’, gives the P-value of an analogous Wilcoxon test for the recall with an FDR of 0.05. cn.MOPS performed significantly better than all other methods.
Real sequencing data with implanted CNVs from the X chromosome
In contrast to simulated read counts, we next considered real reads obtained from the sequencing of a single male HapMap individual (NA20755). This man's genome was sequenced 17 times by the Solexa Genome Analyzer II at the Wellcome Trust Sanger Institute [(26) see Supplementary Table S5]. These 17 samples ensure a constant copy number, as they stem from the same individual. We mapped the reads with Bowtie (25) for paired reads, allowing two mismatches. The numbers of reads range from 12 069 758 to 18 810 212, of which between 10 419 510 and 16 041 464 could be mapped, which corresponds to coverages between 0.13 and 0.21 (see Supplementary Section S3.3, for details on read mapping and the number of reads).
We created 110 benchmark data sets by choosing each of human chromosomes 1–22 five times, implanting 20 random CNV regions in each chromosome data set. The lengths of these implanted CNV regions were chosen to be 75, 100, 150, and 200 kb (5 each), and, for each of the regions, a random segment on the X chromosome which supplied reads for the region was selected. CNV region types and individual copy numbers were determined according to the procedure and distributions described in the first experiment except that we only considered CNV copy numbers 1 and 3 since they are the most difficult to distinguish from copy number 2. We assigned CNV region types such that 80% are of ‘loss region’ type, 15% of ‘gain region’ type, and 5% of ‘mixed region’ type. For a CNV region of ‘loss region’ type, a sample has probabilities 0.8 and 0.2 of having copy number 2 and 1, respectively, for a CNV region of ‘gain region’ type, 0.85 and 0.15 of having copy numbers 2 and 3, respectively, and for a CNV region of ‘mixed region’ type, 0.2, 0.67 and 0.13 of having copy numbers 1, 2 and 3, respectively. Finally, the read counts of the 17 samples were computed in the following way: outside CNVs the original reads counts were used; within CNVs, we added as many read counts as there are copies from the corresponding segment on the X chromosome, taking independent read counts from the considered sample and other random samples.
The CNV detection results were evaluated as described in the ‘Evaluation of CNV Detection Results’ section. The number of evaluation segments ranges from around 32 000 for chromosome 21 to around 168 000 for chromosome 1. On average, 0.1% of the evaluation segments are gains and 0.4% are losses.
Table 2 reports the performance of the compared copy number detection methods separately for gains and losses. As before, we use the area under the precision-recall curve and the recall for the FDR fixed at 0.05. Again, all methods performed better at detecting losses. If we adhere to the Poisson assumption, the reasons for this performance difference are the same as those stated in the first experiment. cn.MOPS significantly outperformed all other methods with respect to both the area under the precision-recall curve (PR AUC) and the recall for FDR at 0.05. No method considers the variation of the read counts across samples, except cn.MOPS, which estimates this variation via its Poisson parameter, thus achieving superior performance.
Table 2.
Performance of the compared copy number detection methods on real sequencing data with implanted CNVs from the X chromosome
| PR AUC | P-value | Recall | P-value | |
|---|---|---|---|---|
| Gains | ||||
| cn.MOPS | 0.70 | – | 0.65 | – |
| MOFDOC | 0.20 | 1.12e-17 | 0.10 | 2.31e-17 |
| EWT | 0.22 | 1.95e-16 | 0.13 | 8.70e-17 |
| JointSLM | 0.06 | 1.94e-19 | 0.03 | 7.00e-18 |
| CNV-Seq | 0.13 | 1.74e-19 | 0.13 | 5.75e-18 |
| FREEC | 0.49 | 1.22e-12 | 0.30 | 4.41e-15 |
| Losses | ||||
| cn.MOPS | 0.89 | – | 0.88 | – |
| MOFDOC | 0.57 | 3.78e-15 | 0.21 | 2.48e-18 |
| EWT | 0.62 | 1.77e-12 | 0.34 | 2.02e-17 |
| JointSLM | 0.17 | 4.43e-20 | 0.08 | 4.43e-20 |
| CNV-Seq | 0.50 | 4.43e-20 | 0.50 | 4.43e-20 |
| FREEC | 0.52 | 7.05e-17 | 0.36 | 4.56e-20 |
‘PR AUC’ gives the average area under the precision-recall curve of 100 experiments. The second column, ‘P-value’, reports the P-value of a Wilcoxon signed-rank test (over the 100 experiments) with the null hypothesis that cn.MOPS (in bold) and another method have the same area under the curve. ‘Recall’ reports the recall at a precision of 0.95, that is, an FDR of 0.05. The last column, ‘P-value’, gives the P-value of an analogous Wilcoxon test for the recall with an FDR of 0.05. cn.MOPS outperformed all other methods significantly.
Rediscovery of known CNVs in HapMap sequencing data
Next, we compared how well the methods are able to rediscover known CNVs of HapMap individuals whose DNA was sequenced by the Solexa Genome Analyzer II at the Wellcome Trust Sanger Institute (26). We focused on 18 individuals for each of whom the reads were produced on one lane (one sequencing run contains seven lanes). The reads were mapped by Bowtie (25) for paired reads, allowing three mismatches. The numbers of reads range from 12 442 124 to 31 977 690, of which 7 498 420–22 217 020 could be mapped, which lead to a coverage between 0.20 and 0.60 (see Supplementary Section S3.4, for details on individuals, read mapping and the number of reads). We considered the CNVs of these 18 individuals, determined previously by means of microarrays (17), to be the true CNVs. They were detected by the Affymetrix Human SNP array 6.0 and reconfirmed with the Illumina Human1M-single BeadChip. After filtering for CNVs larger than 75 kb, we obtained 170 CNVs, of which 66 are gains and 104 are losses, with lengths ranging from 76 kb to 457 kb. Though some of these CNVs might still be false positives, the double confirmation and considering only CNVs of vast lengths approaches a golden standard. The CNV detection results were evaluated as described in the ‘Evaluation of CNV Detection Results’ section with evaluation segments of length 25 kb. In total, we have 2 064 906 evaluation segments, of which 450 are labeled as losses, as they lie within one of the 104 loss CNVs, and 469 are labeled as gains, as they lie within one of the 66 gain CNVs.
Table 3 shows the performance of the six compared methods at rediscovering known CNVs for the 18 HapMap individuals, where the average area under the precision-recall curve is used as evaluation criterion. As found in previous experiments, all methods perform better at detecting losses. cn.MOPS performs significantly better than its competitors in terms of both the PR AUC and the recall for an FDR of 0.05, although FREEC performs equally well for gains.
Table 3.
Performance of the compared copy number detection methods on HapMap individuals, where known CNVs should be rediscovered
| PR AUC | P-value | Recall | P-value | |
|---|---|---|---|---|
| Gains | ||||
| cn.MOPS | 0.35 | – | 0.24 | – |
| MOFDOC | 0.13 | 1.17e-03 | 0.06 | 1.95e-03 |
| EWT | 0.16 | 5.34e-04 | 0.10 | 1.86e-02 |
| JointSLM | 0.08 | 3.81e-05 | 0.05 | 7.81e-03 |
| CNV-Seq | 0.22 | 1.74e-02 | 0.21 | 3.61e-01 |
| FREEC | 0.35 | 8.68e-01 | 0.17 | 2.38e-01 |
| Losses | ||||
| cn.MOPS | 0.53 | – | 0.45 | – |
| MOFDOC | 0.40 | 2.67e-04 | 0.33 | 3.42e-03 |
| EWT | 0.36 | 7.63e-06 | 0.23 | 6.10e-05 |
| JointSLM | 0.15 | 3.81e-06 | 0.06 | 1.53e-05 |
| CNV-Seq | 0.32 | 7.63e-05 | 0.27 | 3.66e-04 |
| FREEC | 0.42 | 2.37e-03 | 0.26 | 1.01e-03 |
‘PR AUC’ gives the average area under the precision-recall curve of 18 samples. The second column, ‘P-value’, reports the P-value of a Wilcoxon signed-rank test (over the 18 samples) with the null hypothesis that cn.MOPS (in bold) and another method have the same area under the curve. ‘Recall’ reports the recall at a precision of 0.95, that is, an FDR of 0.05. The last column, ‘P-value’, gives the P-value of an analogous Wilcoxon test for the recall with an FDR of 0.05. cn.MOPS rediscovered known CNVs most reliably. Only for gains the performance of FREEC is similar to that of cn.MOPS, whereas cn.MOPS performs significantly better than all its competitors at losses.
So far we have considered CNV detection as a classification task whose goal was to detect CNVs in individual samples. In order to assess the quality of the CNV calling across HapMap samples, we also investigated the performance of each method at a different task—detecting segments in which at least one CNV occurs in one sample. For this task, we did not obtain segments from a segmentation algorithm, but we computed a CNV call for each evaluation segment. The CNV calls must be defined depending on the method. For cn.MOPS, we utilized the I/NI call. For z-score based methods, namely MOFDOC, EWT and JointSLM, we used the mean of the z-score on the evaluation segment. For the ratio-based methods, CNV-Seq and FREEC, we took the mean log-ratios of the evaluation segments. The area under precision-recall curve was 0.18 for the I/NI call, 0.02 for the mean z-score, and 0.14 for the mean log-ratio. The area under curve values are lower than in the other experiments because outliers were not filtered out by a segmentation algorithm. Alternative CNV calls such as variance and maximum-based values are reported in Supplementary Section S3.5.
Figure 4 visualizes the results of this comparison in the form of whole-genome CNV calling plots along all evaluation segments. cn.MOPS separates true CNVs (indicated by red dots) from non-CNV segments (blue dots) more successfully than the other methods. Furthermore, cn.MOPS has lower FDRs for different calling thresholds, as can be seen from the lower variance of the blue dots at the bottom. The superior performance of cn.MOPS at CNV calling across samples is the reason why cn.MOPS outperformed the other methods in previous experiments.
Figure 4.

Whole-genome CNV calling plots that visualize the performance of cn.MOPS, MOFDOC, EWT, JointSLM, CNV-Seq, and FREEC at rediscovering known CNVs of HapMap individuals. The plots visualize CNV calling values (vertical axis) along chromosomes 1–22 of the human genome without segmentation. The first panel shows the I/NI call used for cn.MOPS. The second panel provides mean z-scores used by EWT, JointSLM, while the last panel depicts mean log-ratios used by CNV-Seq and FREEC. We called the largest 0.5% of the CNV calling values (blue dots) and scaled them to maximum one. Darker shades of blue indicate a high density of calling values. True CNV regions are displayed as light red bars, and the corresponding CNV calls are indicated by red dots. Segments without calling values (white segments) correspond to assembly gaps in the reference genome. A perfect calling method would call all segments in true CNV regions (red dots) at maximum 1 and would call others (blue dots) at minimum 0. Arrows indicate segments in true CNV regions that are called by one method group but not by the other method groups. A threshold of 0.6 for log-ratios-based methods, namely CNV-Seq and FREEC, and a threshold of 0.8 for cn.MOPS would lead to the same true positive rate, while cn.MOPS yields fewer false discoveries (lower FDR). cn.MOPS is better at separating segments of true CNV regions from non-CNV segments than the other methods, as indicated by the lower variance of I/NI values (see blue area at the bottom of the first panel). The better separation by cn.MOPS results in FDRs lower than those of other methods, regardless of the calling thresholds.
High coverage data sets
Finally, we compared the performance of CNV detection methods on two high coverage data sets. The first data set is simulated analogously to our previous simulated data but now with high coverage. On this data set we first show that, if we fix the resolution, higher coverage leads to better performance in terms of precision and recall. Next we show that, if we fix the performance in terms of precision and recall, higher coverage allows for higher resolution. The second data set consists of six high coverage samples from the 1000 Genomes Project on which we show that cn.MOPS is well suited for high coverage data sets (for details see Supplementary Section S3.6).
Simulated Data: Coverage versus Performance and Resolution. The first data set was simulated as described in the ‘Simulated Data with Constructed CNVs’ section, but now with different depths of coverage including high coverage. These data allow for investigating the impact of increasing coverage on the performance and on the resolution of CNV detection methods. In order to evaluate the methods as realistically as possible, we drew small blocks of consecutive λ values from data of the 1000 Genomes Project to include mutual dependencies between adjacent segments like in real data.
First, in order to analyze the dependencies between coverage and performance, we implanted short CNVs with lengths 1–5 kb in a 25 Mb chromosome. To be able to detect CNVs of these lengths, we chose a segment length of 250 bp for all compared methods. For each of the coverages 1×, 5×, 10×, 25× and 50×, we generated 10 data sets and determined the recall of each method at a fixed FDR of 0.05. Figure 5 shows that the average performance of all methods increases with the depth of coverage. Again, cn.MOPS outperforms the other methods at all coverages.
Figure 5.

CNV detection performance for different levels of coverage. Each curve in the two panels corresponds to the recall of one method at detecting short CNVs of lengths 1–5 kb (left panel: gains; right panel: losses). The FDR was fixed at 0.05.
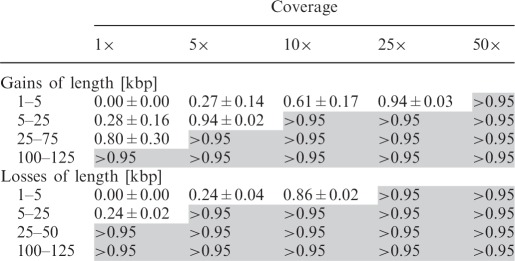
Second, we analyzed the dependencies between coverage and resolution for cn.MOPS. We implanted CNVs of different ranges of lengths 1–5 kb, 5–25 kb, 25–75 kb and 100–125 kb into chromosomes of lengths 25, 125, 250 and 250 Mb, respectively. For each of the coverages 1×, 5×, 10×, 25× or 50× and each range of CNV lengths, cn.MOPS is evaluated on 10 simulated data sets. In each run, we chose the segment size as a fifth of the minimal CNV length. Table 4 shows the recall of cn.MOPS for different coverages, again at a fixed FDR of 0.05. Obviously, for a given performance threshold of 0.95, higher coverage allows for higher resolution.
Table 4.
Average recall values of cn.MOPS at an FDR of 0.05 for different levels of coverage and different CNV lengths, along with their standard deviations over the 10 runs
 |
High Coverage Real World Data: Performance Comparison
On the second high coverage data set from the 1000 Genomes Project we compare the performance of CNV detection methods. The data consist of alignment files of chromosome 1 of two trios that were sequenced at a coverage of 20–60×. A segment length of 500 bp led to 498 502 segments. The International HapMap 3 Consortium (17) found 68 CNVs of ‘loss’ type and 4 of ‘gain’ type, which we considered as true CNVs. Using these true CNVs, out of 2 991 012 evaluation segments, 192 were gains and 2016 losses.
Again, the performance of the CNV detection methods was evaluated by the area under the precision-recall curve and the recall at a fixed FDR. For this experiment, however, we report the recall value at an FDR of 0.9, since all methods detect a large number of new CNVs thus resulting in indistinguishable small recalls at an FDR of 0.05. Table 5 shows the results. cn.MOPS performs best, where EWT performs equally well for losses in terms of the area under the precision-recall curve.
Table 5.
Performance of the compared copy number detection methods for six high coverage samples from the 1000 Genomes Project
| Gains |
Losses |
|||
|---|---|---|---|---|
| PR AUC | Recall | PR AUC | Recall | |
| cn.MOPS | 0.34 | 0.92 | 0.33 | 0.59 |
| MOFDOC | 0.00 | 0.00 | 0.21 | 0.28 |
| EWT | 0.00 | 0.00 | 0.30 | 0.37 |
| JointSLM | 0.01 | 0.00 | 0.26 | 0.26 |
| CNV-Seq | 0.14 | 0.79 | 0.26 | 0.38 |
| FREEC | 0.26 | 0.92 | 0.22 | 0.25 |
‘PR AUC’ gives the area under precision-recall curve. ‘Recall’ reports the recall at a precision of 0.1. cn.MOPS performs best, where FREEC performs equally well for gains in terms of recall.
In Supplementary Section S3.7, we additionally provide experiments on a 58 sample data set of medium sequencing coverage from the 1000 Genomes Project. cn.MOPS performs well on this data set with considerable more samples than the current data set, too.
We have shown that cn.MOPS is also well suited for high coverage data sets on which it outperformed its competitors.
DISCUSSION
Data Access
The data of the second, third and fourth experiment are part of the 1000 Genomes Project. For the second experiment we used one chromosome from a Tuscany sample (NA20755), for the third experiment 18 samples from Pilot Phase 1, and for the fourth experiment chromosome 1 of 6 high coverage samples in order to comply with the Ft. Lauderdale principle for use of unpublished data for method development.
Limitations
cn.MOPS cannot be applied to a single sample because it decomposes variations along samples into those stemming from copy numbers and those from noise. The quality of this decomposition increases with the number of samples. We recommend to use at least 6 samples for proper parameter estimation (see Supplementary Section S3.9). If the majority of samples has a copy number different from 2, then the cn.MOPS model regards this copy number as copy number 2. However, this incorrect assignment of components to copy numbers can readily be corrected by comparing the expected read counts (the parameter λ) along the chromosome.
Computational Costs
With massively growing amounts of NGS data, computation time is becoming an increasingly important, and possibly limiting, factor in CNV analysis. To create an impression of the computational cost of cn.MOPS, we report the computation times for a medium coverage data set (see Supplementary Section S3.7 and Supplementary Table S17). The data set consists of chromosome 20 of 58 samples from the 1000 Genomes Project with coverages ranging from 2.5× to 8×. Not surprisingly, the model-free approaches MOFDOC (110s), EWT (239s) and CNV-Seq (96s) were faster than the model-based approaches cn.MOPS (250s), JointSLM (1001s), and FREEC (693s), where cn.MOPS was the fastest among the model-based methods. All computations were done on a Linux server with Intel® Xeon® CPU with 2.27GHz. In order to facilitate a fair comparison, all computations were performed on single processors only. Note, however, that cn.MOPS can be parallelized easily since it models each genomic location independently. The parallelization is already implemented in the R package cn.mops (but was not used in the above comparison).
NGS-based versus array-based CNV detection
For the HapMap data set, microarray-based techniques missed CNVs that are clearly identified by sequencing techniques. This entails that CNV detection in NGS data will be important in the future to complement and confirm CNVs previously detected by microarray techniques. In contrast to microarrays, NGS allows estimation of allele-specific copy numbers without a priori allele selection, which, in the context of diseases, is especially relevant for determining whether an allele is fully functional.
Exon sequencing
We are currently adapting cn.MOPS to analyze data from exon sequencing (ExonSeq), where DNA fragments are first captured by hybridization to probes attached to baits and then sequenced. For exon sequencing, the read counts show higher variation along the chromosome because hybridization and cross-hybridization effects are introduced via the baits. Thus, cn.MOPS is even better suited to this task than other methods. First results are very promising.
CONCLUSION
We have introduced cn.MOPS—a novel method and pipeline for the detection of copy number variations in NGS data. cn.MOPS incorporates a probabilistic model that decomposes read variations across samples into integer copy numbers and noise by means of its mixture components and its Poisson distributions, respectively. cn.MOPS is able to control the FDR for CNV detection via a Dirichlet prior on the model's mixture components. The Dirichlet prior prefers a constant copy number of 2 for all samples, which corresponds to the null hypothesis. The more the data drag the posterior away from the Dirichlet prior, the more likely a CNV is present in the data.
We compared cn.MOPS with the five most popular CNV detection methods using four benchmark data sets. For all benchmarks, cn.MOPS outperformed its competitors, especially in terms of FDR.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables S1–S17, Supplementary Figures S1–S14 and Supplementary Sections S1–S4.
FUNDING
Funding for open access charge: Funds from the Institute of Bioinformatics, Johannes Kepler University Linz.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 3.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 4.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Łabaj PP, Leparc GG, Linggi BE, Markillie LM, Wiley SH, Kreil DP. Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics. 2011;27:i383–i391. doi: 10.1093/bioinformatics/btr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang DY, Getz G, Jaffe DB, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods. 2008;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen Y-J, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 13.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 14.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magi A, Benelli M, Yoon S, Roviello F, Torricelli F. Detecting common copy number variants in high-throughput sequencing data by using JointSLM algorithm. Nucleic Acids Res. 2011;39:e65. doi: 10.1093/nar/gkr068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. The International HapMap 3 Consortium (2010) Integrating common and rare genetic variation in diverse human populations. Nature, 467, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim T-M, Luquette LJ, Xi R, Park PJ. rSW-seq: algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinformatics. 2010;11:432. doi: 10.1186/1471-2105-11-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivakhno S, Royce T, Cox AJ, Evers DJ, Cheetham RK, Tavar S. CNAseg–a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics. 2010;26:3051–3058. doi: 10.1093/bioinformatics/btq587. [DOI] [PubMed] [Google Scholar]
- 20.Xie C, Tammi MT. CNV-Seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boeva V, Zinovyev A, Bleakley K, Vert J-P, Janoueix-Lerosey I, Delattre O, Barillot E. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics. 2011;27:268–269. doi: 10.1093/bioinformatics/btq635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harchaoui Z, Levy-Leduc C. Catching Change-points with Lasso. In: Platt JC, Koller D, Singer Y, Roweis S, editors. Advances in Neural Information Processing Systems. Vol. 20. Cambridge, MA: MIT Press; 2008. pp. 617–624. [Google Scholar]
- 23.Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M. Detecting copy number variation with mated short reads. Genome Res. 2010;20:1613–1622. doi: 10.1101/gr.106344.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clevert D-A, Mitterecker A, Mayr A, Klambauer G, Tuefferd M, Bondt AD, Talloen W, Göhlmann H, Hochreiter S. cn.FARMS: a latent variable model to detect copy number variations in microarray data with a low false discovery rate. Nucleic Acids Res. 2011;39:e79. doi: 10.1093/nar/gkr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. The 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown L, Zhao L. A new test for the Poisson distribution. Sankhya Ser. A. 2002;64:611–625. [Google Scholar]
- 28.Sathirapongsasuti FJ, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–2654. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hochreiter S, Clevert D-A, Obermayer K. A new summarization method for Affymetrix probe level data. Bioinformatics. 2006;22:943–949. doi: 10.1093/bioinformatics/btl033. [DOI] [PubMed] [Google Scholar]
- 30.Talloen W, Clevert D-A, Hochreiter S, Amaratunga D, Bijnens L, Kass S, Göhlmann H. I/NI-calls for the exclusion of non-informative genes: a highly effective filtering tool for microarray data. Bioinformatics. 2007;23:2897–2902. doi: 10.1093/bioinformatics/btm478. [DOI] [PubMed] [Google Scholar]
- 31.Talloen W, Hochreiter S, Bijnens L, Kasim A, Shkedy Z, Amaratunga D. Filtering data from high-throughput experiments based on measurement reliability. Proc. Natl Acad. Sci. USA. 2010;107:173–174. doi: 10.1073/pnas.1010604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 33.Le SQ, Durbin R. SNP detection and genotyping from low-coverage sequencing data on multiple diploid samples. Genome Res. 2011;21:952–960. doi: 10.1101/gr.113084.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.