

Abstract
Mutational activation of growth factor signaling pathways is commonly observed and often necessary for oncogenic transformation. Under physiologic conditions, these pathways are subject to tight regulation via negative feedback which limits the extent and duration of signaling events after physiologic stimulation. Until recently, the role of these negative feedback pathways in oncogene driven cancers has been poorly understood. In this review, I will discuss the evidence for the existence and relevance of negative feedback pathways within oncogenic signaling networks, the selective advantages such feedback pathways may confer and the effects such feedback might have upon therapies aimed at inhibiting oncogenic signaling.
Biological systems must maintain homeostasis in the face of various physiologic perturbations and environmental stresses. One of the main mechanisms employed to enable homeostasis is ‘negative feedback’. Negative feedback may simply be thought of as a direct output of a given stimulus that serves to deactivate that stimulus. The role of negative feedback in generating stability in complex systems is observed at all levels of biologic organization: in ecosystems (e.g. predator-prey relationships), multisystem organ regulation (e.g. thyroid hormone production), and intracellular function (e.g. transcriptional regulation via operons). The regulation of growth factor signaling pathways by negative feedback is a universal mechanism for limiting the extent and duration of signaling output. The manifold roles of negative feedback loops on signaling systems in normal cell physiology and developmental biology have been well appreciated (1, 2). More recently, several studies have sought to determine the implications of feedback regulation of pathways that are driven by constitutively activated oncoproteins in tumor cells. In this review, I will discuss the relevance of negative feedback pathways within oncogenic signaling networks, the selective advantages these pathways may confer and the implications of feedback regulation for therapies aimed at inhibiting growth factor signaling.
Signal activation and negative feedback
The binding of epidermal growth factor (EGF) to the EGF receptor triggers a rapid succession of intermolecular binding and phosphorylation steps that induce and amplify the signal to ultimately drive procession through cell cycle checkpoints, transcription factor activation of gene expression modules, assembly of macromolecules to promote protein translation, and alterations in choice of nutrient utilization programs (Figure 1). On a timescale of minutes to hours, these signaling events also induce negative regulatory events. These events cause the signal to be self-limited and help specify its strength and duration (Figure 1, last panel). These negative regulatory loops are initiated as a direct consequence of EGFR activation. Several mechanisms for feedback regulation of the EGFR pathway have been identified: (1) the GRB2 adaptor protein recruits the CBL E3 ubiquitin ligase to mediate endocytosis and downregulation of EGFR, (2) the ERK1/2 kinase phosphorylates upstream pathway components SOS and RAF at sites that promote their inactivation, (3) the MAPK phosphatase that inhibits ERK1/2 function is transcriptionally upregulated in response to ERK1/2 activation, (4) activation of AKT and mTOR results in downregulation of cooperating RTKs like HER3 and IGF1R (3, 4). These represent only a fraction of the negative feedback events that limit the effects of growth factor stimulation.
Figure 1. Growth factor activation of signaling and negative feedback.
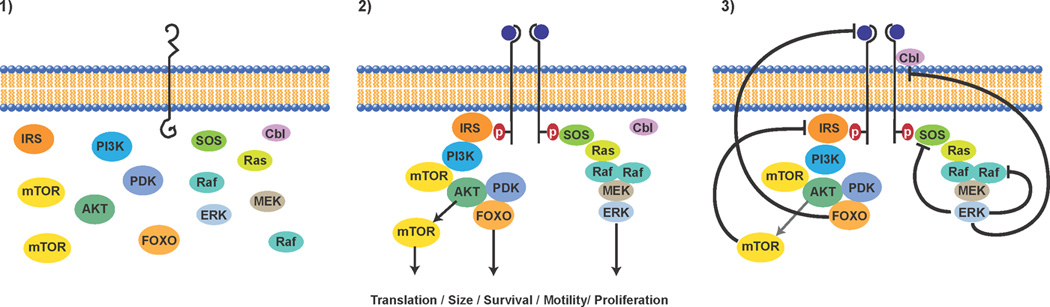
Depicted in the first panel are major elements of the EGFR signaling transduction apparatus in a disassembled state in the absence of growth factor stimulation. In the second panel, addition of growth factor triggers receptor conformational change, receptor dimerization, receptor transphosphorylation, binding of adaptor proteins, activation of kinase cascades, and stimulation of cellular programs involved in transformation (cell cycle progression, evasion from apoptosis, motility and invasion, increases in cell size, stimulation of protein translation). In the third panel, negative feedback programs are depicted including ERK phosphorylation of SOS and RAF that downregulates their activity, induction of the CBL E3 ligase that lowers EGFR expression, FOXO mediated repression of expression of receptor tyrosine kinases such as HER3, and mTOR mediated destabilization of the IRS1 adaptor protein via S6K activation.
Theoretical advantages of negative feedback
The presence of negative feedback modules in signaling systems from yeast to drosophila to man suggests that they confer selective advantages. Biochemical and computational methods have identified several possible mechanisms whereby regulation through negative feedback improves cellular fitness. One major consequence of feedback inhibition of growth factor signaling by negative feedback is that it increases the `robustness’ of the system: preserving network stability in the face of environmental and genetic stress (5). This property was illustrated by an analysis of the effects of different feedback states on the response of the MAPK signaling pathway to growth factor (6). Iyengar and colleagues compared a ‘low feedback state’ (depressed MAPK phosphatase expression) and a ‘feedback present state’ for ERK response to PDGF and found that the low feedback state exhibited a bistable response to growth factor. In this case, in the absence of PDGF, there was no ERK signaling, whereas ERK signaling was maximally stimulated over a wide range of PDGF concentrations ranging from low to high. By contrast, the ‘feedback present state’ exhibited a proportional response with different amounts of growth factor inducing different levels of ERK activation. The experiments suggest that the presence of feedback could potentially provide advantages in terms of fitness and appropriate response relative to the condition of the environment. A related advantage of feedback regulation of signaling systems is that it allows diversification in signal response. Induction of negative feedback by morphogens such as Sonic Hedgehog plays a role in cell fate specification by enabling unique cellular responses to subtle differences in ligand concentration (7). As such, the presence of negative feedback can potentially allow the diversity necessary for multicellular structures and organs that may be selectively advantageous in less favorable environments. Finally, feedback inhibition plays an integral role in most signaling pathways because it serves to dampen and turn off the signal thereby preventing potentially toxic overactivation of signaling output in response to growth factors or other stimuli. Moreover, in systems regulated by positive feedback loops, the presence of a negative feedback loop could prevent runaway activation in response to a signal.
While it is clear that negative feedback is a key component of normal cellular signaling pathways and provides selective advantages that ensure its preservation, the role of negative feedback in cancer cells is more complex. The unregulated proliferation of cancer cells is driven in many cases by constitutively activated oncoproteins that cause hyperactivation of growth factor signaling pathways. The selective advantages of feedback modules that can downregulate such signals are less obvious. Perhaps mutational events that attenuate negative feedback are a critical step of the process of transformation. On the other hand, loss of negative regulation with complete dysregulation of signaling output is likely to result in cell death. We may ask whether oncoproteins induce constitutive negative feedback in cancer cells or whether these pathways are inactivated during carcinogenesis.
Negative feedback is associated with oncogene induced senescence
Several common, oncogenic lesions in growth factor signaling pathways (e.g. mutant RAS or loss of PTEN) fail to fully transform normal cells. Oncogene induced senescence is among the mechanisms that is thought serve as a safeguard against transformation (8). Studies by Courtois-Cox et al. described how constitutive activation of RAS signaling due to loss of the RAS-GAP NF1 or overexpression of a constitutively active C-RAF allele leads to transient activation of AKT and ERK pathways. Activation of these pathways results in induction of the expression of several families of proteins (Sproutys, Dual specificity phosphatases (DUSPs), and RAS-GAPs) that function in part by feedback inhibiting RAS signaling (9). Induction of their expression was associated with profound downregulation of AKT and ERK activity and stimulation of a senescence phenotype. Senescence was thought to be related to induction of the activity of the FOXO tumor suppressor in response to feedback inhibition of AKT and ERK signaling. While these studies did not define the loss of the negative feedback as sufficient for ultimate transformation, they did describe a set of negative feedback responses that emerged from oncogene activation and associated their presence with tumor suppressive functions.
EGFR signaling is regulated by negative feedback
The role of negative feedback in regulating the EGFR-ERK pathway was explored by Amit et al. (10). They studied EGF-dependent induction of transcription in HeLa cells and demonstrated that EGF induces numerous negative feedback components including proteins that can downregulate ERK (e.g. DUSP2/3/4/6/7) and ERK transcriptional programs (e.g. JUNB, ATF3, FOSL, ID1, KLF2). While many of the previously described negative feedback pathways governing EGFR signaling are preserved in cancers, when expression of negative feedback regulators was compared between normal and cancer cells, several were found to be underexpressed in tumors. These data helped confirm the continued existence of negative feedback modules in transformed cells while also suggesting that loss of some of the negative feedback components might contribute to tumor progression.
Persistence of negative feedback in tumors
In tumors characterized by the presence of a mutationally activated oncoprotein in the signaling pathway, one might expect the persistently elevated signaling to induce persistently high levels of negative feedback. Alternatively, the expectation might be that the persistently elevated signaling was enabled by the loss or suppression of negative feedback. Pratilas et al. evaluated ERK dependent transcriptional output and feedback signaling in tumor cells in which ERK signaling is driven by mutant BRAF and in tumor cells in which ERK is driven by upstream receptors like EGFR or HER2 (11). Comparison of the tumors in the steady state and under conditions of MEK inhibition revealed that a negative feedback program (DUSPs, Sproutys) is expressed at a considerable higher level in the BRAF-mutant tumors. The observation is in line with the first expectation, an activating oncoprotein in the signaling pathway leads to persistently elevated feedback. However, the presence of high level feedback in these tumors raises a conundrum, how is the mutant oncoprotein able to signal and generate oncogenic output in the face of elevated and persistent negative feedback? The findings in the paper on relative MEK and ERK phosphorylation as well as more recent studies suggest that, at least in the case of mutant BRAF in melanoma, the oncoprotein is insensitive to the feedback. The BRAF V600E mutant can signal independently of the constraints imposed by negative feedback regulating RTKs and RAS (12). This of course does not rule out the possibility that individual components of the entire negative feedback program are lost or modulated during transformation with mutant BRAF as well, but much of the feedback program is evidently preserved. While in the case of mutant BRAF melanoma, insensitivity to the feedback appears to be critical to oncogenic signaling, in other tumor types, downregulation of the feedback or additional mutational hits to bypass the feedback may be essential. Indeed, as the cancer genome is studied with greater breadth and depth, concurrent mutational lesions (e.g. loss of PTEN and activating PIK3CA mutations in uterine cancer) within an individual growth factor signaling pathway are being increasingly found and one suggestion is that these lesions may arise in response to the negative feedback loops to enhance pathway output. In general, the negative feedback may be one of the major selection pressures for the specific type(s) of oncogenic hits that arise in a given tumor.
Relief of feedback uncovered by mTORC1 inhibitors
The concept that oncoproteins induce high levels of feedback inhibition of the signaling network has important implications for targeted therapy. Tumor cells have been said to be `addicted’ to driver oncoproteins. Indeed, drugs that inhibit oncoprotein activated pathways have been aggressively developed as therapeutics and some of these have remarkable clinical effects. This oncoprotein dependence may be due to the feedback inhibition of important parallel and upstream pathways that cause the cell to become hyperdependent on the oncoprotein alone. Inhibition of oncoprotein-signaling by these drugs not only downregulates the pathway, but also relieves feedback inhibition of other pathways in the network. The consequences of such loss of negative feedback have been most clearly described in the case of drugs targeting the PI3K/AKT/mTOR pathway (Figure 2). It had been previously established in normal cells that activation of the IGF/insulin signaling is regulated and limited by feedback inhibition of the expression of insulin receptor substrates (IRS1,2). This is mediated by activation of the PI3K/AKT/mTOR pathway. mTOR phosphorylates and activates S6K1 which phosphorylates the IRS proteins and both induces their degradation and reduces their interaction with the IGF1R and insulin receptors. This serves to inhibit and self-limit IGF1R and insulin signaling (13).
Figure 2. Negative feedback regulation of PI3K/AKT/mTOR signaling.
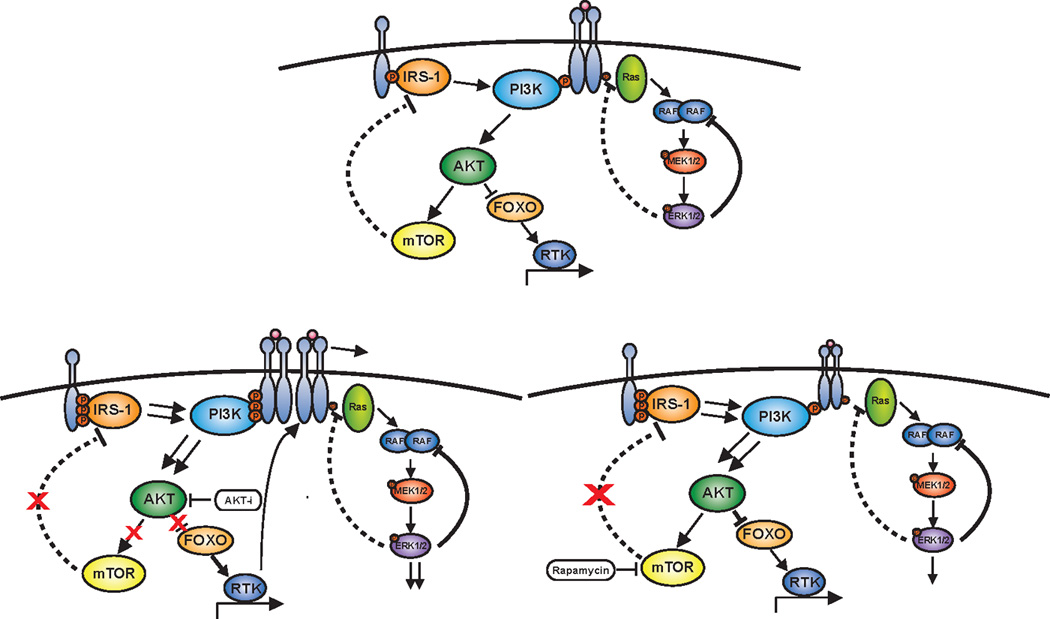
Depicted in the top panel is negative feedback regulation of PI3K/AKT/mTOR signaling through two major pathways from mTOR and AKT. mTOR regulates adaptor proteins such as IRS1 while AKT regulates the expression of receptor tyrosine kinases (RTK) through the FOXO transcription factors. The consequence of drug inhibition of AKT is shown in the bottom left panel with inhibition of AKT causing loss of negative feedback on RTK expression and so inducing RTK expression. In addition, AKT in many cells activates mTOR and so drug inhibition of AKT leads to inhibition of mTOR leading to loss of negative feedback on IRS1. The sum consequence of AKT inhibition is to activate RTK function through adaptors and increases in RTK expression. Whereas, mTORC1 inhibition with rapamycin in the bottom right panel predominantly impacts RTK function through the effects on adaptor proteins without the effects on RTK expression.
The PI3K/AKT pathway is constitutively activated in many human tumors. This suggests that S6K-dependent feedback inhibition of signaling might be an important characteristic of these tumors. Rapamycin is an inhibitor of the mTORC1 complex of mTOR and has been tested as an antitumor drug that would inhibit PI3K/AKT/mTOR signaling. Several groups found, however, that rapamycin relieved S6K dependent feedback and activated AKT in tumor cell lines and in human tumors (14–16). As the action of the drug led to potent and prolonged inhibition of S6K, IRS1 was stabilized and this was associated with an induction of PI3K and AKT kinase activity. Thus, relief of the negative feedback caused activation of a segment of the pathway the drug was meant to inhibit. This specific finding of AKT induction by rapamycin was further confirmed in patients in a neoadjuvant trial of rapamycin in PTEN-deficient glioblastoma where upregulation in P-AKT by IHC was observed in 7/10 patients and correlated with shorter time to progression after surgery (17). With more comprehensive profiling of tumors, additional mechanisms of negative feedback regulation of mTOR signaling are now being identified. For instance, it was recently shown that the adaptor protein GRB10 is directly phosphorylated by mTORC1 (18, 19). Phosphorylation stabilizes GRB10 protein thereby enhancing its suppression of PI3K. So, inhibitors of mTORC1 function relieve negative feedback in at least two key ways; they result in enhanced stability of IRS1 and diminished stability of GRB10. In both cases, these actions result in enhanced PI3K-AKT activity. While these are two important mechanisms of mTOR regulation, it appears likely that several other forms of negative feedback regulation of mTOR exist as well. The redundancy with which mTOR is feedback regulated raises the possibility that this phenomenon will occur with many inhibitors of oncoprotein activated pathways and that relief of feedback could attenuate or prevent the expected therapeutic effects of such drugs.
Negative feedback regulation is multifaceted
Hyperactivation of the mTOR program in cancer is commonly observed in numerous cancers through mutational events such as loss of the PTEN and INPP4B phosphatases, mutational activation of receptor tyrosine kinases such as EGFR and HER2, mutational activation of the small G protein RAS, and mutational activation of the lipid kinase PI3K. A major route of activation of the mTOR kinase involves activation of the PI3K which generates the second messenger PIP3 (20). Increased levels of PIP3 lead to membrane recruitment and activation of AKT through phosphorylation by PDK1 and mTORC2 on T308 and S473 respectively. Activated AKT then signals to mTORC1 through at least two routes. First, AKT phosphorylates PRAS40 which helps relieve its inhibitory binding to mTORC1. Second, AKT phosphorylates and inhibits TSC2 resulting in activation of mTORC1 by the RHEB GTPase negatively regulated by TSC2. As the lesions activating this pathway in cancer are predominantly upstream of AKT, direct therapeutic targeting of AKT was envisioned as an approach to block pathway activation while avoiding the consequences of loss of the S6K-IRS1 feedback loop. However, this approach rested on the supposition that the predominant mechanism of negative feedback suppression of the PI3K/AKT/mTOR pathway is mediated by the mTOR regulated IRS1/GRB10 loops. This did not turn out to be the case. To look for negative feedback modules that might regulate AKT function, a panel of breast tumors was screened for the response of upstream signaling components to AKT pathway inhibition (Figure 2). Across a wide variety of tumor types, AKT inhibition, but not mTORC1 inhibition, was shown to induce the RNA expression of a set of receptor tyrosine kinases (HER3, IGF1R, insulin receptor) with known functions in activating PI3K/AKT signaling (4). This was found to be mediated through the FOXO family of transcription factors which are direct AKT substrates. As such, an mTOR independent function of AKT is to exert negative feedback on receptor tyrosine kinases. From the different effects of mTORC1 versus AKT inhibition upon upstream signaling components comes an additional lens through which we might view targeted therapy of growth factor signaling, part of the action of the drug is to relieve the specific program of negative feedback that normally regulates that molecule. The implications of the drug/target-specific feedback profile will vary based on a number of factors (tumor genotype, lineage, and microenvironment most obviously), but the data suggest it would be insufficient to view the drug as only modulating the oncogenic functions of the target.
Relief of feedback can re-activate the inhibited pathway
Activation of the AKT kinase has been shown to require a series of steps. Binding of the pleckstrin homology domain with PIP3 poises the kinase for processive phosphorylations that occur in the regulatory domain (S473) and catalytic domain (T308). Mutagenesis studies suggest that all of these steps are required for the oncogenic functions of AKT (21). An alternative approach to simultaneously inhibit AKT and mTOR was hypothesized through use of an mTOR kinase inhibitor. Such an inhibitor that binds to the ATP pocket of mTOR would block both mTORC1 complex that regulates S6K1 and 4EBP1 as well as mTORC2 that regulates S473 phosphorylation on AKT. At a minimum, such an inhibitor would be predicted to relieve the negative feedback that regulates the mTORC1 (IRS/GRB10) and the negative feedback that regulates AKT (HER3/IGF1R/IR). Rodrik-Outmezguine et al. analyzed the impact of mTOR kinase inhibition and found that indeed the effect of the mTOR kinase inhibitor was to activate upstream PI3K/RTK signaling among other components yet to be fully elucidated (22). The activation of PI3K through adaptors and RTKs did not alter the ability of the inhibitor to block mTORC2 phosphorylation of S473 on AKT. The surprising finding was that under these conditions of hyperactivated upstream signaling, T308 on AKT was phosphorylated independently of S473 and led to an activated AKT that could resume its oncogenic functions. Inhibition of this S473 inhibited/T308 phosphorylated AKT species could be achieved through inhibitors of the induced RTKs or of the pleckstrin homology domain of AKT and this could durably inhibit AKT’s oncogenic functions. These findings illustrate an important concept about the consequence of negative feedback relief, that hyperactivated signals can re-activate the signaling pathway despite continued presence and action of the inhibitor. In this case, the enzymatic function of AKT could be decoupled from its normal regulatory constraints. The loss of negative feedback becomes a tool the network utilizes to adapt to the pressure of the drug and generate a new steady state that preserves the essential output of the network.
Relief of feedback can activate parallel growth factor pathways
The eukaryotic cell possesses a series of growth factor signaling cascades that it utilizes to respond to extracellular cues. In many cases, modules such as RAS/RAF/MEK/ERK or PI3K/AKT/mTOR can be adapted onto the same receptor complex through adaptor proteins so enabling multiple pathways to be activated by a single growth factor. For instance, heterodimers of the ERBB family can activate the PI3K/AKT cascade directly through sites on HER3 or adaptors such as GRB2 while also directly activating the RAS/RAF/MEK/ERK cascade through sites on EGFR/HER2/HER4 or adaptors such as SHC. Indeed, oncogenic activation of EGFR or HER2 is associated with hyperactivated signaling through both the ERK and AKT pathways. Similarly, activated RAS can activate several signaling modules important for transformed phenotypes including the RAF/MEK/ERK, PI3K/AKT/mTOR, and RAL/CDC42 pathways. As these upstream effectors can simultaneously activate multiple pathways that may promote tumor growth, conditions that promote loss of negative feedback and activate the upstream components will activate multiple growth factor signaling modules. For instance, Carracedo et al. demonstrated that inhibition of mTORC1 with rapamycin not only activated PI3K-AKT signaling but also induced ERK phosphorylation in cell lines and tumor biopsies from patients treated with the drug (23). This activation of ERK was demonstrated to be PI3K dependent and associated with upregulation of CRAF. The specific mechanism of upstream activation was not identified in this analysis, but demonstrated the point that persistent inhibition of one pathway could feedback upregulate signaling through another pathway and this could limit the antitumor benefit of the drug. Serra et al. demonstrated that inhibition of PI3K/AKT/mTOR signaling could induce the activity and expression of HER family receptors through some of the aforementioned mechanisms of RTK feedback regulation and this resulted in activation of ERK signaling (24). In this case, simultaneous inhibition of HER kinases blocked the induction in ERK phosphorylation. While much of the early work on such signaling cross-talk has focused on the canonical PI3K/AKT/mTOR and RAS/RAF/MEK/ERK modules, other signaling modules (e.g. JAK/STAT) have been described to be interconnected and are predicted to be involved. The precise outputs that these pathways can transduce under conditions of potent inhibition of one pathway are unknown. In the case of the AKT and ERK pathways, several key mediators of the transformed phenotype are co-regulated by these pathways and the activity of both may be necessary for output. For instance, in tumors with RAS and PIK3CA mutations, downregulation of both AKT and ERK is necessary to fully recruit the 4EBP1 suppressor to the eIF4E mRNA cap complex to block cap-dependent translation (25). In breast tumors with EGFR overexpression and PTEN loss, both ERK and AKT signaling are activated and inhibition of both pathways is necessary for dephosphorylation and activation of the pro-apoptotic BH3 containing protein, BAD (26). While these models for dual pathway regulation of tumorigenic functions may not be true for all tumor types, they imply another tumor escape mechanism where upregulation of crosstalk as a consequence of loss of negative feedback engages these mediators and prevents the antitumor action of the drug.
Relief of feedback activating hormone signaling pathways
Growth factor signaling pathways not only have well established roles collaborating with other polypeptide growth factor signaling pathways, they also interface with non-polypeptide growth factor signaling pathways such as hormone receptor signals. Cooperative roles for the AKT and ERK pathways with estrogen and androgen receptor signaling in breast and prostate cancer have been well established. Recent work has highlighted another potential implication for loss of negative feedback with androgen receptor signaling. Carver et al. utilized fine transcriptome analyses of prostate cancer to identify cross talk pathways between androgen receptor and PI3K/AKT signaling (27). In this work, they define “reciprocal feedback” wherein loss of negative feedback is identified under conditions of either androgen deprivation or PI3K inhibition and result in activation of the reciprocal pathway (Figure 3). So, inhibition of PI3K/AKT signaling drives androgen signaling in part through upregulation of ERBB kinase expression and activity. Meanwhile androgen deprivation induces PI3K/AKT activity through repression of the PHLPP phosphatase that regulates AKT. The mechanistic understanding of how these discrete signaling pathways can be linked raises a theory about how such negative feedback might play a role in the evolution of tumors. Mutational activation of one signaling network will repress lateral signaling pathways through induction of negative feedback programs. For instance, loss of PTEN ought to suppress androgen receptor signaling in part through negative feedback on RTKs. The pressure of this relative androgen deprivation for key androgen outputs may help select for mutational events that drive up androgen receptor expression or androgen production. The tumor cell with coexistent mutations is then buffered against scarcity of signals from either pathway. Periods of relative androgen deprivation are compensated by hyperactivation of PI3K/AKT signaling through further loss of PHLPP. Therapeutic efforts at targeting such mutationally mature tumors with monotherapies will similarly drive the cell to dependence on the other through hyperactivation from loss of negative feedback. To the degree this logic holds, rational combinations of therapies must target both the pathway and the loss of negative feedback signal in order to achieve durable antitumor benefits.
Figure 3. Reciprocal feedback regulation of PI3K/AKT and AR signaling.
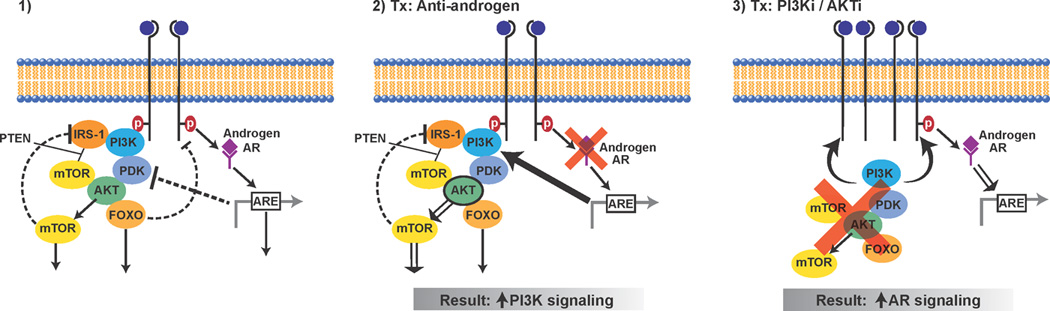
On the left panel is a model depicting crosstalk between the PI3K/AKT pathway and the androgen receptor (AR) signaling pathway with each pathway negatively regulating the other. In the middle panel is the consequence of inhibiting AR signaling with downregulation of AR causing lower levels of FKBP5 and thus impairing the function of the AKT phosphatase PHLPP. The result is that AR inhibition causes an upregulation in AKT activity. In the right panel is the consequence of inhibiting PI3K/AKT signaling with downregulation of AKT causing an induction of RTKs such as HER3 and this causing an induction of AR signaling.
Antitumor effects of targeting relief of negative feedback
A key prediction of the hypothesis that loss of negative feedback diminishes the antitumor benefit of targeted therapy is that combinations that can successfully target the loss of negative feedback signal should add meaningful benefit. In a variety of laboratory models this has proven to be the case although it has been experimentally difficult to prove that the loss of negative feedback signal was the sole reason. Sergina et al. defined how HER2 inhibition with the HER1/2 kinase inhibitor lapatinib led to loss of negative feedback on HER3 and this mediated resistance to the inhibitor (28). Knocking down the induced HER3 indeed augmented the apoptotic response, but was this through blockade of the induced HER3 or simply superior HER2/HER3 inhibition in the first place? In a similar vein, our group utilized an ineffective as monotherapy schedule of HER1/2 inhibitor to block the feedback signal induced by AKT inhibition and found this to clearly improve the antitumor effects beyond what either inhibitor alone achieved (4). The studies defining upregulation of AKT or ERK in response to mTORC inhibition also demonstrated the added antitumor benefit of blocking ERK with a MEK inhibitor or AKT with a PI3K inhibitor. However, such experiments cannot exclude the possibility that the benefits arise from inhibiting more pathways or more potent inhibition of the pathway upfront. In all cases, the experiments published to date have been highly consistent with the concept that strategies that incorporate blocking the loss of negative feedback are superior in depth and duration of antitumor benefit; nevertheless a definitive set of experiments to selectively isolate the impact of loss of negative feedback remains to be demonstrated.
Clinical efforts at targeting relief of negative feedback
Given the possible synthetic lethal effects of targeting the oncogenic pathway and one of the loss of feedback signals, several groups are now testing combinations designed to block loss of feedback signals. As some of the initial findings of loss of negative feedback were identified with rapamycin, several trials have examined combinations with this compound. In HER2 amplified breast cancer, the rapamycin induced activation of AKT is HER2 dependent (SC, unpublished). Moreover, one of the primary modes of resistance to HER2 targeted therapies has been mutational activation in PI3K or loss of PTEN, suggesting an epistatic role for mTOR inhibitors. As such, combinations of rapamycin with HER2 targeted therapy represent a rational approach. Morrow et al. combined everolimus with the HER2 antibody trastuzumab and observed a 15% response rate in trastuzumab refractory patients which is higher than either single agent might be predicted in this setting (29). This study can only incompletely model loss of negative feedback given that trastuzumab is a weak inhibitor of HER2 signaling, particularly in trastuzumab refractory disease, and so the drug may not have been sufficient to block the rapamycin feedback. Our group has recently reported on a Phase 1/2 study of trastuzumab refractory HER2+ breast cancers combining temsirolimus with an irreversible HER1/2 inhibitor, neratinib. Although patient numbers are small, 9/12 patients treated at the maximally tolerated dose had a partial response with two of these responses lasting for more than 16 months (30). Such activity has not been without some toxicity and raises the concern that these negative feedback pathways represent mechanisms that normal cells employ to avoid death from targeted therapy. While neither study has reported untoward or magnified toxicities beyond what has been seen for the single agents, this concern may represent an obstacle to efforts at such combinations. Two understandings needed in this regard are a fuller description of which negative feedback pathways are cancer cell specific for survival as well as knowledge of the dose and schedule of the combinations which maximize therapeutic index.
Future directions in targeting adaptive resistance
Overall, the findings from both the laboratory and the clinic implicate negative feedback as an important and targetable mechanism of drug resistance. At a minimum there are four areas in which this concept needs to be advanced in order to move beyond pioneering examples. First, a more comprehensive view of the feedback program regulating a given oncoprotein-driven network is needed. Studies examining the effects of drugging the PI3K/AKT/mTOR and RAS/RAF/MEK pathways have revealed dense layers of negative feedback rather than singular feedback loops. Some of these pathways are unique to specific cell lineages while others are unique to specific tumor genotypes. The powerful technologies for global assessment of gene expression/modification, protein expression/modification, etc. need to be employed to identify characteristic responses to selective inhibitors of these signaling networks. The ability to mine such data will depend heavily on the use of inhibitors that are highly selective for their target and cancer models that effectively mimic the genetic complexity of the diseases being studied. As common feedback modules are identified, a second key area for research on negative feedback in cancer is to define the relevance of the individual feedback programs for adaptive phenotypes. Based on the findings to date, it is very likely that numerous negative feedback loops will be found for a given oncoprotein. It may not be practical or desirable to block all of the feedback programs induced by the drug. Indeed some of these feedback loops may be utilized by normal cells to survive a drug and avert toxicity. Nevertheless, cancer cells may have an Achilles heel in terms of specific feedback loops that are essential for survival. Apart from inducing cell death, some of the adaptive resistance phenotypes are likely to engender other changes in cell fate. For instance, drug induced feedback may enable a metastatic or quiescence program rather than activating continued proliferation. Careful analysis of the cell biology of adaptive programs will be essential to identifying combination therapeutic strategies that effectively translate to patients. While such rational combination strategies to target the oncoprotein and the negative feedback program are being identified, it will be essential to simultaneously identify biomarkers that predict which tumor types will most benefit. To the degree that mTORC1 inhibition induces AKT and ERK, in which patients should we add a MEK inhibitor and in which patients should we add an AKT inhibitor? The third major area of understanding needed to advance effective translation of this concept will be development of robust predictors of response for such combinations. In this regard, it will be necessary for studies performed on feedback responses to extend beyond 1 or 2 cell lines and identify genotype, cell of origin, chromatin state, or other marks to ultimately guide patient selection. Finally, while much of this work will be most realistically achieved using cancer cell lines, mouse models, and patient derived xenografts, there remain reasons to be concerned that these will not fully capture the clinical feedback response. A critical component of the feedback response relies on upstream components like receptors that are strongly influenced by the microenvironment and ligand milieu in which they are found. Moreover, the pharmacodynamics of drug inhibition in the tumor are not well modeled outside the host due to specific circumstances such drug metabolism, tumor vasculature, etc. Despite the hurdles involved, there is still no effective substitute for the gold standard of the patient tumor biopsy. As drugs are developed in the clinic, carefully planned tumor biopsies prior to therapy and while on therapy will yield vital information towards understanding and validating the feedback responses.
The oncoprotein driven signaling pathway can be more realistically envisioned as a web with multiple interconnected inputs. Pressuring the web at a singular point will move some aspects of it in a specified direction, but other significant regions will run counter to this in part due to negative feedback. Synthetic lethality may be an achievable outcome by pressuring the network at both a key node and the major negative feedback signal that is relieved in response. Exploiting such vulnerabilities may greatly improve the chances of therapeutic success. Avant-garde trial designs incorporating tumor biopsies to identify adaptive responses and drug them in patient specific ways may ultimately represent the idealized way forward but will necessarily involve the commitment of investigators, philanthropists, and pharmaceutical companies to understanding and modulating these networks in all their complexity and heterogeneity.
Significance.
Negative feedback pathways are ubiquitous features of growth factor signaling networks. As growth factor signaling networks play essential roles in the majority of cancers, their therapeutic targeting has become a major emphasis of clinical oncology. Drugs targeting these networks are predicted to inhibit the pathway but also to relieve the negative feedback. This loss of negative feedback can itself promote oncogenic signals and cancer cell survival. Drug-induced relief of feedback may be viewed as one of the major consequences of targeted therapy and a key contributor to therapeutic resistance.
Acknowledgements
I would like to thank Neal Rosen, Poulikos Poulikakos, Marie Will, Brett Carver, Charles Sawyers, Maurizio Scaltriti, and Jose Baselga for their insights and helpful discussions and Eric Joseph for his comments and assistance with illustrations. S.C. is supported by a National Institutes of Health Mentored Clinical Scientist Award K08-CA134833 and the Geoffery Beene Cancer Center.
References
- 1.Alon U. An Introduction To Systems Biology: Design Principles Of Biological Circuits. Boca Raton, FL: Chapman & Hall/CRC; 2007. [Google Scholar]
- 2.Ferrell JE., Jr. Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Current opinion in cell biology. 2002;14:140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- 3.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nature reviews. 2011;12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 4.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stelling J, Sauer U, Szallasi Z, Doyle FJ, 3rd, Doyle J. Robustness of cellular functions. Cell. 2004;118:675–685. doi: 10.1016/j.cell.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogenactivated protein kinase signaling network. Science. 2002;297:1018–1023. doi: 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- 7.Ribes V, Briscoe J. Establishing and interpreting graded Sonic Hedgehog signaling during vertebrate neural tube patterning: the role of negative feedback. Cold Spring Harbor perspectives in biology. 2009;1:a002014. doi: 10.1101/cshperspect.a002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 9.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer cell. 2006;10:459–472. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, et al. A module of negative feedback regulators defines growth factor signaling. Nature genetics. 2007;39:503–512. doi: 10.1038/ng1987. [DOI] [PubMed] [Google Scholar]
- 11.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, et al. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–794. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- 14.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Molecular cancer therapeutics. 2005;4:1533–1540. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 16.Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26:1603–1610. doi: 10.1200/JCO.2007.14.5482. [DOI] [PubMed] [Google Scholar]
- 17.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 21.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. The EMBO journal. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 22.Rodrik-Outmezguine V, Chandarlapaty S, Pagano N, Poulikakos PI, Scaltriti M, Moskatel E, et al. mTOR Kinase Inhibition causes Feedback-Dependent Biphasic Regulation of AKT Signaling. Cancer Discovery. 2011;1:248–259. doi: 10.1158/2159-8290.CD-11-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of clinical investigation. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011 doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.She QB, Solit DB, Ye Q, O'Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer cell. 2005;8:287–297. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol. 2011;29:3126–3132. doi: 10.1200/JCO.2010.32.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gajria D, King T, Pannu H, Sakr R, Seidman A, Modi S, et al. Combined inhibition of mTORC1 with Temsirolimus nad HER2 with Neratinib: A Phase I/II study in patients with metastatic HER2-amplified or triple-negative breast cancer. San Antonio Breast Cancer Symposium; San Antonio. AACR; 2011. [Google Scholar]