

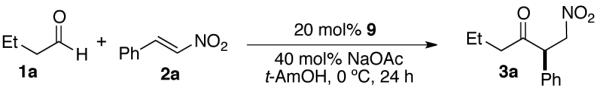
Over the past decade, N-heterocyclic carbenes (NHCs) have been used as catalysts in a variety of C-C bond forming reactions.[1] Our group has been interested in the development of chiral NHCs as catalysts for the asymmetric intra-molecular Stetter reaction[2,3] and more recently the inter-molecular variant.[4,5] We recently reported that hetaryl aldehydes and enals react efficiently with nitroalkenes in the Stetter reaction, leading to β-nitro ketones with high enantioselectivity.[4d] Crucial to the success of this method was the development of a fluorinated triazolium salt pre-catalyst that provides significantly enhanced enantioselectivity over des-fluoro analogues.[6] Although this new catalyst system greatly expands the scope of this method, these conditions are not amenable to the use of unactivated aliphatic aldehydes. Due to their lower electrophilicity than aryl aldehydes, aliphatic aldehydes have rarely been used successfully in the asymmetric inter-molecular Stetter reaction.[7,8]
Our initial attempts at rectifying this problem began by evaluating more reactive Michael acceptors such as β-nitrostyrenes. The unstable nature of the reaction components mandated milder reaction conditions,9 and a brief screen revealed tertiary alcohol solvents and weak inorganic bases as being optimal. Under these conditions, pre-catalyst 5, which previously demonstrated high reactivity and enantioselectivity for hetaryl aldehydes and enals, affords only modest yield (53%) with low enantioselectivity (48%) (Table 1). Interestingly, this reaction affords the opposite major enantiomer to that observed in our previous work using the same pre-catalyst with aliphatic nitroalkenes.[4d] Surprisingly, trans-fluorinated pre-catalyst 6 provides substantial increases in both yield (78%) and enantioselectivity (74%). In order to further increase selectivity we evaluated the more sterically demanding pre-catalyst 7, derived from t-leucine. This pre-catalyst displays low reactivity compared to the valine derived pre-catalysts (4-6), but with greatly increased enantioselectivity (80%). Further evaluation of this scaffold shows the same trends in reactivity and selectivity to that of the valine derived series; trans-fluorinated pre-catalyst 9 provides drastically better selectivity (93% ee) than both cis-fluoro (74%) and des-fluoro (80%) catalysts.
Table 1.
Catalyst and optimization studies.a
 | |||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) b | ee(%) c |
| 1 | none | 80 | 93 |
| 2 | i-PrOH as solvent | 42 | 88 |
| 3 | THF as solvent | 0 | nd |
| 4 | PhMe as solvent | 0 | nd |
| 5 | i-Pr2NEt instead of NaOAc | trace | nd |
| 6 | 1.0 equiv NaOAc | 78 | 93 |
| 7 | 23 °C | 42 | 90 |
Reactions conducted with 1.5 equiv 1a and 1.0 equiv 2a.
Isolated yield after chromatography.
Enantiomeric excess determined by HPLC analysis on a chiral stationary phase. BF4 counterions omitted for clarity.
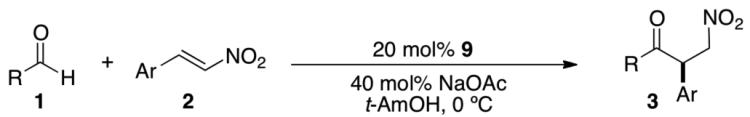
Under optimized conditions the scope of this transformation was evaluated with respect to both the aldehyde and nitrostyrene derivative (Table 2). Using β-nitrostyrene 2a as the Michael acceptor a variety of aliphatic aldehydes were examined. Straight-chain aliphatic substitution provides products in high yield (80-87%) and excellent enantioselectivities (92-93%) with the exception of acetaldehyde, which gives good yield (71%) but is only modestly selective (62% ee). β-Branched aldehydes are tolerated and provide excellent enantioselectivity (95%), albeit in lower yield, while α-branched aldehydes do not participate. A variety of functional groups are well-tolerated including thio-ethers, silyl ethers, alkyl halides and terminal olefins. Substitution on the aryl ring of the nitroalkene leads to fairly invariant results. Ortho-, meta-, and para-substitution was examined providing fair to good yields (50-83%) in all cases as well as excellent enantioselectivities (91-94%). A 2.5 mmol scale experiment was performed using 10 mol% 9, providing the product in 84% yield and 93% ee.
Table 2.
Reaction Scopea
 | ||||
|---|---|---|---|---|
| Entry | R | Ar | yileld % b | ee %c |
| 1 | n-pr | ph | 80% | 93% |
| 2 | Et | ph | 87% | 92% |
| 3 | Me | ph | 71% | 62% |
| 4 | i-Bu | ph | 32% | 95% |
| 5 | ph | 68% | 87% | |
| 6 | ph | 67% | 92% | |
| 7 |  |
ph | 76% | 93% |
| 8 | ph | 83% | 93% | |
| 9 | 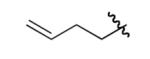 |
ph | 83% | 93% |
| 10 | Cy | ph | <5% | N/A |
| 11 | n-pr | 2-Cl-C6H4 | 70% | 91% |
| 12 | n-pr | 2-F-C6H4 | 75% | 93% |
| 13 | n-pr | 2-Meo-C6H4 | 83% | 94% |
| 14 | n-pr | 3-Meo-C6H4 | 63% | 91% |
| 15 | n-pr | 3-Br-C6H4 | 50% | 91% |
| 16 | n-pr | 4-Cl-C6H4 | 70% | 92% |
| 17 | n-pr | 4-Me-C6H4 | 81% | 92% |
| 18 | n-pr | 4-(B(pin))-C6H4 | 62% | 91% |
Reactions conducted with 1.5 equiv 1 and 1.0 equiv 2 for 24-48 h.
Isolated yield after chromatography.
Enantiomeric excess determined by HPLC analysis on a chiral stationary phase.
Derivatization of product 3a was performed to demonstrate the synthetic utility of β-nitro ketone products (Scheme 1). Reduction with NaBH4 leads to the desired nitro alcohol in quantitative yield and 8:1 dr. The major diastereomer is easily isolated by chromatography providing 82% of 10 in 93% ee. Reduction of the nitro group is accomplished using NiCl2/NaBH4 which, following a simple workup, is transformed to the corresponding benzamide in 81% yield and 98% ee.
Scheme 1.
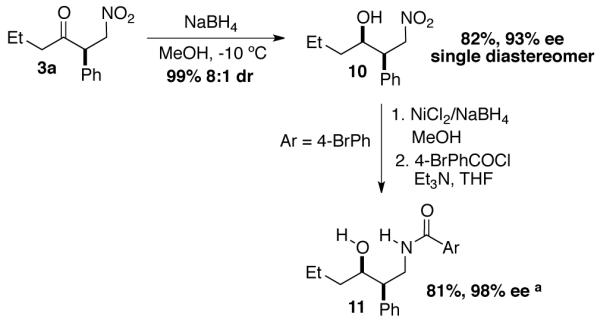
Derivatization of β-nitro ketone products. [a] Absolute and relative configuration determined by x-ray analysis. See supporting information.
We were intrigued by the large difference in both reactivity and enantioselectivity among diastereomeric catalysts 8 and 9. To further probe this variance, a series of competition experiments were performed between catalysts that allowed us to assess the relative rates of product formation for each catalyst.[10] By means of this assessment, trans-fluorinated pre-catalyst 9 is drastically (~13 fold) more active than cis-fluorinated pre-catalyst 8. Even more remarkable is the difference in reactivity between pre-catalyst 9 and achiral catalyst 12. Pre-catalyst 9 is the most sterically encumbered scaffold that we have examined to date, yet is still more reactive than achiral pre-catalyst 12 which lacks a bulky directing group.
In our initial report of the asymmetric intermolecular Stetter using fluorinated triazolium salt pre-catalyst 9, we proposed that backbone fluorination of the triazolium salt results in a conformational change due to the gauche effect.[4c] Our recent DFT study has provided evidence that an attractive electrostatic interaction between the C-F dipole in the catalyst and the developing nitronate in the transition state is the source of increased selectivity.[11] Due to the divergence in both the stereochemical outcome of this reaction as well as the relative stereochemistry of the fluorinated catalyst architecture required for good selectivity, a different effect may be operative.
To further understand the effect of fluorination of the catalyst architecture in this system we undertook a DFT study. Reactions with catalysts 7-9 were quantum mechanically investigated in order to resolve the origin of selectivity. The focus of our investigation is to determine why the R enantiomer of the product is favoured, and why trans-fluorinated catalyst 9 is more selective than cis-fluorinated catalyst 8 and des-fluoro catalyst 7.
Calculations were performed with Gaussian09.[ 12 ] All geometries were optimized with B3LYP/6-31G(d) with the CPCM solvation model[13] for methanol (UAKS radii, methanol, ε = 32.6). Single point calculations were performed on the B3LYP geometries with M06-2X/6-31+G(d,p), again using the CPCM model for methanol. Aldehyde 1a was modelled with propionaldehyde. DFT calculations predict good transition state geometries[14] and have been used to study the Stetter reaction and related reactions.[15,11] M06-2X provides more accurate selectivities and thermochemistries and incorporates dispersion effects. [16] Including the implicit solvation model was important for predicting accurate selectivities.
Catalysts 7-9 react with propionaldehyde to form acyl anion equivalents 13-15, respectively (Figure 2). The favoured conformations of intermediates 13-15 are important for determining the stereocenter that is formed in the product. No minima for the endo conformers could be located as all optimizations beginning from endo conformations converge to the exo conformations. Optimizations with the triazolium ring frozen in the endo conformation predict that these conformations are disfavoured by 5.2-7.5 kcal/mol and are therefore too high in energy to be involved in the reaction. As shown in our previous computational study,[11] the A1,3-like strain involving the alkyl substituent (here t-Bu) is extremely large in the endo conformers of the enol intermediates, and these conformers are not involved in subsequent reactions steps.
Figure 2.
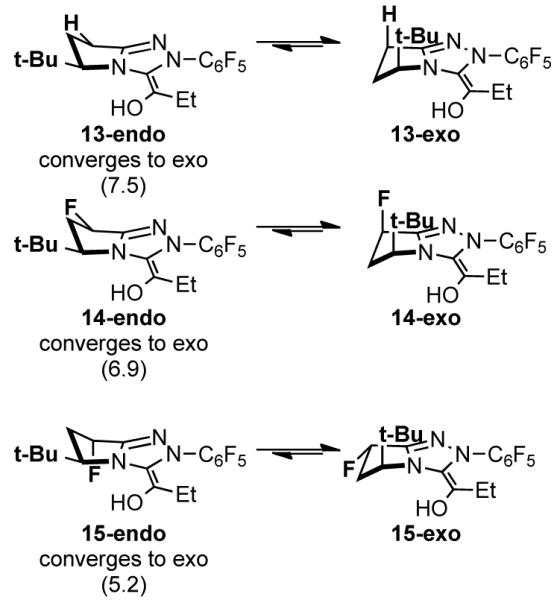
Relative free energies (M06-2X/6-31+G(d,p),CPCM(MeOH)//B3LYP/6-31G(d),CPCM(MeOH) of acyl anion equivalents 13-15. Endo conformations were located by freezing the triazolium ring.
Acyl anion equivalents 13-15 exo attack either the Si or Re face of the nitrostyrene 2 leading to transition structures TS1, TS3, TS5 or TS2, TS4, TS6, respectively (Figure 3). This step determines the stereocenter formed, but is not the rate-determining step.[17] Conformations resulting from the rotation about the forming carbon-carbon bond were considered. The optimized geometries for the most favourable transition structures are shown in Figure 3. Unlike previous studies on similar Stetter reactions,[11] TS1-TS6 do not all follow Seebach’s topological rule,[18] which describes a preference for a synclinal orientation of the double bonds of donors and acceptors in Michael addition transition states.
Figure 3.

Transition structures TS1-TS6 and energies (M06-2X/6-31+G(d,p),CPCM(MeOH)//B3LYP/6-31G(d),CPCM(MeOH). The dipole moments are based on Mulliken charges and are given in Debye.
In agreement with experiment, addition to the Re-face of the nitrostyrene is favoured (TS2, TS4, TS6) and catalyst 9 is computed to be the most selective. A gauche – orientation of the double bonds in the Si-face attack (TS1, TS3, TS5) places the nitro group under the catalyst ring. This is electrostatically favourable for TS1 and TS3. However, for TS5 the negative electrostatic interactions between the fluorine, which points down in catalyst 9, and the nitro group disfavours this conformation. For TS5 the gauche + conformation is favoured because it is well solvated due to its large dipole moment. The anti conformation for the Re-face attack is favoured[19] due to the stabilizing interaction between the hydrogen of the hydroxyl group and the carbon α to the nitro group,[20] favourable electrostatic interactions between the alkyl group of the aldehyde and the nitro group and because it is well solvated. These combined effects are enough to favour the Re-face attack over the Si-face attack. Additionally, TS6 is especially favourable because the negative π cloud of the phenyl ring is near the electropositive catalyst ring and the positive part of the phenyl ring is near the electronegative fluorine. Therefore, TS6 has the lowest barrier, making catalyst 9 the most selective.
The experimental and calculated % ee are given in Table 3. The calculated % ee is determined by a Boltzman distribution of all optimized transition structures.[21] These values correlate well with the experiment but are slightly underestimated.
Table 3.
Calculated and experimental percent enantiomeric excesses.
| Catalyst | Calculated[a] %ee |
Experimental %ee |
|---|---|---|
| 7 | 62 | 80 |
| 8 | 51 | 74 |
| 9 | 80 | 93 |
M06-2X/6-31+G(d,p),CPCM(MeOH)//B3LYP/6-31G(d), CPCM(MeOH).
In conclusion, we have identified a novel catalyst, capable of inducing high levels of enantio-induction in the intermolecular Stetter reaction of aliphatic aldehydes and nitrostyrenes. Trans-fluorination of the catalyst architecture promotes unparalleled reactivity and enantioselectivity in this transformation compared to previously known scaffolds. Computations show that catalyst 9 is the most stereoselective because the Re-face attack (TS6) is stabilized by favourable electrostatic interactions between the phenyl group and the fluorine on the catalyst backbone. Further computational studies on relative reaction rates and on selectivities of the triazolium catalysts 7-9 are underway.
Experimental Section
To a dry 4 mL vial, with a magnetic stir bar, was added triazolium salt 9 (22 mg, 0.05 mmol, 0.2 equiv), β-nitrostyrene (38 mg, 0.25 mmol, 1.0 equiv), sodium acetate (8 mg, 0.10 mmol, 0.4 equiv) and tert-amyl alcohol (2 mL, 0.125 M). The vial was cooled to 0 °C in a cooling bath with stirring and purged with argon. Butyraldehyde (34 μl, 0.375 mmol, 1.5 equiv) was added dropwise and the reaction was stirred at 0 °C until TLC indicated consumption of the nitrostyrene (24-48 h), at which point the reaction was concentrated in vacuo. The residue was purified by flash chromatography (hexanes:ether) which provided the desired β-nitro ketone as a colorless oil.
Supplementary Material
Figure 1.
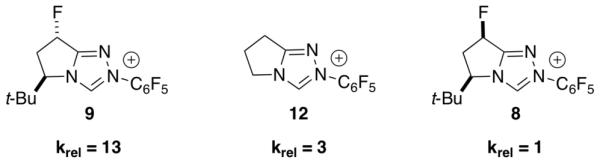
Influence of catalyst structure on relative rate. See supporting information for details. BF4 counterions omitted for clarity.
Footnotes
We thank NIGMS for generous support of this research (GM 36700 to K.N.H. and GM 72586 to T.R.). T.R. thanks Amgen and Roche for unrestricted support. KNH is grateful to the National Institute of Health Chemistry-Biology Interface Training Program Grant (T32GM008496) and the National Science Foundation (CHE-0548209) for financial support, TeraGrid resources provided by NCSA (CHE-0400414) and the UCLA Academic Technology Services (ATS) Hoffman2 and IDRE clusters for computational resources.
Contributor Information
Daniel A. DiRocco, Department of Chemistry, Colorado State University, Fort Collins, CO 80526 (USA)
Elizabeth L. Noey, Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095
Prof. K. N. Houk, Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095.
Prof. Tomislav Rovis, Department of Chemistry, Colorado State University, Fort Collins, CO 80526 (USA).
References
- [1].For reviews see: Marion N, Díez-González S, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. Moore J, Rovis T. Top. Curr. Chem. 2009;290:77–144. doi: 10.1007/978-3-642-02815-1_18. Vora HU, Rovis T. Aldrichimica Acta. 2011;44:3–11.
- [2].a) Stetter H, Schrecke M. Angew. Chem. Int. Ed. 1973;12:81. [Google Scholar]; b) Stetter H. Angew. Chem. Int. Ed. Engl. 1976;15:639–647. [Google Scholar]; c) Stetter H, Kuhlmann H. Org. React. 1991;40:407–496. [Google Scholar]; d) Christmann M. Angew. Chem., Int. Ed. 2005;44:2632–2634. doi: 10.1002/anie.200500761. [DOI] [PubMed] [Google Scholar]; e) Rovis T. Chem. Lett. 2008;37:2–7. [Google Scholar]
- [3].For a review of the asymmetric intramolecular Stetter reaction, see: Read de Alaniz J, Rovis T. Synlett. 2009:1189–1207. doi: 10.1055/s-0029-1216654.
- [4].a) Liu Q, Perreault S, Rovis T. J. Am. Chem. Soc. 2008;130:14066–14067. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Q, Rovis T. Org. Lett. 2009;11:2856–2859. doi: 10.1021/ol901081a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) DiRocco DA, Oberg KM, Dalton DM, Rovis T. J. Am. Chem. Soc. 2009;131:10872–10874. doi: 10.1021/ja904375q. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) DiRocco DA, Rovis T. J. Am. Chem. Soc. 2011;133:10402–10405. doi: 10.1021/ja203810b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For other contributions to the asymmetric intermolecular Stetter reaction, see: Enders D, Han J, Henseler A. Chem. Commun. 2008:3989–3991. doi: 10.1039/b809913h. Enders D, Han J. Synthesis. 2008:3864–3868. Jousseaume T, Wurz NE, Glorius F. Angew. Chem. Int. Ed. 2011;50:1410–1414. doi: 10.1002/anie.201006548. Sanchez-Larios E, Thai K, Bilodeau F, Gravel M. Org. Lett. 2011;13:4942–4945. doi: 10.1021/ol202040b. Fang X, Chen X, Lv H, Chi YR. Angew. Chem. Int. Ed. 2011;50 doi: 10.1002/anie.201105812. doi: 10.1002/anie.201105812.
- [6].For reviews of fluorine’s effect on molecular conformation see: Hunter L. Beilstein J. Org. Chem. 2010;6(38) doi: 10.3762/bjoc.6.38. Zimmer LE, Sparr C, Gilmour R. Angew. Chem. Int. Ed. 2011 doi: 10.1002/anie.201102027. DOI: 10.1002/anie.201102027.
- [7].Enders has reported the asymmetric Stetter of butanal and chalcone with two different chiral thiazolium catalysts (4% yield, 39% ee and 29% yield, 30% ee); see: Enders D, Bockstiegel B, Dyker H, Jegelka U, Kipphardt H, Kownatka D, Kuhlmann H, Mannes D, Tiebes J, Papadopoulos K. Wege zu neuen Verfahren und Produkten der Biotechnologie. DECHEMA-Monographie. 1993;129:209. Enders D. In: Stereoselective Synthesis. Ottow E, Schöllkopf K, Schulz B-G, editors. Springer-Verlag; Berlin: 1994. p. 63. Enders D, Breuer K. Comprehensive Asymmetric Catalysis. Springer; Berlin: 1999. pp. 1093–1104. Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534–541. doi: 10.1021/ar030050j.
- [8].a) Glorius et al reported a single example of an asymmetric intermolecular Stetter reaction of an aliphatic aldehyde adding to a β-unsubstituted Michael acceptor; see ref. 5c. b) An enzyme catalyzed asymmetric Stetter reaction has recently been published providing Stetter products of acetaldehyde: Dresen C, Richter M, Pohl M, Lüdeke S, Müller M. Angew. Chem. Int. Ed. 2010;49:6600–6603. doi: 10.1002/anie.201000632.
- [9].The use of stronger bases and/or more polar solvents leads to base-induced decomposition products.
- [10].See supporting information for complete experimental details.
- [11].Um JM, DiRocco DA, Noey EL, Rovis T, Houk KN. J. Am. Chem. Soc. 2011;133:11249–11254. doi: 10.1021/ja202444g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Frisch MJ, et al. Gaussian 09, revision A.1. Gaussian, Inc.; Wallingford, CT: 2009. [Google Scholar]
- [13].(a) Barone V, Cossi M. J. Phys. Chem. A. 1998;102:1995. [Google Scholar]; (b) Cossi M, Rega N, Scalmani G, Barone V. J. Comput. Chem. 2003;24:669. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- [14].Simon L, Goodman JM. Org. Biomol. Chem. 2011;9:689. doi: 10.1039/c0ob00477d. [DOI] [PubMed] [Google Scholar]
- [15].(a) Dudding T, Houk KN. PNAS. 2004;101:5770. doi: 10.1073/pnas.0307256101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hawkes KJ, Yates BF. Eur. J. Org. Chem. 2008;33:5563. [Google Scholar]
- [16].(a) Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215. [Google Scholar]; (b) Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- [17].Moore JL, Silvestri AP, de Alaniz JR, DiRocco DA, Rovis T. Org. Lett. 2011;13:1742. doi: 10.1021/ol200256a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Seebach D, Golinski J. Helv. Chim. Acta. 1981;64:1413. [Google Scholar]
- [19].The anti conformation is favoured for TS4 and TS6. The gauche + conformation is only slightly favored (0.5 kcal) over the anti conformation for TS2.
- [20].The addition of the acyl anion equivalent to the olefin is a concerted reaction reminiscent of the reverse-Cope elimination, as we and others have previously proposed; see: Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2005;127:6284–6289. doi: 10.1021/ja0425132. Piel I, Steinmetz M, Hirano K, Fröhlich R, Grimme S, Glorius F. Angew. Chem. Int. Ed. 2011;50:4983–4987. doi: 10.1002/anie.201008081. DiRocco DA, Rovis T. Angew. Chem. Int. Ed. 2011;50:7982–7983. doi: 10.1002/anie.201102920.
- [21].A table of the all transition structure energies is given in SI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.