

Background: 2-(Quinolin-3-yl)-acetic-acid derivatives target HIV-1 integrase and inhibit viral replication.
Results: The compounds are allosteric integrase inhibitors (ALLINIs) that block integrase interactions with viral DNA and its cellular cofactor LEDGF and cooperatively inhibit HIV-1 replication.
Conclusion: ALLINIs block multiple steps of HIV-1 integration.
Significance: These new properties of ALLINIs will facilitate their further development as potent antiretroviral compounds.
Keywords: Allosteric Regulation, HIV-1, Integrase, Protein-DNA Interaction, Protein-Protein Interactions
Abstract
The multifunctional HIV-1 enzyme integrase interacts with viral DNA and its key cellular cofactor LEDGF to effectively integrate the reverse transcript into a host cell chromosome. These interactions are crucial for HIV-1 replication and present attractive targets for antiviral therapy. Recently, 2-(quinolin-3-yl) acetic acid derivatives were reported to selectively inhibit the integrase-LEDGF interaction in vitro and impair HIV-1 replication in infected cells. Here, we show that this class of compounds impairs both integrase-LEDGF binding and LEDGF-independent integrase catalytic activities with similar IC50 values, defining them as bona fide allosteric inhibitors of integrase function. Furthermore, we show that 2-(quinolin-3-yl) acetic acid derivatives block the formation of the stable synaptic complex between integrase and viral DNA by allosterically stabilizing an inactive multimeric form of integrase. In addition, these compounds inhibit LEDGF binding to the stable synaptic complex. This multimode mechanism of action concordantly results in cooperative inhibition of the concerted integration of viral DNA ends in vitro and HIV-1 replication in cell culture. Our findings, coupled with the fact that high cooperativity of antiviral inhibitors correlates with their increased instantaneous inhibitory potential, an important clinical parameter, argue strongly that improved 2-(quinolin-3-yl) acetic acid derivatives could exhibit desirable clinical properties.
Introduction
HIV-1 integrase is an important antiretroviral target due to its essential role in virus replication (1). Multimeric integrase functions within the context of the preintegration complex to catalyze pair-wise integration of the linear viral DNA ends synthesized by reverse transcription into a host chromosome in a two-step reaction (2). In the first step, termed 3′-processing, integrase cleaves a GT dinucleotide from each 3′ terminus of viral DNA. Concerted transesterification reactions (DNA strand transfer) subsequently integrate both viral DNA ends into the host genome in a staggered fashion. Raltegravir, the clinically approved integrase inhibitor, specifically impairs the second step of integration. Although raltegravir results in significant reduction of viral loads in patients (3), HIV phenotypes resistant to this inhibitor evolve comparatively rapidly in the clinic (4). Therefore, there is a continued need for developing novel integrase inhibitors with alternative mechanisms of action.
We previously proposed one such alternative mechanism with a small molecule inhibitor that stabilizes interacting integrase subunits into an inactive multimeric form (5). Our biochemical studies indicated that highly dynamic individual subunits of integrase correctly assemble in the presence of viral DNA to form the functional nucleoprotein complex or intasome (6, 7). Restricting the molecular movement of individual integrase subunits within the multimer during its assembly with DNA compromised integrase enzyme activity (5–7). These observations were further supported by detailed analysis of the available crystal structure of the prototype foamy virus intasome (8) and corresponding molecular models of HIV-1 integrase-viral DNA complexes (6, 9). The organization of the individual integrase subunits within the intasome indicates that cognate viral DNA plays a crucial role in their assembly into the functional complex. In contrast, the preformed integrase tetramer in the absence of viral DNA would not allow binding of the two viral DNA ends as seen in both the crystal structures and molecular models (6, 8, 9). Therefore, premature multimerization of integrase before it encounters cognate DNA presents an attractive avenue for antiviral drug development.
Recently, inhibitors targeting the interaction between HIV-1 integrase and its key cellular cofactor LEDGF2 have been reported (10). LEDGF directly engages integrase through its C-terminal integrase binding domain (IBD) and tethers the viral protein to chromatin (11, 12). Principal protein-protein contacts of the integrase catalytic core domain (CCD) and N-terminal domain bound to the LEDGF IBD have been revealed in co-crystal structures (13, 14), with the extended interacting interfaces between full-length HIV-1 integrase and LEDGF further defined by MS-based protein footprinting (7). Christ et al. (10) exploited the co-crystal structure of the HIV-1 integrase CCD bound to the LEDGF IBD (14) to rationally design inhibitors of this central protein-protein contact. That study revealed several 2-(quinolin-3-yl) acetic acid derivatives that potently inhibited the integrase-LEDGF interaction in vitro as well as HIV-1 replication in infected cells (10). This class of compounds was termed LEDGINs, with one of the more potent inhibitors designated compound 6 (herein referred to as LEDGIN-6). Co-crystal structures of the LEDGIN-CCD complexes revealed that the compounds bind to the CCD dimer at the LEDGF binding pocket. Furthermore, selection of HIV-1 strains resistant to LEDGIN-6 identified an A128T resistance mutation that localized to the same pocket (10).
Our interest in LEDGINs and hence the present studies were prompted by the observation that they bind at the integrase dimer interface (10) adjacent to where we had previously mapped other small molecule inhibitors of integrase multimerization (5). We accordingly sought to test the hypothesis that LEDGINs could allostrically modulate the dynamic interplay between integrase subunits. In parallel experiments, we investigated the mechanism of action of another 2-(quinolin-3-yl) acetic acid derivative (Fig. 1A), which was patented by Boehringer Ingelheim as HIV replication inhibitor 1001 (15) (herein referred to as BI-1001). Remarkably, BI-1001 was derived from compounds identified via a fluorescence based high throughput screen for integrase 3′-processing activity, whereas LEDGIN-6, which was reported to be highly selective for disrupting integrase-LEDGF binding (IC50 = 1.37 μm), exhibited IC50 values of >250 μm and 19.5 μm for 3′-processing and strand transfer activities, respectively (10). We have analyzed these two compounds in parallel experiments, and our data clarify that LEDGIN-6 and BI-1001 have identical antiviral mechanisms. These compounds potently inhibit not only integrase-LEDGF binding but also LEDGF-independent integrase catalytic function. Furthermore, we demonstrate that the key to inhibiting integrase activities is through compound-mediated premature protein multimerization. Finally, we show that the inherent multimode mechanism of action of this class of inhibitors results in cooperative inhibition of concerted DNA integration in vitro and HIV-1 replication in infected cells.
FIGURE 1.

Effects of LEDGIN-6 and BI-1001 on the integrase-LEDGF binding. A, chemical structures of LEDGIN-6 and BI-1001. B, representative raw data of the inhibition dose response of LEDGIN-6 on the integrase-LEDGF interaction using the integrase-LEDGF HTRF assay. Each data point represents the mean of three independent experiments. C, curve fitting of dose-dependent inhibition of integrase-LEDGF binding by LEDGIN-6 (black squares) and BI-1001 (gray circles). The average values from three independent experiments are shown.
EXPERIMENTAL PROCEDURES
Chemical Synthesis of Integrase Inhibitors
2-(6-Chloro-2-methyl-4-phenylquinolin-3-yl)pentanoic acid (LEDGIN-6) was prepared in six steps from commercially available 2-amino-5-chloro-benzonitrile (Sigma-Aldrich) according to the scheme provided by Debyser and co-workers (10). 2-(6-Bromo-4-(4-chlorophenyl)-2-methylquinolin-3-yl)-2-methoxyacetic acid (BI-1001) was synthesized in five steps from commercially available 2-amino-4′-chlorobenzophenone (TCI America) through slight modification of the procedures reported in the patent (15). The chemical structures of these compounds are shown in Fig. 1A. Full experimental procedures and characterization data (1H and 13C NMR spectra) for the preparation of the compounds are provided in the supplemental data.
Construction of FLAG-tagged Proteins
C-terminally FLAG-tagged LEDGF was constructed as described previously (16). N-terminally FLAG-tagged integrase was constructed by PCR amplification of C-terminally His-tagged integrase construct pKBIN6Hthr (7) with T7T and InFlagN (5′-ggaattccatatggactacaaagacgatgatgacaaatttttagatggaatagataaggccc-3′) primers. The C-terminal His tag was then removed by insertion of a stop codon using site-directed mutagenesis. Sequences of PCR-generated regions of plasmid DNA were verified by Sanger sequencing.
Preparation of Recombinant Proteins and DNA Substrates
Full-length proteins were expressed in Escherichia coli strain BL21 (DE3). FLAG-tagged and tagless INs were purified by loading the ammonium sulfate precipitate of cell lysate onto a phenyl-Sepharose column (GE Healthcare) and eluting bound integrase with a decreasing ammonium sulfate gradient (800 mm to 0 mm) in a 50 mm HEPES (pH 7.5) buffer containing 200 mm NaCl, 7.5 mm CHAPS, 2 mm β-mercaptoethanol. Peak fractions were pooled and loaded onto a heparin column (GE Healthcare), and integrase was eluted with an increasing NaCl gradient (200 mm to 1 m) in a 50 mm HEPES (pH 7.5) buffer containing 7.5 mm CHAPS and 2 mm β-mercaptoethanol. Fractions containing integrase were pooled and stored in 10% glycerol at −80 °C. His-tagged integrase was purified as described previously (7, 17). Purified recombinant wild-type and FLAG-tagged LEDGF/p75 were obtained as described previously (18). The blunt-end viral DNA substrate (∼1 kb) for stable integrase-viral DNA complex formation was obtained by PCR and purified by agarose gel electrophoresis as described previously (6).
In Vitro Integration Assays
Integrase 3′-processing and strand transfer activities were assayed using 32P-labeled blunt ended 21-mer or recessed end 19-mer synthetic double-stranded U5 DNA, respectively. 500 nm integrase was preincubated with LEDGIN-6 or BI-1001 for 30 min on ice in 50 mm MOPS (pH 7.2) buffer containing 2 mm β-mercaptoethanol, 50 mm NaCl and 10 mm MgCl2. Then, 50 nm DNA substrate was added to the reaction and incubated at 37 °C for 1 h. The reactions were stopped with 50 mm EDTA. The reaction products were subjected to denaturing polyacrylamide gel electrophoresis and visualized using a Storm 860 Phosphorimager (Amersham Biosciences).
LEDGF-dependent concerted integration assays were carried out as described previously (13, 17). Briefly, 2 μm integrase was preincubated with increasing concentrations of LEDGIN-6 or BI-1001 at room temperature for 30 min in 22 mm HEPES (pH 7.4) buffer containing 25.3 mm NaCl, 5.5 mm MgSO4, 11 mm DTT, 4.4 μm ZnCl2. To this mixture, 1 μm viral donor DNA (32-mer blunt-ended U5) and 600 ng of target (pBR322) DNAs were added. Samples were incubated at 25 °C for 5 min, and then LEDGF was added at a final concentration of 2 μm, after which reactions proceeded for 90 min at 37 °C. Integration reactions stopped by addition of 0.5% SDS and 25 mm EDTA were deproteinized by digestion with 40 μg of proteinase K (Roche Applied Science) for 60 min at 37 °C. DNA products were separated in 1.5% agarose gels in Tris acetate-EDTA buffer and visualized by staining with ethidium bromide.
HTRF-based Integrase-LEDGF Interaction Assay
A previously described homogeneous time resolved fluorescence (HTRF) assay (16) was modified for the testing of inhibitors. Briefly, 10 nm N-terminally His-tagged integrase was pre-incubated in a binding buffer (150 mm NaCl, 2 mm MgCl2, 0.1% Nonidet P-40, 1 mg/ml BSA, 25 mm Tris (pH 7.4)) with the tested compound for 30 min at room temperature, and then 10 nm C-terminally FLAG-tagged LEDGF was added to the reaction. 6.6 nm anti-His6-XL665 and 0.45 nm anti-FLAG-EuCryptate antibodies (Cisbio, Inc., Bedford, MA) were then added to the reaction. After 4 h at 4 °C, the HTRF signal was recorded using a Molecular Devices M5 plate reader using 314 nm for excitation wavelength and 668 and 620 nm for the wavelength of the acceptor and donor emission, respectively. The HTRF signal is defined as the emission ratio 665 nm/620 nm multiplied by 10,000.
HTRF-based Integrase Multimerization Assay
Two separate preparations of His-tagged and FLAG-tagged INs (each at 10 nm final concentration) were mixed in 25 mm Tris (pH 7.4) buffer containing 150 mm NaCl, 2 mm MgCl2, 0.1% Nonidet P-40, 1 mg/ml BSA. Test compounds were then added to the mixture and incubated for 2.5 h at room temperature. 6.6 nm anti-His6-XL665 and 0.45 nm anti-FLAG-EuCryptate antibodies (Cisbio, Inc., Bedford, MA) were then added to the reaction and incubated at room temperature for 3 h. The HTRF signal was recorded as above.
Stable Synaptic Complex (SSC) Formation and SSC-LEDGF Binding Inhibition Assays
The previously reported methods (6) for assembly of the SSC and SSC-LEDGF binding were used for compound testing. Briefly, integrase was pre-incubated with the compound for 30 min at room temperature before adding viral DNA to assemble the SSC (6). In the SSC-LEDGF assay, the purified SSC was pre-incubated with inhibitor for 30 min at room temperature before addition of LEDGF. SSC-associated integrase and LEDGF proteins were separated by spin-size exclusion chromatography and analyzed by SDS-PAGE. Proteins were visualized by Western blot using monoclonal antibodies against integrase (8G4, National Institutes of Health AIDS Research and Reference Reagent Program (19)) and against human LEDGF (BD Biosciences).
Differential Scanning Fluorimetry
Differential scanning fluorimetry was performed on a LightCycler 480 96-well plate, real-time PCR instrument (Roche Applied Science) according to Nettleship et al. (20). Sypro orange was purchased from Invitrogen. Differential scanning fluorimetry is based on denaturation of the protein in the absence or presence of a ligand, exposing hydrophobic residues that can be detected with high sensitivity with a fluorescent dye. The melting temperature (Tm) of the protein is calculated from these data. After incubation of HIV-1 integrase and inhibitor at room temperature for 1 h in 50 mm MOPS (pH 7.2) 50 mm NaCl, 10 mm MgCl2, 2 mm β-mercaptoethanol, and 1% dimethyl sulfoxide, sypro orange was added to the final concentration of 0.1% (v/v). The mixture was subsequently heated in a LightCycler 480 from 30 to 90 °C in increments of 0.11 °C/s. Fluorescence intensity was measured using excitation/emission wavelengths of 483 and 610 nm, respectively. Changes in protein thermal stability (ΔTm) upon inhibitor binding were analyzed by using LightCycler 480 Software provided by the manufacturer. All assays were performed in duplicate.
Crystallization and X-ray Structure Determination
The HIV-1 integrase CCD (residues 50–212 containing the F185K mutation) was expressed and purified as described (21). The protein was concentrated to ∼8 mg/ml and crystallized at 4 °C using the hanging drop (2 μl) vapor diffusion method. The crystallization buffer contained 10% PEG 8K, 0.1 m sodium cacodylate (pH 6.5) 0.1 m ammonium sulfate, and 5 mm DTT, and cubic-shaped crystals reached 0.1- 0.2 mm within 4 weeks. A soaking buffer containing 5 mm BI-1001 was prepared by dissolving the compound in crystallization buffer supplemented with 10% dimethyl sulfoxide. The protein crystal was soaked in the buffer for 12 h at 4 °C before flash-freezing it in liquid N2. Diffraction data were collected at 100 F on a Rigaku Raxis 4++ image plate detector at the Ohio State University Crystallography Facility. The intensity data integration and reduction were performed with HKL2000 program (22). Molecular replacement program Phaser (23) in the CCP4 package method was used to solve the structure. Coot (24) was used for the subsequent refinement and building of the structure. Refmac5 (25) of the CCP4 package was used for the restraint refinement. TLS (26) and restraint refinement was applied for the last step of the refinement. The crystal belonged to space group P3121 with cell dimensions a = b = 73.082 Å and c = 64.808 Å, with one 18-kDa monomer in the asymmetric unit. The structure was refined to 2.45 Å with Rcryst/Rfree = 0.2308/0.2763.
Antiviral Activity Assays
CD4-positive SupT1 T cells were grown in RPMI 1640 medium supplemented to contain 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin, whereas HEK293T cells were maintained in Dulbecco's modified Eagle's medium modified to contain the same supplements. The concentration of HIV-1NL4-3 in the supernatant of plasmid pNL4–3-transfected HEK293T cells was determined using a radionuclide-based exogenous assay for reverse transcriptase (RT) activity (27), and SupT1 cells (4 × 104 per well of a 96-well plate) were infected with 5 × 105 32P counts per minute in 200 μl. The effective concentration of compound required to inhibit 50% (EC50) of HIV-1 replication was determined after 5 days using the WST-1 assay (Roche Applied Science) to quantify cell viability. Control compounds raltegravir and saquinavir were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program.
Curve Fittings
The fitted dose-response curves were obtained as follows. Reaction yields in the absence of the inhibitors were considered 100%. The IC50 and the Hill slope parameter m (28), which is analogous to the Hill coefficient n (29), for each reaction were determined from a respective dose-response curve using a modified Hill equation (Equation 1) and Origin software (OriginLab, Inc.). All fitted curves displayed a R2 of 0.97 or greater,
 |
where x is the inhibitor concentration, y is the percentage of inhibition, k is IC50 (or EC50 for antiviral measures), and n (or m for the virus data) is the Hill slope (28).
RESULTS
Two 2-(quinolin-3-yl) acetic acid derivatives, LEDGIN-6 and BI-1001, which potently inhibit HIV-1 replication, were discovered using two different approaches. LEDGIN-6 emerged through rational structure-based design to spatially mimic the interactions of LEDGF IBD hot spot residues Ile-365 and Asp-366 in their contacts with the integrase-CCD dimer interface and has been reported to selectively inhibit the integrase-LEDGF interaction (IC50 = 1.37 μm) but not integrase 3′-processing activity (IC50 > 250 μm) (10). Paradoxically (Fig. 1A), BI-1001 was identified via a high throughput screen for integrase 3′-processing activity (15). Therefore, it was important to evaluate LEDGIN-6 and BI-1001 in parallel to dissect their mechanism of action.
We first compared the compounds for their ability to inhibit the integrase-LEDGF interaction using the HTRF-based assay (Fig. 1B). Both compounds effectively impaired integrase-LEDGF binding. In triplicate repeats of these experiments, BI-1001 consistently was severalfold more potent than LEDGIN-6 (Fig. 1C). The compounds were next evaluated for their ability to inhibit integrase catalytic activities in the absence of LEDGF. For these experiments, we chose to employ commonly used 3′-processing and strand transfer activity assays that enable reliable quantitation of 32P-labeled DNA substrates and reaction products. The 3′-processing assays, which were conducted with a 21-mer blunt-ended DNA, revealed that LEDGIN-6 and BI-1001 inhibited integrase activity with IC50 values of 3.9 and 2.3 μm, respectively (Fig. 2, A and B; results summarized in Table 1). The strand transfer reactions with pre-processed donor DNA substrates were also inhibited by LEDGIN-6 (IC50 = 4.2 μm) and BI-1001 (IC50 = 1.7 μm) (Fig. 2, C and D; Table 1). Our results differ significantly from the previously reported LEDGIN-6 IC50 values of >250 and 19.5 μm for inhibiting integrase 3′-processing and strand transfer activities, respectively (10).
FIGURE 2.

Effects of LEDGIN-6 and BI-1001 on integrase 3′-processing and strand transfer activities. A, a representative gel image for 3′-processing inhibition by LEDGIN-6. The 21-mer DNA substrate (sub.) and 19-mer reaction product (prod.) are indicated. Lane 1, DNA load; lane 2, DNA+integrase without inhibitor; lane 3, 25 mm EDTA was included in the reaction; the remaining lanes contained the following concentrations of LEDGIN-6. Lane 4, 1 mm; lane 5, 500 μm; lane 6, 250 μm; lane 7, 125 μm; lane 8, 62.5 μm; lane 9, 31.25 μm; lane 10, 15.6 μm; lane 11, 7.8 μm; lane 12, 3.9 μm; lane 13, 1.95 μm; lane 14, 977 nm; lane 15, 488 nm; lane 16, 240 nm. B, curve fitting of the dose-dependent inhibition of integrase 3′-processing activity by LEDGIN-6 (black squares) and BI-1001 (gray circles). The average values from three independent experiments are shown. C, a representative gel image for strand transfer inhibition by LEDGIN-6. The 19-mer DNA substrate and strand transfer (ST) products are indicated. Lane 1, DNA load; lane 2, DNA+integrase without inhibitor; lane 3, 25 mm EDTA was included in the reaction; the remaining lanes contained the following concentrations of LEDGIN-6. Lane 4, 1 mm; lane 5, 500 μm; lane 6, 250 μm; lane 7, 125 μm; lane 8, 62.5 μm; lane 9, 31.25 μm; lane 10, 15.6 μm; lane 11, 7.8 μm; lane 12, 3.9 μm; lane 13, 1.95 μm; lane 14, 977 nm; lane 15, 488 nm; lane 16, 240 nm. D, curve fitting of the dose-dependent inhibition of integrase strand transfer activity by LEDGIN-6 (black squares) and BI-1001 (gray circles). The average values from three independent experiments are shown.
TABLE 1.
Activities of LEDGIN-6 and BI-1001
Average values with S.E. from the mean are shown for two or three independent experiments.
| Integrase-LEDGF binding | 3′-Processing | Strand transfer | Integrase multimerization | Concerted integration | Antiviral activity | |
|---|---|---|---|---|---|---|
| LEDGIN-6 IC50 (μm) | 10.0 ± 0.4 | 3.9 ± 0.5 | 4.2 ± 0.6 | 11.3 ± 1.1 | 12.9 ± 0.8 | 12.2 ± 2.9 |
| LEDGIN-6 Hill coefficient | 1.0 ± 0.03 | 0.9 ± 0.1 | 0.95 ± 0.1 | 1.7 ± 0.2 | 1.9 ± 0.2 | 3.9 ± 0.6 |
| BI-1001 IC50 (μm) | 1.0 ± 0.1 | 2.3 ± 0.1 | 1.7 ± 0.1 | 4.9 ± 0.3 | 5.4 ± 0.5 | 5.8 ± 0.1 |
| BI-1001 Hill coefficient | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.0 ± 0.1 | 1.9 ± 0.2 | 1.8 ± 0.3 | 3.7 ± 0.2 |
Our observations that the compounds inhibited integrase-LEDGF binding and inherent, LEDGF-independent integrase function equally well raised the question regarding the structural basis for their multimode mechanisms of action. We therefore solved the x-ray crystal structure of BI-1001 bound to the HIV-1 integrase CCD (Fig. 3A) and compared it with the previously reported co-crystal structure with LEDGIN-6 (Fig. 3B) (10). Comparative analysis revealed nearly overlapping drug binding. However, we note one important difference: the BI-1001 methoxy group, which is absent in LEDGIN-6, forms an additional H-bond with integrase residue Thr-174. This interaction is likely to account for the superior potency of BI-1001 over LEDGIN-6 in both LEDGF-dependent and -independent assays (Fig. 1 and 2; Table 1). Our discovery of relative potent inhibition of inherent integrase catalytic activities, and confirmation that BI-1001 binds to the CCD dimer interface at the same position as LEDGIN-6, defines both compounds as bona fide allosteric inhibitors of the HIV-1 integrase enzyme. We next sought to determine the mechanistic basis for allosteric inhibition.
FIGURE 3.

Structural analysis of the inhibitor-CCD complexes. A, the crystal structure of BI-1001 bound to the integrase CCD dimer. Surface views of individual integrase subunits are depicted in magenta and cyan. B, overlay of CCD-LEDGIN-6 (Protein Data Bank code 3LPU) and CCD-BI-1001 co-crystal structures. Schematic views of integrase subunits are colored as described in A; LEDGIN-6 and BI-1001 backbones are green and yellow, respectively. Compound oxygen and nitrogen atoms, as well as those of integrase residues Thr-174, Glu-170, and His-171, are colored red and blue, respectively (for simplicity, only main chain Glu-170 and His-171 atoms are shown). The inhibitor carboxyl groups H-bond (green and orange dashed lines for LEDGIN-6 and BI-1001, respectively) with the main chain nitrogens of Glu-170 and His-171, and to the Thr-174 side chain. The BI-1001 methoxy group forms an additional H-bond (black dashed line) with Thr-174.
Co-crystal structures (Fig. 3) (10) revealed that both compounds recapitulate the function of LEDGF hot spot residue Asp-366, in that they engage main chain nitrogens of integrase residues Glu-170 and His-171, thus elucidating the mechanism for inhibition of integrase-LEDGF binding. However, this site is removed significantly from the presumed viral donor or host chromosomal target DNA binding sites on HIV-1 integrase (6, 9). Although the Fig. 3 structures lack the DNA substrates, comparing the inhibitor-CCD complexes with the apo-CCD dimer did not indicate any gross differences in the positions of the integrase active site residues. Instead, because both inhibitors establish extensive interactions with both CCD subunits of the dimer, we hypothesized that the compounds might deregulate integrase-integrase interactions critical for enzyme function.
To test this hypothesis, we designed an HTRF-based assay to monitor the integrase-integrase interaction. Anti-His6-XL665 and anti-FLAG-EuCryptate antibodies allow fluorescence energy transfer upon interaction of two full-length, wild type HIV-1 integrase proteins, one containing an N-terminal His6 tag and the other, an N-terminal FLAG tag (Fig. 4A). Compounds that inhibit integrase-integrase binding would accordingly decrease the HTRF signal, whereas those that promote multimerization by stabilizing the interacting integrase subunits would increase the signal. Representative data with LEDGIN-6 revealed a striking dose-dependent increase of the HTRF signal (Fig. 4B). As a control, we used raltegravir, which targets the integrase active site distal from the CCD dimer interface. The HTRF signal predictably remained at the background level with increasing raltegravir concentrations (Fig. 4B). The data in Fig. 4C show that LEDGIN-6 and BI-1001 promoted integrase multimerization with IC50 values of 11.3 and 4.9 μm, respectively (Table 1).
FIGURE 4.

Effects of LEDGIN-6 and BI-1001 on integrase multimerization. A, HTRF assay design. The assay monitors the interaction between two integrase molecules: one containing His6 and the other containing the FLAG tag. The antibodies conjugated with Europium cryptate (Eu) and XL665 yield HTRF signal upon the protein-protein interaction. Europium cryptate is excited at 320 nm, and emissions at 665 and 620 nm are measured. The HTRF signal is calculated from the 665:620 nm ratio. B, representative raw data for affects of LEDGIN-6 (black bars) and raltegravir (gray bars) on integrase (IN) multimerization. Each data point represents the mean of three independent reactions. C, curve fittings of dose-response affects of LEDGIN-6 (black squares) and BI-1001 (gray circles) on integrase multimerization. The maximal HTRF signal, obtained at high compound concentrations, was set to 100%. The average values from three independent experiments are shown.
To further test the notion that LEDGIN-6 and BI-1001 stabilize interacting integrase subunits, we monitored the melting temperatures of free integrase protein and protein-inhibitor complexes. Fig. 5 shows that integrase complexes with LEDGIN-6 and BI-1001 were significantly more stable to thermal denaturation than free integrase. The kd values obtained from these experiments for LEDGIN-6 and BI-1001 were 8.0 and 5.3 μm, respectively. In control experiments, increasing concentrations of raltegravir did not affect integrase stability (Fig. 5A). A logical interpretation of data of Fig. 5 is that LEDGIN-6 and BI-1001 stabilize interacting integrase subunits (Fig. 4) and thus increase thermostability of the multimer.
FIGURE 5.

Effects of LEDGIN-6 and BI-1001 on integrase thermostability. A, representative raw data with LEDGIN-6 (black bars) and raltegravir (light bars). B, curve fitting of the dose-response effects of LEDGIN-6 (black squares) and BI-1001 (gray circles) on integrase (IN) thermostability. The average values from two independent experiments are shown. Temp., temperature.
Integration proceeds through the SSC or intasome comprising a tetramer of integrase acting on the two ends of linear viral DNA substrate that we and others (6, 8, 30) have shown is resistant in vitro to chaotropic agents such as high concentrations of salt. We therefore next tested whether integrase multimers assembled in the presence of LEDGIN-6 and BI-1001 retained the ability to form the SSC. In low ionic strength buffers, integrase forms both stable and nonspecific complexes with viral DNA and subsequent treatment of the reaction mixture with high NaCl, followed by spin-column chromatography yields the purified SSC (6). Because the limited amounts of the SSC recovered from these experiments did not permit us to carry out extensive dose-dependent analysis of LEDGIN-6 and BI-1001 activities, we tested two relatively high inhibitor concentrations (100 and 200 μm). We first pre-incubated LEDGIN-6 and BI-1001 with integrase and then supplied viral DNA to the reaction. Data of Fig. 6A show that both compounds effectively inhibited SSC formation. Collectively, our findings argue that LEDGF-6 and BI-1001 stabilize integrase multimers (Figs. 4 and 5), which are then incapable of forming the SSC (Fig. 6A) and accordingly lack catalytic function (Fig. 2).
FIGURE 6.

Effects of LEDGIN-6 and BI-1001 on SSC formation (A) and the SSC-LEDGF interaction (B). A, SDS-PAGE analysis of SSCs. Lane 1, 1/10 of integrase (IN) load; lane 2, protein markers (MagicMark XP Western Protein Standard (Invitrogen)); lane 3, SSC assembly without compound; lane 4, SSC assembly with 100 μm LEDGIN-6; lane 5, SSC assembly with 200 μm LEDGIN-6; lane 6, SSC assembly with 100 μm BI-1001; lane 7, SSC assembly with 200 μm BI-1001. Integrase was visualized by Western blotting. B, SDS-PAGE analysis of LEDGF interactions with the SSC. Lane 1, 1/100 of LEDGF load; lane 2, SSC load; lane 3, protein markers (MagicMark XP Western Protein Standard (Invitrogen)); lane 4, SSC plus LEDGF; lane 5, SSC incubated with 100 μm LEDGIN-6 plus LEDGF; lane 6, SSC incubated with 200 μm LEDGIN-6 plus LEDGF. Integrase and LEDGF were visualized by Western blot.
Effective pair-wise integration of HIV-1 DNA ends during infection requires the interaction of the SSC with LEDGF. Although data of Fig. 1 show that LEDGIN-6 and BI-1001 impaired integrase-LEDGF binding, it was important to examine whether these compounds also inhibited the interaction between LEDGF and the pre-assembled integrase-viral DNA complex. Both compounds effectively impaired this interaction (Fig. 6B and data not shown). Data of lanes 5 and 6 of Fig. 6B moreover revealed that the treatment of the preassembled SSC with LEDGIN-6 did not dissociate integrase from viral DNA. Comparative analysis of the data in Fig. 6, A and B, thus reveals the importance of order of addition. Addition of inhibitors to free integrase impair its ability to assemble with viral DNA (Fig. 6A, lanes 4–7), whereas the preformed integrase-viral DNA complex remains stable upon treatment with LEDGIN-6 or BI-1001 (Fig. 6B, lanes 4–6). The inhibitors, however, still effectively block SSC-LEDGF binding (Fig. 6B, lanes 4–6). Taken together, our results reveal that LEDGIN-6 and BI-1001 can disrupt at least two intermediate steps along the pathway of concerted HIV-1 DNA integration, namely: (i) proper integrase-integrase multimerization and hence formation of the basic catalytic SSC and (ii) the downstream interaction of the SSC with the LEDGF integration targeting host factor.
We next examined the compounds in an in vitro integration assay dependent on the LEDGF-integrase interaction for effective stimulation of concerted integration (13, 17). Although inhibition of pair-wise integration products was expected, the dose-response curves yielded Hill coefficients of ∼2, indicating cooperative modes of action for LEDGIN-6 and BI-1001 under these reaction conditions (Fig. 7 and Table 1). As alluded to above, we propose that such cooperativity is likely due to the ability of these compounds to impair two steps in the reaction pathway: inhibition of SSC formation and subsequent SSC-LEDGF binding. Conversely, in reactions where integrase-LEDGF binding (Fig. 1) or integrase catalytic function (Fig. 2) were monitored separately, Hill coefficients were ∼1 (Table 1). To make sure that a cooperative mode of action in the concerted integration reaction was specific to LEDGIN-6 and BI-1001, we conducted control experiments with raltegravir. No cooperativity was observed under these conditions (data not shown), due presumably to the fact that raltegravir impairs only the strand transfer step of HIV-1 integration.
FIGURE 7.

Effects of LEDGIN-6 and BI-1001 on integrase concerted integration activity. A, representative raw data of the inhibition dose response of LEDGIN-6 on LEDGF-dependent concerted integration activity. Positions of supercoiled (SC) target and 32-mer donor DNA substrates as well as half-site (HS) and full-site (FS) integration products are indicated. Lane 1, DNA markers (BIOLINE Quanti-Marker, 1 kb); lane 2, target DNA load; lane 3, integrase activities in the presence of LEDGF and target DNA and donor DNA substrates. The remaining lanes contained the following concentrations of LEDGIN-6: lane 4, 1 mm; lane 5, 500 μm; lane 6, 250 μm; lane 7, 125 μm; lane 8, 62.5 μm; lane 9, 31.25 μm; lane 10, 15.6 μm; lane 11, 7.8 μm; lane 12, 3.9 μm; lane 13, 1.95 μm; lane 14, 977 nm; lane 15, 488 nm; lane 16, 240 nm. Curve fitting of the inhibition dose responses of LEDGIN-6 (black squares) and BI-1001 (gray circles) on LEDGF-dependent concerted integration activity. The average values from two independent experiments are shown.
To determine whether cooperative inhibitor action in vitro extended to the physiologically relevant condition of HIV-1 replication, LEDGIN-6 and BI-1001 EC50 and Hill coefficient values were determined and compared with those of control compounds raltegravir and saquinavir. As established previously (28), the protease inhibitor saquinavir displayed cooperative inhibition (m = 2.6) under conditions where raltegravir failed to reveal evidence of cooperativity (m = 1.1). Both LEDGIN-6 and BI-1001 displayed highly cooperative inhibition, yielding m values of 3.9 and 3.7, respectively (Fig. 8 and Table 1).
FIGURE 8.
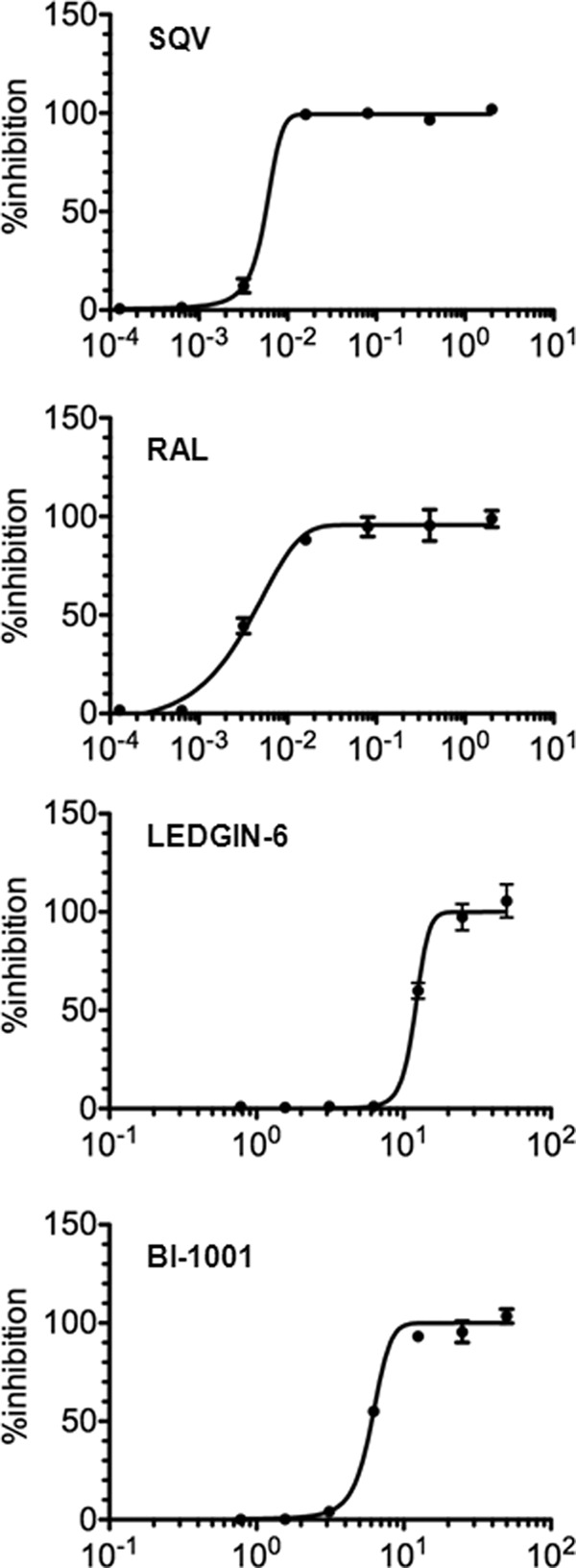
Dose-response curves of antiviral activities of saquinavir, raltegravir, LEDGIN-6, and BI-1001. The average values from two to three independent experiments are indicated. RAL, raltegravir; SQV, saquinavir.
DISCUSSION
Here, we investigated the mechanism of action of two prototypes of a growing number of small molecule compounds that bind HIV-1 integrase distal from the enzyme active site. In contrast to the previous report (10) indicating that LEDGIN-6 specifically inhibited the integrase-LEDGF interaction, we show conclusively that this inhibitor impairs both integrase-LEDGF binding and the inherent catalytic activities of integrase, which do not rely on LEDGF, with very similar IC50 values. Although structurally similar, BI-1001 was more potent than LEDGIN-6, and the two compounds displayed an overlapping mechanism of action. We therefore conclude that LEDGIN-6 and BI-1001 belong to the same class of inhibitors. Because these 2-(quinolin-3-yl) acetic acid derivatives allosterically modulate integrase structure, we propose to name this class of compounds allosteric integrase inhibitors.
We show that the underlying basis of inhibition of LEDGF-independent integrase catalytic function by allosteric integrase inhibitors is premature integrase multimerization (Figs. 4 and 5). Although the functional intasome or SSC contains a tetramer of integrase stably bound to the two viral DNA ends (8, 31), the highly dynamic interplay between free integrase subunits is critical for their productive assembly with viral DNA and hence functional SSC formation (7). In the absence of cognate DNA, free integrase can also multimerize, but these preformed integrase multimers do not form the SSC and accordingly lack integrase catalytic function (6). Previously, we showed that the LEDGF IBD promotes formation of integrase multimers (6, 7). Remarkably, the conformations of integrase multimers within integrase-LEDGF complexes formed in the absence of viral DNA and SSCs differ significantly, and the preassembled integrase-LEDGF IBD complex moreover lacks the ability to functionally integrate viral DNA ends (6, 32). Our findings that allosteric integrase inhibitors also modulate integrase multimerization and impair the formation of the SSC argue further for exploiting integrase multimerization as a novel therapeutic target. Due to their molecular mimicry of LEDGF hot spot residues Ile-365 and Asp-366, we postulate that allosteric integrase inhibitors recapitulate the inhibitory activities of anti-integrase peptides derived from the tip of the corresponding helix-hairpin-helix IBD structure (33).
The allosteric integrase inhibitor mode of action of stabilizing rather than inhibiting integrase subunit-subunit interactions has a major advantage in that these small molecules do not have to overcome the high energy barrier created by large interfaces between interacting protein subunits. Instead, they stabilize the interacting subunits to promote premature integrase multimerization. The substituted benzene ring of the compounds primarily engages one integrase monomer through hydrophobic interactions, whereas the carboxylic acid group, and in the case of BI-1001, the nearby methoxy moiety, hydrogen bond with the second integrase molecule (Fig. 3). Such compounds are likely to be effective during the early stages of HIV-1 replication when cognate DNA is unavailable for integrase until after viral DNA synthesis by RT is completed. LEDGIN-6 accordingly blocked the integration step of HIV-1 replication, and mutations in integrase conferred resistance to the compound (10).
In vitro concerted integration and ex vivo experiments have revealed a cooperative mechanism of action of allosteric integrase inhibitors. High cooperativity of antiviral compounds is important because it strongly influences the instantaneous inhibitory potential, the key clinical parameter for a retroviral drug that indicates the log reduction in a single round infectivity assay at clinical drug concentrations (28, 34). Inhibitors with high cooperativity or high instantaneous inhibitory potentials are particularly desirable for superior clinical outcomes. Comparative analysis (28, 34) of current HIV therapies under clinically relevant conditions have revealed that protease inhibitors and non-nucleoside RT inhibitors, which affect large pools of protease or RT, respectively, exhibit intermolecular cooperativity due to the importance of multiple copies of these proteins for virus maturation and reverse transcription. Consequently, these inhibitors exhibit high instantaneous inhibitory potential values, whereas inhibitors such as nucleoside RT inhibitors and raltegravir, that specifically target the active enzyme complexes, do not display cooperativity and thus have low instantaneous inhibitory potential values (∼1).
Based on our observations, it is logical to propose that allosteric integrase inhibitors could affect the entire population of integrase molecules (estimated to be ∼40 to 100 copies) produced in a single infectious cycle by promoting premature protein multimerization and thus impair the multiple functions of this key retroviral protein. Under simplified in vitro conditions, we identified two concerted integration intermediates that are effectively inhibited by allosteric integrase inhibitors, which likely accounts for the Hill coefficient of ∼2 observed in the concerted integration assay. Inhibition of integrase multimerization was also cooperative (Hill coefficient ∼ 2, Fig. 4, Table 1), due presumably to allosteric integrase inhibitors stabilizing integrase dimers and thus shifting the equilibrium toward their further assembly into tetramers. Premature integrase multimerization could be a key contributor of the greater Hill coefficient (∼4) observed under the ex vivo replication conditions, although additional work with viral replication intermediates will be needed to reveal the details. It is nevertheless noteworthy that mutations in integrase can affect a variety of steps along the HIV-1 life cycle, including virus assembly and release from virus producer cells, and subsequent viral core uncoating, reverse transcription, preintegration complex nuclear import, and integration in challenged target cells (reviewed in Refs. 35 and 36)).
Our findings, together with published results (10), argue strongly for further development of allosteric integrase inhibitors as well as studies to discover new inhibitors targeting integrase multimerization. Although the original goal of the rational design of small molecule inhibitors was to effectively compete with integrase-LEDGF binding (10), future efforts can consider enhancing allosteric integrase inhibitor properties to more tightly bridge the two integrase subunits that meet at the LEDGF binding cleft (14). As an example, our co-crystal structure (Fig. 3) indicates the significance of the BI-1001 methoxy group for establishing a unique H-bond with one of the integrase subunits. In general, structural analysis of the pocket at the integrase dimer interface reveals ample opportunities for further enhancing the ability of allosteric integrase inhibitors to more effectively engage both integrase subunits.
Discovery of new integrase multimerization inhibitors could proceed through a high throughput screen. Our method of identifying compounds that stabilize interacting integrase subunits (Fig. 4) exhibits excellent statistical parameters (Z′ = 0.87) and could be exploited for screening large chemical libraries. The rationale for pursuing these studies is provided by the present and prior findings that a number of small molecule inhibitors interact with the integrase CCD dimer interface (5, 37, 38). Significantly, two additional integrase domains (N-terminal and C-terminal domains) also are essential for functional protein multimerization (39, 40), and new inhibitors targeting these unexploited protein-protein interfaces are likely to emerge from high throughput screen. As established here, integrase multimerization inhibitors can be expected to behave cooperatively to disarm integrase molecules in excess of the four that compose the heart of the DNA recombination machine. They moreover can be expected to be active against raltegravir-resistant virus and hence complementary to current antiretroviral therapies (10). Our clarification of a cooperative mode of ALLINI action argues strongly that improved integrase multimerization inhibitors could exhibit desirable clinical properties.
Supplementary Material
Acknowledgment
We are grateful to Dr. Ross Larue for critical reading of the manuscript and helpful comments.
This work was supported by National Institutes of Health Grants AI081581, AI062520, and CA100730 (to M. K.), AI097044 (to J. J. K. and J. R. F.), AI039394 (to A. E.); the Intramural Research Program of the NCI, National Institutes of Health, Department of Health and Human Services (to H. T. S. and S. F. J. L. G.).

This article contains supplemental data.
- LEDGF
- lens epithelium-derived growth factor
- CCD
- catalytic core domain
- SSC
- stable synaptic complex
- IBD
- integrase binding domain
- HTRF
- homogeneous time resolved fluorescence.
REFERENCES
- 1. Johnson A. A., Marchand C., Pommier Y. (2004) HIV-1 integrase inhibitors: A decade of research and two drugs in clinical trial. Curr. Top Med. Chem. 4, 1059–1077 [DOI] [PubMed] [Google Scholar]
- 2. Brown P. O. (1997) in Retroviruses (Coffin J. M., Hughes S. H., Varmus H. E., eds), pp. 161–204, Cold Spring Harbor Laboratory, Plainview, NY [Google Scholar]
- 3. Murray J. M., Emery S., Kelleher A. D., Law M., Chen J., Hazuda D. J., Nguyen B. Y., Teppler H., Cooper D. A. (2007) Antiretroviral therapy with the integrase inhibitor raltegravir alters decay kinetics of HIV, significantly reducing the second phase. Aids 21, 2315–2321 [DOI] [PubMed] [Google Scholar]
- 4. Malet I., Delelis O., Valantin M. A., Montes B., Soulie C., Wirden M., Tchertanov L., Peytavin G., Reynes J., Mouscadet J. F., Katlama C., Calvez V., Marcelin A. G. (2008) Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob Agents Chemother 52, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kessl J. J., Eidahl J. O., Shkriabai N., Zhao Z., McKee C. J., Hess S., Burke T. R., Jr., Kvaratskhelia M. (2009) An allosteric mechanism for inhibiting HIV-1 integrase with a small molecule. Mol. Pharmacol. 76, 824–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kessl J. J., Li M., Ignatov M., Shkriabai N., Eidahl J. O., Feng L., Musier-Forsyth K., Craigie R., Kvaratskhelia M. (2011) FRET analysis reveals distinct conformations of IN tetramers in the presence of viral DNA or LEDGF/p75. Nucleic Acids Res. 39, 9009–9022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McKee C. J., Kessl J. J., Shkriabai N., Dar M. J., Engelman A., Kvaratskhelia M. (2008) Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. J. Biol. Chem. 283, 31802–31812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hare S., Gupta S. S., Valkov E., Engelman A., Cherepanov P. (2010) Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krishnan L., Li X., Naraharisetty H. L., Hare S., Cherepanov P., Engelman A. (2010) Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 15910–15915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Christ F., Voet A., Marchand A., Nicolet S., Desimmie B. A., Marchand D., Bardiot D., Van der Veken N. J., Van Remoortel B., Strelkov S. V., De Maeyer M., Chaltin P., Debyser Z. (2010) Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 6, 442–448 [DOI] [PubMed] [Google Scholar]
- 11. Maertens G., Cherepanov P., Pluymers W., Busschots K., De Clercq E., Debyser Z., Engelborghs Y. (2003) LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 278, 33528–33539 [DOI] [PubMed] [Google Scholar]
- 12. Cherepanov P., Devroe E., Silver P. A., Engelman A. (2004) Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 279, 48883–48892 [DOI] [PubMed] [Google Scholar]
- 13. Hare S., Shun M. C., Gupta S. S., Valkov E., Engelman A., Cherepanov P. (2009) A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog 5, e1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cherepanov P., Ambrosio A. L., Rahman S., Ellenberger T., Engelman A. (2005) Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U.S.A. 102, 17308–17313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsantrizos Y. S., Boes M., Brochu C., Fenwick C., Malenfant E., Mason S., Pesant M. (2007), pp. 1–116, International Patent Application PCT/CA2007/000845
- 16. Tsiang M., Jones G. S., Hung M., Mukund S., Han B., Liu X., Babaoglu K., Lansdon E., Chen X., Todd J., Cai T., Pagratis N., Sakowicz R., Geleziunas R. (2009) Affinities between the binding partners of the HIV-1 integrase dimer-lens epithelium-derived growth factor (IN dimer-LEDGF) complex. J. Biol. Chem. 284, 33580–33599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cherepanov P. (2007) LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic Acids Res. 35, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Turlure F., Maertens G., Rahman S., Cherepanov P., Engelman A. (2006) A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 34, 1653–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nilsen B. M., Haugan I. R., Berg K., Olsen L., Brown P. O., Helland D. E. (1996) Monoclonal antibodies against human immunodeficiency virus type 1 integrase: Epitope mapping and differential effects on integrase activities in vitro. J. Virol. 70, 1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nettleship J. E., Brown J., Groves M. R., Geerlof A. (2008) Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol. Biol. 426, 299–318 [DOI] [PubMed] [Google Scholar]
- 21. Dyda F., Hickman A. B., Jenkins T. M., Engelman A., Craigie R., Davies D. R. (1994) Crystal structure of the catalytic domain of HIV-1 integrase: Similarity to other polynucleotidyl transferases. Science 266, 1981–1986 [DOI] [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W. (1997) in Methods in Immunology (Carter C. W., Jr., Sweet R. M., eds) Part A, pp. 307–326, Academic Press, New York [Google Scholar]
- 23. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 26. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 27. Li X., Koh Y., Engelman A. (2012) Correlation of recombinant integrase activity and functional preintegration complex formation during acute infection by replication-defective integrase mutant human immunodeficiency virus. J. Virol. 86, 3861–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen L., Peterson S., Sedaghat A. R., McMahon M. A., Callender M., Zhang H., Zhou Y., Pitt E., Anderson K. S., Acosta E. P., Siliciano R. F. (2008) Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 14, 762–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hill A. (1910) J. Physiol. (Lond.) 40, iv–vii [Google Scholar]
- 30. Li M., Mizuuchi M., Burke T. R., Jr., Craigie R. (2006) Retroviral DNA integration: Reaction pathway and critical intermediates. EMBO J. 25, 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Michel F., Crucifix C., Granger F., Eiler S., Mouscadet J. F., Korolev S., Agapkina J., Ziganshin R., Gottikh M., Nazabal A., Emiliani S., Benarous R., Moras D., Schultz P., Ruff M. (2009) Structural basis for HIV-1 DNA integration in the human genome, role of the LEDGF/P75 cofactor. EMBO J. 28, 980–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pandey K. K., Sinha S., Grandgenett D. P. (2007) Transcriptional coactivator LEDGF/p75 modulates human immunodeficiency virus type 1 integrase-mediated concerted integration. J. Virol. 81, 3969–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hayouka Z., Rosenbluh J., Levin A., Loya S., Lebendiker M., Veprintsev D., Kotler M., Hizi A., Loyter A., Friedler A. (2007) Inhibiting HIV-1 integrase by shifting its oligomerization equilibrium. Proc. Natl. Acad. Sci. U.S.A. 104, 8316–8321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen L., Rabi S. A., Siliciano R. F. (2009) A novel method for determining the inhibitory potential of anti-HIV drugs. Trends Pharmacol. Sci. 30, 610–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Briones M. S., Chow S. A. (2010) A new functional role of HIV-1 integrase during uncoating of the viral core. Immunol. Res. 48, 14–26 [DOI] [PubMed] [Google Scholar]
- 36. Engelman A. (2011) in HIV-1 Integrase: Mechanism and Inhibitor Design (Neamati N., ed), pp. 67–81, John Wiley & Sons, Hoboken, NJ [Google Scholar]
- 37. Al-Mawsawi L. Q., Fikkert V., Dayam R., Witvrouw M., Burke T. R., Jr., Borchers C. H., Neamati N. (2006) Discovery of a small-molecule HIV-1 integrase inhibitor-binding site. Proc. Natl. Acad. Sci. U.S.A. 103, 10080–10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Molteni V., Greenwald J., Rhodes D., Hwang Y., Kwiatkowski W., Bushman F. D., Siegel J. S., Choe S. (2001) Identification of a small-molecule binding site at the dimer interface of the HIV integrase catalytic domain. Acta Crystallogr. D Biol. Crystallogr. 57, 536–544 [DOI] [PubMed] [Google Scholar]
- 39. Cai M., Zheng R., Caffrey M., Craigie R., Clore G. M., Gronenborn A. M. (1997) Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol. 4, 567–577 [DOI] [PubMed] [Google Scholar]
- 40. Eijkelenboom A. P., Lutzke R. A., Boelens R., Plasterk R. H., Kaptein R., Hård K. (1995) The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nat. Struct. Biol. 2, 807–810 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.