

Background: Calcium-binding proteins bind to intracellular termini of TRP channels.
Results: Two Ca2+-dependent binding sites for CaM/S100A1 were revealed by biophysical methods on TRPM3 N terminus.
Conclusion: CaM and S100A1 serve as ligands for TRPM3 channel.
Significance: Two clusters of positively charged residues form mutual binding sites for CaM and S100A1 proteins on TRPM3 N terminus.
Keywords: Calmodulin, Fluorescence, S100 Proteins, Surface Plasmon Resonance (SPR), TRP Channels
Abstract
Transient receptor potential melastatin 3 ion channel (TRPM3) belongs to the TRP family of cation-permeable ion channels involved in many important biological functions such as pain transduction, thermosensation, and mechanoregulation. The channel was reported to play an important role in Ca2+ homeostasis, but its gating mechanisms, functions, and regulation are still under research. Utilizing biophysical and biochemical methods, we characterized two independent domains, Ala-35–Lys-124 and His-291–Gly-382, on the TRPM3 N terminus, responsible for interactions with the Ca2+-binding proteins calmodulin (CaM) and S100A1. We identified several positively charged residues within these domains as having a crucial impact on CaM/S100A1 binding. The data also suggest that the interaction is calcium-dependent. We also performed competition assays, which suggested that CaM and S100A1 are able to compete for the same binding sites within the TRPM3 N terminus. This is the first time that such an interaction has been shown for TRP family members.
Introduction
The transient receptor potential melastatin 3 (TRPM3) ion channel is a member of the melastatin TRP2 subfamily. It is closely related to the first known member of the TRPM subfamily, TRPM1 (1). TRPM3 shares typical structural features with other TRP family members, such as six putative transmembrane-spanning domains, intracellular N and C termini, and a conserved TRP box and coiled-coil region in its C terminus (2, 3). There are no ankyrin repeat domains present on its N-tail; instead, there is a so-called TRPM homology region, a roughly 700-amino acid-long domain only found in TRPM family members (4).
The channel is proposed to mediate a Ca2+ concentration-dependent Ca2+ entry pathway in kidney cells and thus is thought to play an important role in renal Ca2+ homeostasis (2, 5). Recently, TRPM3 was also reported to serve as a nociceptor channel involved in the detection of noxious heat (6).
Calcium entry pathways make TRPM3 a probable target for Ca2+-binding proteins such as calmodulin and S100 proteins. Calmodulin (CaM) is a small cytoplasmic protein (16.7 kDa) that is ubiquitously expressed in all eukaryotic organisms. It contains four EF-hand motifs that bind Ca2+ ions cooperatively. When binding Ca2+, CaM undergoes a conformational change that increases its affinity for its target proteins (7). CaM was found to regulate the function of numerous TRP channels via binding to their intracellular termini. Multiple CaM binding sites were reported on either the N-tail or the C-tail of TRPC, TRPV, and TRPM ion channel family members (7–10). These domains conform to at least one of the consensus CaM recognition motifs 1-5-10 or 1-8-14 and often contain more than one of these binding motifs (7–10).
The S100 proteins are a group of proteins with at least 25 members found in humans. Most of these proteins bind calcium and undergo a conformational change enabling them to interact with specific target proteins and control cellular activity (11). The proteins contain two EF-hand calcium binding domains, and in contrast to CaM, the first EF-hand motif binds Ca2+ with a lower affinity than the second EF-hand (12, 13). S100 proteins have been shown to bind similar structural motifs to CaM (14), and the Ca2+-S100A1 complex also interacts with target proteins via a Ca2+-dependent mechanism to elicit biological responses. One of the first characterized proteins of this family was S100A1. It is a small (10.5-kDa) protein that forms dimers containing four EF-hand Ca2+ binding motifs.
Here we show that CaM and S100A1 bind to the N terminus of the TRPM subfamily. Using two recombinant protein constructs corresponding to the TRPM3 sequences 35–124 and 291–382, we were able to find CaM/S100A1 binding sites using a combination of biophysical and bioinformatical tools. Furthermore, we proved that these two Ca2+-binding proteins compete for these binding sites. Our results suggest that CaM and S100A1 serve as important regulators of TRPM3, which is known to play an important role in Ca2+ homeostasis. Whether CaM and S100A1 act as activators, inhibitors, or bidirectional regulators still remains elusive and will require further investigation.
EXPERIMENTAL PROCEDURES
Cloning and Site-directed Mutagenesis of Protein Constructs on N terminus of Human TRPM3
The N-terminal sequence of human TRPM3 was analyzed for a presence of potential CaM binding motifs with the Calmodulin Target Database (15). Based on this analysis, two putative CaM binding domains were found, Ala-35–Lys-124 (TRPM335–124) and His-291–Gly-382 (TRPM3291–382). cDNA from the human TRPM3 N terminus corresponding to these sequences was cloned into the bacterial expression vector pET32b (Novagen). Using the QuikChange (Stratagene) method, point mutations were created in both N-terminal protein constructs. The integrity of the coding region and the results of the mutagenesis were verified by sequencing.
Expression and Purification of N-terminal Human TRPM3 Protein Constructs
Protein constructs Ala-35–Lys-124 and His-291–Gly-382 of the human TRPM3 ion channel were expressed as fusion proteins with thioredoxin and a His tag on the N terminus to increase their solubility in Escherichia coli Rosetta cells. Protein expression was induced by isopropyl-1-thio-β-d-galactopyranoside (Carl-Roth) for 12 h at 25 °C. The cells were pelleted by centrifugation and resuspended in 1× PBS buffer (pH 8.0) containing 1 m NaCl, 10 mm imidazole, 0.1 mm PMSF, 1 mm β-mercaptoethanol, and 0.05% Nonidet P-40. The cells were disrupted by sonication and centrifuged. The proteins were purified using affinity chromatography in a chelating Sepharose fast flow column (Amersham Biosciences) where 1× PBS buffer (pH 8.0) containing 0.5 m NaCl, 2 mm β-mercaptoethanol, and 400 mm imidazole was used for elution (see supplemental Fig. 1, A and B). Gel permeation chromatography in a Superdex 75 column (Amersham Biosciences) was used as a final purification step. The protein was eluted with 50 mm Tris-HCl buffer (pH 7.5) containing 250 mm NaCl, 2 mm β-mercaptoethanol, 0.05% Nonidet P-40, and 10% glycerol (see supplemental Figs. 1B and 2B). Protein samples were concentrated using spin columns for protein concentration (Millipore). Protein concentration was assessed by measuring absorption at 280 nm and by Bradford assay (16). The purity was verified using 15% SDS-polyacrylamide gel electrophoresis (PAGE). The integrity of the protein constructs was verified by MS analysis.
Mass Spectrometric Analysis
Two excised protein bands from SDS-PAGE, TRPM335–124 and TRPM3291–382, were digested with trypsin endoprotease (Promega) directly in the gel after destaining and cysteine modification by iodoacetamide (17). The resulting peptide mixtures were extracted and loaded onto the MALDI-TOF/TOF target with α-cyano-4-hydroxycinnamic acid as the matrix, and positively charged spectra or MS/MS were acquired with an UltraFLEX III mass spectrometer (Bruker-Daltonics, Bremen, Germany) (see supplemental Fig. 3).
CaM Expression, Purification, and Labeling
CaM was expressed and purified as described previously (8). The protein was then dialyzed overnight into 10 mm NaHCO3 (pH 10.0) at 4 °C. The protein sample was mixed with 0.6 m dansyl chloride (DNS) solution (Sigma) at a molar ratio of 1:1.5 and incubated at room temperature for 8 h. The sample was dialyzed overnight at 4 °C against 20 mm Tris-HCl buffer (pH 8.0) containing 250 mm NaCl and 2 mm CaCl2 to remove the free dansyl chloride. The degree of protein labeling was checked by measuring the ratio of the fluorescence intensities of the unbound and bound states (excitation at 340 nm, emission at 500 nm).
S100A1 Cloning, Expression, Purification, and Labeling
cDNA coding the human S100A1 protein was cloned into the pET28b expression vector. The protein was expressed in BL21 E. coli cells. Protein expression was induced by isopropyl-1-thio-β-d-galactopyranoside (Carl-Roth) for 12 h at 25 °C. The cells were pelleted by centrifugation and resuspended in 50 mm Tris-HCl buffer (pH 7.5) containing 2 mm EDTA and 0.2 mm PMSF. The cells were disrupted by sonication and centrifuged. CaCl2 was added to the supernatant (final concentration 5 mm). The protein was purified using affinity chromatography on phenyl-Sepharose CL4B (Amersham Biosciences), where 50 mm Tris-HCl buffer (pH 7.5) containing 1.5 mm EDTA and 100 mm NaCl was used for the elution. Gel permeation chromatography in a Superdex 75 column (Amersham Biosciences) was used as a final purification step. The protein was eluted with 50 mm HEPES buffer (pH 7.0) containing 250 mm NaCl, 2 mm CaCl2, 2 mm β-mercaptoethanol, and 10% glycerol. Protein samples were concentrated using spin columns for protein concentration (Millipore). Protein concentration was assessed by measuring absorption at 280 nm. The purity was verified using 15% SDS-PAGE. The protein was labeled with the fluorescent probe DNS, as described for CaM. The degree of protein labeling was checked by measuring the ratio of the fluorescence intensities of the unbound and bound states (excitation at 340 nm, emission at 500 nm).
TRPM335–124 and TRPM3291–382-CaM and S100A1 Binding Assays
Steady-state fluorescence anisotropy measurements were performed on an ISS PC1TM photon counting spectrofluorometer at room temperature in a buffer containing 20 mm Tris-HCl (pH 7.5), 6 mm CaCl2, and 5.4 mm DNS-CaM and DNS-S100A1, respectively. The samples were titrated with increasing amounts of a 200 μm solution of TRPM3 N-terminal protein constructs. At each TRPM3 concentration, the steady-state fluorescence anisotropy of DNS-CaM or DNS-S100A1 was recorded (excitation at 340 nm, emission at 500 nm). The fraction of bound TRPM3 protein (FB) was calculated from Equation 1 (18)
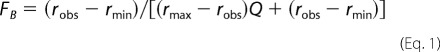 |
where Q represents the quantum yield ratio of the bound to the free form and was estimated by the ratio of the intensities of the bound to the free fluorophore. The parameter rmax is the anisotropy at saturation, robs is the observed anisotropy for any TRPM3 protein concentration, and rmin is the minimum observed anisotropy for the free CaM or S100A1. FB was plotted against TRPM3 protein concentration and fitted using Equation 2 (18) to determine the equilibrium dissociation constant (KD) for TRPM335–124/CaM, TRPM3291–382/CaM, TRPM335–124/S100A1, and TRPM3291–382/S100A1 complex formation.
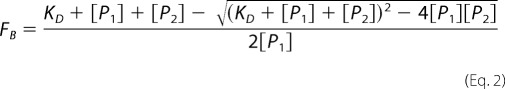 |
[P1] stands for the DNS-CaM or DNS-S100A1 concentration, and [P2] is the TRPM3 protein concentration. Nonlinear data fitting was performed using the program SigmaPlot 10.0. All experiments were carried out in at least triplicate. Control experiments with thioredoxin (Sigma-Aldrich) were performed to show that this protein is not involved in the binding to CaM or S100A1 in the fusion protein (data not shown).
To assess the role of Ca2+ on the binding of TRPM3 proteins to CaM and S100A1, we buffered Ca2+ present in the buffer after CaM and S100A1 purification with 10 mm EDTA. Then we performed the binding assays for the wild types of TRPM335–124 and TRPM3291–382.
Surface Plasmon Resonance
Surface plasmon resonance (SPR) measurements were performed at 25 °C using a ProteOn XPR36 protein interaction array system (Bio-Rad). CaM and S100A1 proteins were diluted to concentrations of 5–50 μg/ml in 10 mm acetate buffer (pH 3.5) and immobilized on a GLC chip using a ProteOn amine coupling kit (Bio-Rad) at a flow rate of 30 μl/min. Nonreacted activated groups were blocked by the injection of 1 m ethanolamine (pH 8.5). The subsequent SPR measurements were carried out in Hanks' balanced salt buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm CaCl2) enriched with 0.005% Tween 20 at a flow rate of 25 μl/min for both the association and the dissociation phase of the sensorgrams. To determine the equilibrium binding, the association phases of five different concentrations of each analyte were brought to near equilibrium values (Req). Surfaces were typically regenerated with 200 μl of buffer containing 50 mm EDTA, 1 m NaCl, and 50 mm NaOH. Sensorgrams were evaluated using the ProteOn Manager software (Bio-Rad). Response curves were generated by subtracting the sensor background signal generated simultaneously in the “interspot” areas (those sites within the 6 × 6 array that were not exposed to ligand immobilization but were exposed to analyte flow). The background-subtracted curves were prepared for fitting by subtracting the signal generated simultaneously by buffer alone in the flow channel (5 test + 1 control channel). Assuming a Langmuir-type binding between the ligand (L) and analyte (A) (i.e. L + A ↔ LA), Req values were then plotted versus protein concentration (P0), and the Kd value was determined by a nonlinear least squares analysis of the binding isotherm using the equation, Req = rmax/(1 + Kd/P0).
Circular Dichroism Spectroscopy
Circular dichroism (CD) experiments were carried out in a JASCO J-815 spectrometer (Tokyo, Japan). The peptide concentration was kept constant for all measured samples and was 0.35 mg/ml. The spectra were collected from 200 to 300 nm using a 0.1-cm quartz cell at room temperature. A 0.5-nm step resolution, 20 nm/min speed, 8 s response time and 1-nm bandwidth were used. After baseline correction, the final spectra were expressed as a molar ellipticity ϴ (degrees cm2/dmol) per residue. Secondary structure content was estimated using Dichroweb software (19).
Computer Homology Modeling
The sequences of human TRPM3 N terminus 41–70 and 304–324 were modeled via homology modeling using the program Modeller 9 version 5 (20). The structure of the voltage-gated calcium channel CaV1.2 IQ domain-Ca2+-CaM complex (Protein Data Bank (PDB) code 2be6) (21), with a 63% sequence similarity, was used as a template for TRPM3 41–70, and the structure of the complex between CaM and αII-spectrin (PDB code 2fot) (22), with a 65% sequence similarity, was used for TRPM3 304–324. The sequences were aligned using CLUSTALX 2.0.10 (see Fig. 1) (23). The models were optimized by energy minimization using the GROMOS96 parameter set (24) (implementation of Swiss-PdbViewer (24)) and checked with ProSA-web (25, 26) for recognizing errors in the three-dimensional protein structure.
FIGURE 1.

Sequence alignment of TRPM3 N terminus 41–70 (A) and 304–324 (B) with their templates voltage-gated calcium channel CaV1.2 IQ domain (PDB code 2be6) and αII-spectrin (PDB code 2fot), respectively. Identical amino acids are marked with an asterisk, similar amino acids with the more important groups are indicated with a colon, and dots indicate similar amino acids of the less important groups that are less likely to influence the protein structure.
RESULTS
Purification and Expression of TRPM335–124 and TRPM3291–382
We used the Calmodulin Target Database (15) to search for potential CaM binding motifs present in the intracellular termini of hTRPM3. Using our results from the database, we predicted two putative CaM binding sites in the regions 35–124 and 291–382 in the N terminus of the TRPM3. We cloned cDNA coding these regions into a suitable vector for protein expression in bacterial cells. To improve the solubility and expression yield of the proteins, we expressed and purified all the wild types and mutants as fusion proteins with the thioredoxin tag and His6 tag on their N terminus. All expressed fusion proteins were soluble and in sufficient amounts to perform the binding experiments. No interference between thioredoxin and the ligands was detected.
CD Spectroscopy
The secondary structure of the studied proteins was checked by CD spectroscopy (Fig. 1). Numerical analysis of the experimental spectra (19) enabled the relative abundance of the various secondary structures to be estimated (Tables 1 and 2). The α-helical conformation was found to be a major secondary structure component for CaM and S100A1 proteins, which is in good agreement with the conformation found in their native states. Although there is no known structure of the TRPM3 N terminus, the structural composition of both TRPM3 fusion proteins TRPM335–124 and TRPM3291–382 is mostly an unordered structure, which is in a good agreement with the theoretical prediction based on their primary structure, suggesting that the proteins adopt their native fold. The experiment was also used to observe changes in secondary structural element composition during the creation of the TRPM3 fusion proteins/CaM or S100A1 complexes (Fig. 2). We compared the CD spectra of the complexes with the CD spectra of the proteins alone. Because the CD spectra of the mixture are the sum of the CD spectra for the TRPM3 protein constructs and CaM or S100A1 individually, this suggests that changes in the secondary structure of TRPM335–124 and TRPM3291–382 have no significant influence on their binding to CaM or S100A1 (Fig. 2).
TABLE 2.
Calculated incidence (%) of secondary structures of S100A1, TRPM335–124, and TRPM3291–382 protein constructs and their complexes with S100A1, determined by CD spectroscopy
| S100A1 | TRPM335–124 | TRPM3291–382 | S100A1/TRPM335–124 complex | S100A1/TRPM3291–382 complex | |
|---|---|---|---|---|---|
| Helix | 66 | 19 | 22 | 36 | 35 |
| Antiparallel | 3 | 12 | 11 | 8 | 8 |
| Parallel | 3 | 12 | 11 | 8 | 8 |
| β-Turn | 12 | 18 | 18 | 16 | 17 |
| Random coil | 15 | 39 | 38 | 31 | 32 |
FIGURE 2.

CD spectra. A, CaM, TRPM335–124 and CaM/TRPM335–124 complex. B, CaM, TRPM3291–382 and CaM/TRPM3291–382 complex. C, S100A1, TRPM335–124 and S100A1/TRPM335–124 complex. D, S100A1, TRPM3291–382 and S100A1/TRPM3291–382 complex. Deg, degree.
TABLE 1.
Calculated incidence (%) of secondary structures of CaM, TRPM335–124, and TRPM3291–382 protein constructs and their complexes with CaM, determined by CD spectroscopy
| CaM | TRPM335–124 | TRPM3291–382 | CaM/TRPM335–124 complex | CaM/TRPM3291–382 complex | |
|---|---|---|---|---|---|
| Helix | 64 | 19 | 22 | 31 | 32 |
| Antiparallel | 4 | 12 | 11 | 9 | 9 |
| Parallel | 4 | 12 | 11 | 9 | 9 |
| β-Turn | 13 | 18 | 18 | 17 | 17 |
| Random coil | 16 | 39 | 38 | 34 | 33 |
CaM Binds to Regions 35–124 and 291–382 on TRPM3 N Terminus
We used two different biophysical methods to test the binding of CaM to the potential binding sites. First, we employed steady-state fluorescence anisotropy measurements for both the wild types and the mutants (Figs. 3A and 4A). We titrated increasing amounts of TRPM3 protein constructs into a solution containing fluorescently labeled CaM. The equilibrium dissociation constant of the TRPM335–124/Ca2+-CaM complex was estimated to be 1.29 ± 0.14 and 0.92 ± 0.17 μm for the TRPM3291–382/Ca2+-CaM complex. To further evaluate the binding sites, we investigated the role of some basic residues present within the binding domains. In the 35–124 region, the K45A/R67A/K71A/R72A mutation caused a 6-fold decrease in binding affinity to CaM. In the 291–382 region, both of the triple mutants K308A/R310A/R311A and K308A/R310A/K315A were unable to bind CaM.
FIGURE 3.

Binding of CaM to TRPM335–124. Columns A and B, summary of equilibrium dissociation constants obtained by steady-state fluorescence anisotropy measurement (column A) and dissociation constants obtained by SPR (column B). A, fluorescence anisotropy binding isotherms of wild type and mutants. B, SPR binding assay of wild type and mutants. RU, relative units.
FIGURE 4.

Binding of CaM to TRPM3291–382. Columns A and B, summary of equilibrium dissociation constants obtained by steady-state fluorescence anisotropy measurement (column A) and dissociation constants obtained by SPR (column B). A, fluorescence anisotropy binding isotherms of wild type and mutants. B, SPR binding assay of wild type and mutants. RU, relative units.
To confirm the data obtained by steady-state fluorescence anisotropy, we used SPR as a different type of binding assay (Fig. 3B and 4B). CaM was bound covalently onto the chip, and the TRPM3 protein constructs and their mutants were washed over the chip. The dissociation constants of the TRPM335–124/Ca2+-CaM and TRPM3291–382/Ca2+-CaM complex were estimated to be 0.198 ± 0.018 and 0.481 ± 0.074 μm, respectively. Mutations of the basic residues in TRPM335–124 caused an up to 4-fold decrease in binding affinity to CaM. The mutations K308A/R310A/R311A and K308A/R310A/K315A in TRPM3291–382 resulted in a complete loss of binding affinity to CaM, as detected by fluorescence anisotropy.
S100A1 Binds to TRPM335–124 and TRPM3291–382
As reviewed in previous studies, S100A proteins have been suggested to bind to similar structural motifs to CaM (11). Thus we used the same experimental approach to investigate whether the S100A1 protein is able to bind to TRPM335–124 and TRPM3291–382 constructs in the same or a similar way to CaM. Equilibrium dissociation constants assessed by steady-state anisotropy measurement for the TRPM335–124/Ca2+-S100A1 and TRPM3291–382/Ca2+-S100A1 complexes were estimated to be 1.43 ± 0.37 and 1.06 ± 0.29 μm, respectively (Figs. 5A and 6A). Dissociation constants determined by SPR for the TRPM335–124/Ca2+-S100A1 and TRPM3291–382/Ca2+-S100A1 complexes were 0.304 ± 0.037 and 1.8 ± 0.5 μm, respectively (Figs. 5B and 6B). Mutant K45A/R67A/K71A/R72A of TRPM335–124 exhibited an inability to bind S100A1, and there was also no interaction detected for this mutant when measured by SPR. Both steady-state anisotropy measurement and SPR confirmed that triple mutations K308A/R310A/R311A and K308A/R310A/K315A caused a total loss of binding affinity for S100A1 in region 291–382 of the TRPM3 N terminus.
FIGURE 5.

Binding of S100A1 to TRPM335–124. Columns A and B, summary of dissociation constants assessed by steady-state anisotropy measurement (column A) and SPR experiment (column B). A, fluorescence anisotropy binding isotherms of wild type and mutants. B, SPR binding assay of wild type and mutants. RU, relative units.
FIGURE 6.

Binding of S100A1 to TRPM3291–382. Columns A and B, summary of dissociation constants assessed by steady-state anisotropy measurement (column A) and SPR experiment (column B). A, fluorescence anisotropy binding isotherms of wild type and mutants. B, SPR binding assay of wild type and mutants. RU, relative units.
Binding of CaM and S100A1 to TRPM335–124 and TRPM3291–382 Is Calcium-dependent
To determine the role of Ca2+ on CaM and S100A1 binding to regions 35–124 and 291–382 in the TRPM3 N terminus, we performed steady-state fluorescence anisotropy binding experiments in a solution lacking calcium (Fig. 7). We observed no increase in fluorescence anisotropy in either of the experiments, and thus we suggest that the binding is calcium-dependent.
FIGURE 7.

Calcium-dependent binding of CaM and S100A1 proteins to TRPM335–124 and TRPM3291–382. A, CaM binding to TRPM335–124 in the presence (circles) and absence of calcium (triangles). B, S100A1 binding to TRPM335–124 in the presence (circles) and absence of calcium (diamonds). C, CaM binding to TRPM3291–382 in the presence (circles) and absence of calcium (squares). D, S100A1 binding to TRPM3291–382 in the presence (circles) and absence of calcium (triangles).
CaM and S100A1 Compete for Overlapping Binding Sites in TRPM3 N Terminus
We mixed the CaM and S100A1 proteins in a 1:1 molar ratio with the TRPM335–124 and TRPM3291–382 protein constructs. To investigate whether the complexes are able to bind CaM or S100A1, we carried out an SPR experiment. We gradually washed the TRPM335–124/Ca2+-CaM, TRPM3291–382/Ca2+-CaM, TRPM335–124/Ca2+-S100A1, and TRPM3291–382/Ca2+-S100A1 complexes over the CaM/S100A1 chip and observed the binding/nonbinding of the complexes to CaM or S100A1 (Fig. 6). Complexes of TRPM335–124 (Fig. 8A) and TRPM3291–382 (Fig. 8C) with Ca2+-CaM did not bind the S100A1 protein. Complexes of TRPM335–124 (Fig. 8B) and TRPM3291–382 (Fig. 8D) with Ca2+-S100A1 were not able to bind CaM. These data might imply that CaM and S100A1 bind to the same or overlapping binding sites within the TRPM3 N terminus.
FIGURE 8.

SPR competition assay sensorgrams. A, binding of TRPM335–124, TRPM335–124/Ca2+-CaM and TRPM335–124/Ca2+-S100A1 to S100A1. RU, relative units. B, binding of TRPM335–124, TRPM335–124/Ca2+-CaM, and TRPM335–124/Ca2+-S100A1 to CaM. C, binding of TRPM3291–382, TRPM3291–382/Ca2+-CaM, and TRPM3291–382/Ca2+-S100A1 to S100A1. D, binding of TRPM3291–382, TRPM3291–382/Ca2+-CaM, and TRPM3291–382/Ca2+-S100A1 to CaM.
Computer Homology Modeling
To visualize the interactions of the CaM binding sites on the TRPM3 N terminus with Ca2+-CaM in more detail, we created computer homology models of the corresponding sequences. A structure of the ankyrin repeat domain on TRPV1 was reported to bind CaM (27), but no structure with this sequence was found in the complex with CaM. Thus we used the structures of other CaM binding peptides in complex with Ca2+-CaM with a high degree of similarity. According to the secondary structure prediction, these sequences adopt the topology of an α-helix. We used other CaM binding peptides, namely the voltage-gated calcium channel CaV1.2 IQ domain (PDB code 2be6) and αII-spectrin (PDB code 2fot), as templates for our models. These templates have a high degree of sequence similarity with the modeled sequences, especially in the positions of hydrophobic residues important for the interactions. Thus we modeled both the sequence 41–70 (Fig. 9A) and the sequence 304–324 (Fig. 9B) as α-helixes running through the central cavity of CaM, as was observed for the majority of such sequences. The model also suggested that basic residues could interact through ionic interactions with the negatively charged residues in CaM.
FIGURE 9.

Computer homology models of TRPM3 41–70 (A) and TRPM3 302–324 (B) with Ca2+-CaM.
DISCUSSION
The TRPM3 ion channel has been proposed to play an important role in Ca2+ homeostasis serving as a Ca2+ entry pathway (2, 5). To date, no regulation via Ca2+-binding proteins have been reported for this channel. CaM was shown to be an important regulator of various TRP ion channels, serving as both an activator and an inhibitor (7). The identification of more CaM binding sites was predicted from other TRP channels, and the functional implications of these interactions were suggested to be very complex, ranging from channel activation, facilitation, and inhibition/desensitization to subunit assembly, surface expression, recycling, and other regulation (7). It is known that the TRP channels are expressed without additional exogenous proteins. The channels require participation of endogenous signaling proteins for their activation and regulation (e.g. Gq protein, phospholipase C, CaM, protein kinases, and phosphatases).
Because Ca2+ and CaM are tightly linked, it is difficult to know whether CaM is involved in TRP channel activation or only in its inhibition. Furthermore, it is unclear whether these two processes act solely on the TRP channel or affect other stages of the signal transduction cascade (28).
Accordingly, TRPV1 channel expressed in Xenopus oocytes shows inhibition of the channel activity by Ca2+. This effect is mediated by CaM as application of Ca2+-CaM, but not of Ca2+ or CaM separately, inhibits the channel. In this system, no activating effects of either Ca2+-CaM or Ca2+ were observed (29). In agreement with the inhibitory effects of Ca2+-CaM, it has been demonstrated that TRPC3 and TRPC4 are inhibited by Ca2+-CaM but not by Ca2+ or CaM alone (7).
In the TRPM subfamily, CaM was demonstrated to be the Ca2+ sensor responsible for Ca2+-dependent activation of TRPM2 via binding to the IQ motif present on the N terminus of this channel. Replacement of the motif disrupts the interaction of CaM and TRPM2, therefore abolishing TRPM2 activation evoked by Ca2+. These results establish that Ca2+ is necessary and sufficient for TRPM2 activation and also elucidate that the Ca2+-CaM binding to TRPM2 is required for Ca2+-mediated TRPM2 activation. The data reveal a gating mechanism of TRPM2 and its alternative spliced isoforms that may represent a major gating mechanism in vivo and therefore confer novel, as yet unknown, physiological and/or pathological functions (30, 31). Moreover, five CaM binding sites were revealed on the N and C termini of TRPM4, and the three binding sites on the C-tail were suggested to be important for the Ca2+-induced activation of this ion channel (32).
The aim of this study was to identify and characterize the putative CaM binding sites within the intracellular amino and carboxyl termini of TRPM3. We used two independent binding assays, steady-state fluorescence anisotropy measurement and SPR, to identify two CaM binding sites in regions Ala-35–Lys-124 and His-291–Gly-382 on the TRPM3 N terminus. These regions contain the 1-14, 1-10, and 1-5-10 consensus CaM recognition motifs with hydrophobic residues located in these positions (7), which is a common feature of CaM binding domains typically containing a basic amphiphilic α-helix, which can be classified into motifs based upon variations in the positions of conserved hydrophobic residues (14). We found several important basic residues in the N-terminal CaM binding domains, namely Lys-45, Arg-56, Lys-62, Arg-67, Lys-71, and Arg-72 on the TRPM335–124 and Lys-308, Arg-310, Arg-311, and Lys-315 on the TRPM3291–382, which are crucial for CaM binding. Replacing these residues had a critical impact on the interaction, leading to a total loss of binding ability when three or more of these residues were replaced. The cluster of basic residues within the CaM binding motif was reported to interact with the negatively charged residues on CaM (33, 34), which was also found with the TRP channel family members TRPV1 (10), TRPV2 and TRPV5 (8).
The Ca2+-binding S100 proteins were shown to interact with similar binding motifs to CaM (14, 35). Here we show that the S100A1 protein binds the same regions with equal affinity on the TRPM3 N terminus to CaM. Moreover, the mutation of positively charged residues within these domains affected the binding to S100A1, as was observed for CaM.
As was shown for the CaM binding sites present on the TRP channels, the binding of CaM to these sequences is calcium-dependent (7). To assess the role of calcium on CaM and S100A1 binding to the TRPM3 Ala-35–Lys-124 and His-291–Gly-382 N-terminal sequences, we performed the steady-state anisotropy measurement in a solution lacking calcium. We observed no changes in the anisotropy values, in contrast to the experiment when calcium was present, and thus we conclude that the binding is calcium-dependent.
To show that the binding sites for CaM/S100A1 on the TRPM3 N terminus overlap, we performed the SPR experiment. It is clear from the data that the complexes of the TRPM3 N-terminal constructs with Ca2+-CaM do not bind S100A1, and similarly, the TRPM3 N-terminal complexes with Ca2+-S100A1 do not bind CaM. A similar interaction was reported at the ryanodine receptor, where CaM and S100A1 compete for the same binding site with the same hydrophobic and basic residues being involved in CaM/S100A1 binding (36, 37). However, such evidence was missing for TRP channels, and here we show that the same basic residues participate in CaM/S100A1 binding.
To explain the interactions of the TRPM3 N-terminal CaM binding sites with CaM in detail, we created homology models of the sequences with CaM. According to the secondary structure prediction, the models are suggested to have the structure of an α-helix running through the central pore of CaM. We expect that the positively charged residues that we found to be crucial for CaM/S100A1 binding interact with the negatively charged residues in CaM.
Undoubtedly, CaM is a very important regulator of TRP channels, and it is therefore highly probable that, together with other Ca2+-binding proteins, it could play an important role in the regulation of the TRPM3 ion channel via binding to its intracellular termini in the same way as was reported for other members of the TRPM subfamily. A possible role of Ca2+-CaM in the gating mechanism was proposed for TRPM2 and TRPM4 (30, 31, 32) channels, and a similar way of regulation of TRPM3 could be assumed, but more functional studies will be required to elucidate the role of these Ca2+-binding proteins in regulation of the channel.
Supplementary Material
Acknowledgments
We thank Dr. S. L. Hamilton from Baylor College of Medicine for providing us with rat CaM cloned into the pET3a vector and also Prof. C. Harteneck from the University of Tubingen for a clone of human TRPM3. We also thank Dr. L. Bednarova from the Institute of Organic Chemistry and Biochemistry of the Academy of Sciences of the Czech Republic for assistance with CD spectra measurements.
This study was supported by the Grant Agency of the Czech Republic (Grants GACR 301/10/1159, GACR P205/10/P308, and GACR 207/11/0717) and the Academy of Sciences of the Czech Republic (Research Project AV0Z50110509).

This article contains supplemental Figs. 1–3.
- TRP
- transient receptor potential
- TRPM
- transient receptor potential melastatin
- TRPC
- transient receptor potential canonical
- TRPV
- transient receptor potential vanilloid
- CaM
- calmodulin
- DNS
- dansyl chloride
- SPR
- surface plasmon resonance
- CaV
- voltage-gated calcium channel.
REFERENCES
- 1. Harteneck C. (2005) Function and pharmacology of TRPM cation channels. Naunyn Schmiedebergs Arch. Pharmacol. 371, 307–314 [DOI] [PubMed] [Google Scholar]
- 2. Lee N., Chen J., Sun L., Wu S., Gray K. R., Rich A., Huang M., Lin J. H., Feder J. N., Janovitz E. B., Levesque P. C., Blanar M. A. (2003) Expression and characterization of human transient receptor potential melastatin 3 (hTRPM3). J. Biol. Chem. 278, 20890–20897 [DOI] [PubMed] [Google Scholar]
- 3. Oberwinkler J., Lis A., Giehl K. M., Flockerzi V., Philipp S. E. (2005) Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J. Biol. Chem. 280, 22540–22548 [DOI] [PubMed] [Google Scholar]
- 4. Oberwinkler J., Phillipp S. E. (2007). Trpm3. Handb. Exp. Pharmacol. 253–267 [DOI] [PubMed] [Google Scholar]
- 5. Grimm C., Kraft R., Sauerbruch S., Schultz G., Harteneck C. (2003) Molecular and functional characterization of the melastatin-related cation channel TRPM3. J. Biol. Chem. 278, 21493–21501 [DOI] [PubMed] [Google Scholar]
- 6. Vriens J., Owsianik G., Hofmann T., Philipp S. E., Stab J., Chen X., Benoit M., Xue F., Janssens A., Kerselaers S., Oberwinkler J., Vennekens R., Gudermann T., Nilius B., Voets T. (2011) TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 70, 482–494 [DOI] [PubMed] [Google Scholar]
- 7. Zhu M. X. (2005) Multiple roles of calmodulin and other Ca2+-binding proteins in the functional regulation of TRP channels. Pflugers Arch. 451, 105–115 [DOI] [PubMed] [Google Scholar]
- 8. Holakovska B., Grycova L., Bily J., Teisinger J. (2011) Characterization of calmodulin binding domains in TRPV2 and TRPV5 C-tails. Amino Acids 40, 741–748 [DOI] [PubMed] [Google Scholar]
- 9. Friedlova E., Grycova L., Holakovska B., Silhan J., Janouskova H., Sulc M., Obsilova V., Obsil T., Teisinger J. (2010) The interactions of the C-terminal region of the TRPC6 channel with calmodulin. Neurochem. Int. 56, 363–366 [DOI] [PubMed] [Google Scholar]
- 10. Grycova L., Lansky Z., Friedlova E., Obsilova V., Janouskova H., Obsil T., Teisinger J. (2008) Ionic interactions are essential for TRPV1 C terminus binding to calmodulin. Biochem. Biophys. Res. Commun. 375, 680–683 [DOI] [PubMed] [Google Scholar]
- 11. Rezvanpour A., Shaw G. S. (2009) Unique S100 target protein interactions. Gen. Physiol. Biophys. 28, F39–F46 [PubMed] [Google Scholar]
- 12. Rustandi R. R., Baldisseri D. M., Inman K. G., Nizner P., Hamilton S. M., Landar A., Landar A., Zimmer D. B., Weber D. J. (2002) Three-dimensional solution structure of the calcium-signaling protein apo-S100A1 as determined by NMR. Biochemistry 41, 788–796 [DOI] [PubMed] [Google Scholar]
- 13. Wright N. T., Varney K. M., Ellis K. C., Markowitz J., Gitti R. K., Zimmer D. B., Weber D. J. (2005) The three-dimensional solution structure of Ca2+-bound S100A1 as determined by NMR spectroscopy. J. Mol. Biol. 353, 410–426 [DOI] [PubMed] [Google Scholar]
- 14. Rhoads A. R., Friedberg F. (1997) Sequence motifs for calmodulin recognition. FASEB J. 11, 331–340 [DOI] [PubMed] [Google Scholar]
- 15. Yap K. L., Kim J., Truong K., Sherman M., Yuan T., Ikura M. (2000) Calmodulin Target Database. J. Struct. Funct. Genomics 1, 8–14 [DOI] [PubMed] [Google Scholar]
- 16. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 17. Prochazkova K., Osicka R., Linhartova I., Halada P., Sulc M., Sebo P. (2005) The Neisseria meningitidis outer membrane lipoprotein FrpD binds the RTX protein FrpC. J. Biol. Chem. 280, 3251–3258 [DOI] [PubMed] [Google Scholar]
- 18. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., pp. 291–318, Springer, New York [Google Scholar]
- 19. Whitmore L., Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 20. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Petegem F., Chatelain F. C., Minor D. L., Jr. (2005) Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat. Struct. Mol. Biol. 12, 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Simonovic M., Zhang Z., Cianci C. D., Steitz T. A., Morrow J. S. (2006) Structure of the calmodulin αII-spectrin complex provides insight into the regulation of cell plasticity. J. Biol. Chem. 281, 34333–34340 [DOI] [PubMed] [Google Scholar]
- 23. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 24. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 25. Sippl M. J. (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362 [DOI] [PubMed] [Google Scholar]
- 26. Wiederstein M., Sippl M. J. (2007). ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lishko P. V., Procko E., Jin X., Phelps C. B., Gaudet R. (2007) The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 54, 905–918 [DOI] [PubMed] [Google Scholar]
- 28. Minke B., Parnas M. (2006) Insights on TRP channels from in vivo studies in Drosophila. Annu. Rev. Physiol. 68, 649–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosenbaum T., Gordon-Shaag A., Munari M., Gordon S. E. (2004) Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J. Gen. Physiol. 123, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tong Q., Zhang W., Conrad K., Mostoller K., Cheung J. Y., Peterson B. Z., Miller B. A. (2006) Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J. Biol. Chem. 281, 9076–9085 [DOI] [PubMed] [Google Scholar]
- 31. Du J., Xie J., Yue L. (2009) Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. U.S.A. 106, 7239–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nilius B., Prenen J., Tang J., Wang C., Owsianik G., Janssens A., Voets T., Zhu M. X. (2005) Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J. Biol. Chem. 280, 6423–6433 [DOI] [PubMed] [Google Scholar]
- 33. Yamauchi E., Nakatsu T., Matsubara M., Kato H., Taniguchi H. (2003) Crystal structure of a MARCKS peptide containing the calmodulin-binding domain in complex with Ca2+-calmodulin. Nat. Struct. Biol. 10, 226–231 [DOI] [PubMed] [Google Scholar]
- 34. Matsuoka Y., Hughes C. A., Bennett V. (1996) Adducin regulation. Definition of the calmodulin-binding domain and sites of phosphorylation by protein kinases A and C. J. Biol. Chem. 271, 25157–25166 [DOI] [PubMed] [Google Scholar]
- 35. Wilder P. T., Lin J., Bair C. L., Charpentier T. H., Yang D., Liriano M., Varney K. M., Lee A., Oppenheim A. B., Adhya S., Carrier F., Weber D. J. (2006) Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim. Biophys. Acta 1763, 1284–1297 [DOI] [PubMed] [Google Scholar]
- 36. Prosser B. L., Hernández-Ochoa E. O., Schneider M. F. (2011) S100A1 and calmodulin regulation of ryanodine receptor in striated muscle. Cell Calcium 50, 323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wright N. T., Prosser B. L., Varney K. M., Zimmer D. B., Schneider M. F., Weber D. J. (2008) S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J. Biol. Chem. 283, 26676–26683 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.