

Abstract
Purpose
To define gene expression changes associated with diabetic retinopathy in a mouse model using next generation sequencing, and to utilize transcriptome signatures to assess molecular pathways by which pharmacological agents inhibit diabetic retinopathy.
Methods
We applied a high throughput RNA sequencing (RNA-seq) strategy using Illumina GAIIx to characterize the entire retinal transcriptome from nondiabetic and from streptozotocin-treated mice 32 weeks after induction of diabetes. Some of the diabetic mice were treated with inhibitors of receptor for advanced glycation endproducts (RAGE) and p38 mitogen activated protein (MAP) kinase, which have previously been shown to inhibit diabetic retinopathy in rodent models. The transcripts and alternatively spliced variants were determined in all experimental groups.
Results
Next generation sequencing-based RNA-seq profiles provided comprehensive signatures of transcripts that are altered in early stages of diabetic retinopathy. These transcripts encoded proteins involved in distinct yet physiologically relevant disease-associated pathways such as inflammation, microvasculature formation, apoptosis, glucose metabolism, Wnt signaling, xenobiotic metabolism, and photoreceptor biology. Significant upregulation of crystallin transcripts was observed in diabetic animals, and the diabetes-induced upregulation of these transcripts was inhibited in diabetic animals treated with inhibitors of either RAGE or p38 MAP kinase. These two therapies also showed dissimilar regulation of some subsets of transcripts that included alternatively spliced versions of arrestin, neutral sphingomyelinase activation associated factor (Nsmaf), SH3-domain GRB2-like interacting protein 1 (Sgip1), and axin.
Conclusions
Diabetes alters many transcripts in the retina, and two therapies that inhibit the vascular pathology similarly inhibit a portion of these changes, pointing to possible molecular mechanisms for their beneficial effects. These therapies also changed the abundance of various alternatively spliced versions of signaling transcripts, suggesting a possible role of alternative splicing in disease etiology. Our studies clearly demonstrate RNA-seq as a comprehensive strategy for identifying disease-specific transcripts, and for determining comparative profiles of molecular changes mediated by candidate drugs.
Introduction
Diabetes has emerged as a major worldwide public health concern, and the number of diabetics is estimated to exceed 400 million by the year 2030 [1]. A side effect of diabetes, namely diabetic retinopathy, is also a leading cause of blindness in working age adults (NIH MedlinePlus the Magazine). Several approaches, including good glycemic control, use of blood pressure medications, and lipid control, have been demonstrated to inhibit diabetic retinopathy in clinical trials, but many patients are not able to maintain these regimens over the long-term. Thus, additional therapeutic approaches are continuously being sought. Several experimental therapies that include vitamin E, aspirin, aminoguanidine, or inhibitors of receptor for advanced glycation endproducts (RAGE) and p38 mitogen activated protein (MAP) kinase [2–6] have shown positive effects at inhibiting the development of diabetic retinopathy lesions in laboratory animals, but the underlying molecular mechanisms are not clear. Given their importance in cellular metabolism and regulatory processes, these therapeutic agents are expected to target distinct pathways either directly or indirectly. Therefore, identification of the targets of these drugs might assist in characterizing their molecular side effects.
Molecular changes accompanying the progression of disease can now be determined by several approaches. Gene expression microarray analysis has been widely used during the past decade for characterizing complete transcriptomes [7–9] and it has yielded global profiles of whole retina or retinal cell types in both wild type and disease models [10–18]. A comparison of the expressed complement of the genome between normal and diabetic retinas has indicated altered abundance of transcripts involved in several key pathways [19,20]. Although microarray strategies have been successful in describing disease- or phenotype-associated expression changes, hybridization-based profiling approaches suffer from technical variations that are difficult to control. For this reason, many expression changes cannot be validated by quantitative reverse transcription polymerase chain reaction (qRT–PCR) [21]. In addition, relevant causative expression changes, such as alternatively spliced variants of transcripts and the expression of novel transcripts in disease samples, may not be comprehensively captured because specific probe sets may not be included on the particular microarrays being used.
Next generation sequencing based on the RNA sequencing (RNA-seq) approach is now gaining prominence as a means of accurate qualitative and quantitative characterization of the expressed complement of a genome [22,23]. This method provides millions of sequences from expressed RNA molecules and can provide relatively unambiguous definition and abundance of transcripts in a given specimen. RNA-seq is therefore expected to reveal a better representation of the transcriptome, and this strategy is also more amenable for the analysis of alternatively spliced transcripts.
We have recently demonstrated the high accuracy and sensitivity of RNA-seq technology with microarray and qRT–PCR methods by profiling the neural retina specific leucine zipper deficient (Nrl−/−) retina [24]. In this paper, we define the changes occurring in the entire retinal transcriptome of streptozotocin (STZ)-induced diabetic mice compared to wild type mice. We also attempt to identify relevant molecular signatures and cellular pathways, with the goal of examining the impact of inhibitors of RAGE and of p38 MAP kinase, two candidate drugs that inhibit retinopathy in experimental models. Our results indicate that these inhibitors induce common, as well as unique, molecular changes that may assist in determining the efficacy and/or side effects of candidate drugs.
Methods
Streptozotocin-induced diabetes in mice
All animal experiments conformed to the ARVO Resolution on the Use of Animals in Research and were approved by the Animal Care and Use Committee of Case Western University. Diabetes was induced in C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) with streptozotocin and a corresponding number of weight and age-matched animals were maintained as normal controls. Mice received five sequential daily intraperitoneal injections of a freshly prepared solution of streptozotocin (Sigma-Aldrich, St Louis, MO) in citrate buffer (pH 4.5) at 60 mg/kg of bodyweight. After hyperglycemia was verified at least three times during the second week after streptozotocin administration, diabetic mice were randomly assigned to remain as untreated diabetic controls or were administered RAGE-Ig fusion protein intraperitoneally at three different concentrations (10, 100, and 300 µg per mouse) three times per week. Insulin was given to diabetics as needed (0–0.3 units of neutral protamine Hagedorn (NPH) insulin subcutaneously, 0–3 times per week) to prevent weight loss without preventing hyperglycemia.
The RAGE fusion protein (provided by L. Brown, Galactica Pharmaceuticals, Inc., Villanova, PA) consists of a RAGE ligand binding element, a heavy chain immunoglobulin of G4 isotype (IgG4) constant domain with a linker connecting the ligand binding element with the constant domain. The RAGE-Fc binds to all of the known ligands of RAGE and acts as a competitive, negative regulator of RAGE signaling by competing with the membrane-bound receptor for binding of ligands. For the present studies, a murine version of hRAGE was employed in which a G2a isotype (IgG2a) constant domain (biologically equivalent to IgG4 in hRAGE) was used. The inhibitor of p38 MAPK (PHA666859) was provided by Pfizer Research Laboratories (Groton, CT). PHA666859 was mixed into powdered diet and replaced weekly, and food consumption was measured to calculate the amount of drug consumed.
We have previously shown that both of these therapies inhibit the development of early stages of diabetic retinopathy, as determined by the abundance of acellular capillaries and pericyte ghosts [3,25]. Glycated hemoglobin (GHb; an estimate of the average level of hyperglycemia over the previous 2–3 months) was measured by affinity chromatography (Glyc-Affin; Pierce, Rockford, IL) every 3 months in each animal, after an overnight fast . Bodyweight and average daily food consumption were measured weekly. Experimental variations were reduced by CO2 asphyxiation of all animals at 8 months of diabetes between 2 – 4 PM
Isolation of RNA
Retinal RNA was extracted with Trizol reagent [26,27]. Three independent retina samples were obtained from groups of animals that were normal, diabetic, treated with RAGE inhibitor, and treated with p38 MAPK inhibitor. The RNA was quantified by absorbance at 260 nm in a spectrophotometer and its integrity was determined using an Agilent Bioanalyzer (Santa Clara, CA) [28].
Illumina RNA-seq
Independent RNA samples were used for the preparation of cDNA libraries using an mRNA-seq Sample Preparation Kit (Illumina, San Diego, CA). Briefly, double stranded cDNA was prepared by random hexamer priming and reverse transcription of chemically sheared total RNA (2 µg). The double stranded cDNA was ligated with adaptor sequences required for bridge amplification using the Illumina flow-cell, and the ligated DNA was amplified before library assessment and quantitation using the BioAnalyzer DNA-1000 chip (Agilent). Cluster generation of 10 pM loads of a single mRNA-seq library per lane was performed on the Cluster Station using an Illumina Single Read Cluster Generation Kit v4 (Illumina). Sequence-by-synthesis single reads of 54 base length using the SBS Sequencing Kit v4 (Illumina) were generated on the Genome Analyzer IIx. This instrument runs real-time analysis SCS2.4 software (Illumina) for base calling.
Real-time qPCR
Quantitative RT–PCR was performed using a Genetic Analyzer 7900HT (Life Technologies, Carlsbad, CA) with Taqman or SYBR green assays, as previously described [24]. Briefly, total RNA (1 µg) was reverse transcribed by Superscript II reagents (Life Technologies, Carlsbad, CA) primed with oligo(dT)20. The cDNA was diluted fivefold and one microliter was used as template in subsequent qRT–PCR reactions. The integrity of the PCR reaction was verified by melt curve analysis. The results of amplifications of three biologic replicates, performed in triplicate, were averaged to produce the qRT–PCR data. Changes in gene expression were determined by the ddCT method using the levels of HPRT (hypoxanthine guanine phosphoribosyl transferase) transcript for normalization.
Bioinformatics analysis of RNA-seq data
The cDNA sequences captured on the Illumina platform were analyzed using the following workflows.
(1) Transcript isoform level analysis was performed by aligning the 54 base cDNA reads against the reference genome mm9 build using a Burrows-Wheeler transform based short read aligner (BWA) [29], and the aligned reads were visualized using Integrated Genome Viewer [30]. The read alignment files were imported into Partek Genomics Suite (Partek® Genomics SuiteTM. 6.3 ed. St. Louis, MO: Partek Inc.; 2008) and RPKM (reads per kilobase of exon model per million mapped reads) counts for each of the 28,157 transcripts defined in the UCSC refflat annotation file were calculated. A stringent filtering criterion with RPKM value of 1.0 [31] was used to obtain 16,577 expressed transcripts. The RPKM values of filtered transcripts were log-transformed using log2 (RPKM + offset) with an offset value of 1.0. This filtering protocol reduced the number of transcripts from 28,157 to 16,577 for further analyses. Fold changes in transcript expression and p-values were computed using ANOVA, and significantly altered transcripts were selected by applying fold change-cutoff of 1.5 and/or p-value cutoff of 0.05. Gene Ontology (GO) pathways that were significantly enriched between each of the sample pairs were identified using ExPlain tool from Biobase International, Beverly, MA.
(2) Alternatively, gene level analysis was performed by aligning the filtered reads to the mouse reference genome build mm9 using the ELAND (Efficient Local Alignment of Nucleotide Data) algorithm (Anthony J Cox, Solexa Ltd Saffron, Walden, UK). First, raw and RPKM normalized counts were calculated for gene models (as defined in UCSC RefGene). Subsequently, we applied SAM (significance analysis of microarrays) to gene level RPKM to identify RNAs with a fold change greater than 1.5 and false discovery rate (FDR) of less than 25%. Briefly, the boundaries of exons were obtained from the RefGene database (Genome), and the numbers of mapped reads on each exon were calculated. The mapped reads as RPKM indicated the expression level of exons, and this value was used to calculate the Log base 2 of the average (or maximum) expression level of exons for each gene. For this alternative analysis, a less stringent cutoff of 0.5 RPKM was used to select expressed genes for further analysis.
Alternatively spliced exons
Alternatively spliced transcripts were detected using the AltAnalyze tool [32], which employs de novo splice junctions to predict alternative splicing events based on the reads spanning the splice junctions. First, TopHat [33] was run independently on sequence reads obtained for each of the 12 RNA samples to identify all novel splice junctions. Second, we combined all predicted splice junctions into a master file and used this as an input option, together with the Ensembl mouse gene transfer format (GTF) annotation file, for a second iteration of TopHat runs on each of the 12 samples. Finally, these 12 junction files were used as input in AltAnalyze to detect alternative splicing using the analysis of splicing by isoform reciprocity (ASPIRE) algorithm.
Briefly, we calculated the number of sequences mapping to a specific exon junction and the numbers of sequences mapping to junctions as well as exons of that specific gene. These two numbers were then used to calculate the fraction of the junction containing sequences relative to the mean of abundance of sequences of all junctions and exons in the gene. The procedure was applied to each reciprocal isoform pair (i.e., an isoform that includes a specific exon and an isoform in which that exon is excluded). The inclusion and exclusion ratios were then used to compute the value for ΔI. The details of these protocols have been described in AltAnalyze workflow [32], as adapted from ASPIRE algorithm [34]. The ratios described above were used to calculate p values for the sample ASPIRE scores relative to controls, and to determine false-discovery rate p values [35].
Results
Glycemia
Diabetic mice from all experimental groups had levels of GHb and blood glucose that were significantly greater (p<0.05) than levels found in appropriately age-matched nondiabetic controls. Average GHb for the nondiabetic control (N), diabetic control (D), diabetic plus PHA666859 (D + PHA666859), and diabetic plus murine RAGE-Fc fusion protein (D + mRAGE-Fc) groups over the entire duration of study are listed in Table 1. Although diabetic mice did not gain weight at the normal rate, none of the animals lost bodyweight, and all of the animals appeared clinically healthy. These results indicate that the therapies involving inhibitors of RAGE and p38 MAP kinase do not alter the diabetic status of the animals.
Table 1. Glycemic status of control and drug-treated animals over the 8 months of the study.
| Control/Treatments* | Glucose | Glycated hemoglobin (GHb) |
|---|---|---|
| Nondiabetic Controls (N) |
130+8 |
3.0+0.1 |
| Diabetic Controls (D) |
400+21 |
10.4+0.9 |
| Diabetic plus p38 MAPK inhibitor (D+PHA666859) |
411+32 |
10.3+0.6 |
| Diabetic plus murine RAGE-Fc Fusion Protein (D + mRAGE-Fc) | 393+31 | 10.6+0.7 |
*The values are reported as mean ± standard deviation, and were calculated for 10, 7, 7, and 8 animals in the four groups, respectively.
Raw read counts and transcripts in sequenced samples
Total raw reads for various samples were in the range of 27.8–35.5 million, which represented ~1.6 gigabases of sequences for each sample. Efficient Local Alignment of Nucleotide Data (ELAND) and Burrows-Wheeler Alignment (BWA) tools allowed the alignment of 13.8- 20.9×106 reads from various samples to mouse genome sequence database (build mm9). These aligned reads were subsequently analyzed by significance analysis of microarrays (SAM) or ANOVA. The sequence alignment files were imported into PARTEK, and the normalized values for numbers of RPKM for 28,157 transcripts defined in the UCSC refflat file were obtained. Among these sequences obtained from nondiabetic and diabetic animals, 16,231 transcripts had RPKM values of greater than one, and included rhodopsin (Rho), guanine nucleotide binding protein (Gnat1), retinol binding protein (Rbp1), phosphodiesterase 6A (Pde6a), and guanine nucleotide binding protein (Gngt1) as the most abundant sequences specific to photoreceptor cells. Similarly, the list also contained other less abundant transcripts such as paired-like homeodomain 3 (Pitx3), PDZ and LIM domain 1(Pdlim1), and meteorin (Metrnl). The presence of these highly abundant as well as rare transcripts in the data set demonstrates that the library preparation and data analysis methods did not introduce any undesirable bias.
Comparison of transcript differences revealed by two analyses
We analyzed the sequence data using ELAND-SAM and BWA-ANOVA methods, and compared the results to confirm method-specific changes in transcript abundance in the various groups of animals. Differential expression in diabetic and normal animals revealed the top 100 transcripts shortlisted by the two methods, as shown in Table 2 (Appendix 1). Of these 100 transcripts, 81 are present in both data sets (Figure 1) including a variety of crystallin genes. However, UDP-glucuronosyl transferase transcripts were detected only by the ELAND-SAM method. A significant difference was also noted in the fold-changes indicated by the two methods. These differences can be attributed to differences in the alignment algorithms, filtering criteria, and calculation of differential gene expression between the two methods, and demonstrate that altered abundance of transcripts can be detected by either one of these two methods.
Table 2. Comparison of transcript abundance changes obtained by two methods*.
| Gene | Fold-method I* | Gene | Fold-method I* | Gene | Fold-method II* | Gene | Fold-method II* |
|---|---|---|---|---|---|---|---|
| Crybb2 |
19.8 |
Tmem40 |
2.4 |
Rnu73a |
93.3 |
Pdlim1 |
3.7 |
| Cryba1 |
19.4 |
Crygc |
2.4 |
Lim2 |
75.7 |
Trim16 |
3.7 |
| Cryba4 |
18.3 |
Acta2 |
2.3 |
Wnt7a |
47.9 |
Lenep |
3.6 |
| Crygs |
18.2 |
Igfbp7 |
2.3 |
Pitx3 |
40.1 |
S100a6 |
3.6 |
| Cryaa |
17.9 |
Lyz2 |
2.3 |
Tmem40 |
38 |
Cd44 |
3.6 |
| Lim2 |
17.6 |
Vit |
2.3 |
Mip |
36 |
Gpd1 |
3.5 |
| Mip |
16.7 |
Tmem27 |
2.2 |
Wnt7b |
26.1 |
Cbr2 |
3.4 |
| Cryba2 |
16.4 |
Gsn |
2.2 |
Gja3 |
23.8 |
Nanos2 |
3.4 |
| Crybb1 |
15.1 |
C1qtnf5 |
2.2 |
Grifin |
23.7 |
Gpnmb |
3.4 |
| Bfsp2 |
10.7 |
Mfrp |
2.2 |
Cryba4 |
23.1 |
Gsta3 |
3.4 |
| Lgsn |
10.1 |
Igfbp7 |
2.1 |
Bfsp2 |
21 |
Ugt1a5 |
3.3 |
| Grifin |
9.8 |
Ucp2 |
2.1 |
Crybb2 |
20.3 |
Ugt1a10 |
3.3 |
| Crybb3 |
9.2 |
Rspo1 |
2.1 |
Crybb1 |
20.2 |
Ugt1a1 |
3.3 |
| Crybb3 |
8.9 |
Crygc |
2.1 |
Crygs |
20 |
Ugt1a9 |
3.3 |
| Dct |
7.2 |
Wnt7a |
2.1 |
Cryba1 |
19.8 |
Ugt1a2 |
3.3 |
| Cd24a |
6.7 |
Trpm1 |
2.1 |
Cryaa |
18.9 |
Bmper |
3.3 |
| Bfsp1 |
6.6 |
Npff |
2.1 |
Cryba2 |
18.5 |
Ugt1a7c |
3.3 |
| Lctl |
6.6 |
Krt15 |
2.1 |
Tyr |
18.5 |
Ugt1a6a |
3.2 |
| Cryab |
6.6 |
Selenbp1 |
2.1 |
Crybb3 |
16.3 |
1700093K21Ri |
3.2 |
| S100a4 |
4.9 |
Tspan10 |
2 |
Lctl |
14.5 |
1110017D15Ri |
3.1 |
| Ttr |
4.8 |
Mgst1 |
2 |
Cd24a |
10.7 |
Ugt1a6b |
3.1 |
| Mlana |
4.8 |
Tmem176a |
2 |
Mlana |
10.7 |
Cox8b |
3.1 |
| Gja3 |
4.6 |
Papss2 |
2 |
Dct |
9.8 |
Acta2 |
3.1 |
| Crygb |
4.5 |
Mlph |
2 |
Krt5 |
9.2 |
Pla2g2f |
3 |
| Rgr |
4.2 |
Gsta3 |
2 |
Sfrp1 |
7.8 |
Ahnak |
3 |
| Crygd |
3.9 |
Gsto1 |
2 |
Lrat |
6.8 |
Lix1 |
2.9 |
| Pmel |
3.9 |
Perp |
2 |
S100a4 |
6.4 |
Adamts4 |
2.8 |
| Sfrp1 |
3.9 |
Srd5a2 |
2 |
Ttr |
6.4 |
Ela1 |
2.7 |
| Egr1 |
3.6 |
Psca |
2 |
Mlph |
6.3 |
Angptl4 |
2.7 |
| Tyrp1 |
3.3 |
C1qtnf5 |
2 |
Rgr |
5.7 |
Galr2 |
2.6 |
| Aldh3a1 |
3.3 |
Mfrp |
2 |
Tgm1 |
5.6 |
Dock5 |
2.6 |
| Fabp5 |
3.2 |
Lgals1 |
1.9 |
Rpe65 |
5.5 |
Scel |
2.6 |
| Lenep |
3.2 |
Islr |
1.9 |
Emp1 |
5.3 |
Ppfibp2 |
2.6 |
| Gpnmb |
3.2 |
Penk |
1.9 |
Srd5a2 |
5.2 |
Pld5 |
2.6 |
| Gja8 |
3.2 |
Oca2 |
1.9 |
Slc6a20a |
5.2 |
Thbs1 |
2.5 |
| Serpina3n |
3.1 |
Aldh3a1 |
1.9 |
Fam46c |
5 |
Selenbp1 |
2.5 |
| Wnt7b | 2.9 | Ak1 | 1.9 | D21Rik | 4.8 | Ucp2 | 2.5 |
*The work-flow, Method I (BWA-ANOVA) and Method II (ELAND-SAM) are described in the Methods’ section. The numbers indicate fold changes between diabetic and nondiabetic controls as determined by two different algorithms.
Figure 1.
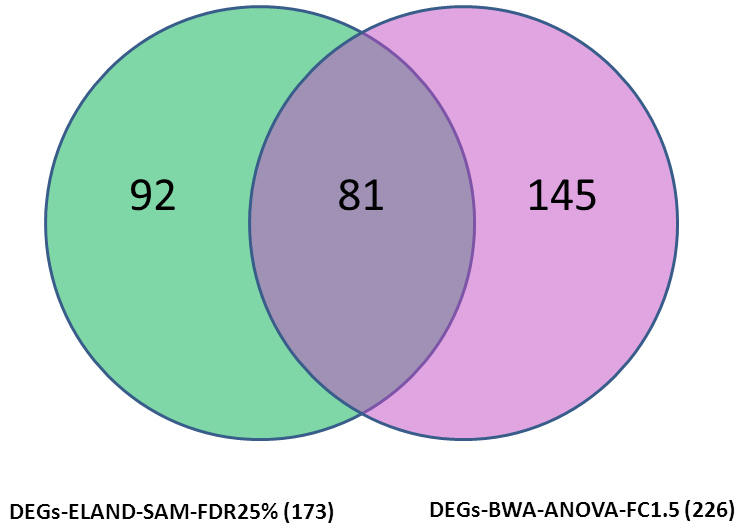
Differentially expressed gene sets were obtained by employing either significance of microarray analysis algorithm or ANOVA method. The gene sets were applied as described in Methods section and the genes unique and common in the two sets are indicated.
Validation of significantly altered transcripts
Some representative transcripts were chosen for validation based on the magnitude of the changes in their abundance and their involvement in various pathways. We performed Taqman real-time PCR to validate 11 transcripts, including Cryga-crystallin gamma A, Crygb-crystallin gamma B, Dct-dopachrome tautomerase, Egr-early growth response, FABP-fatty acid binding protein, Lgsn-lengsin, Lim2-lens intrinsic membrane protein, Nr2c2ap-nuclear receptor 2C2-associated protein, retinal pigment epithelium 65 Kd protein (Rpe65), Sfrp1-secreted frizzled-related protein, and wingless type MMTV integration site, member 7b (Wnt7b). The level of each of these transcripts corresponded to the pattern revealed by RNA-seq, and indicated significant changes in the levels of these transcripts between diabetic and normal mice (Figure 2). However, the magnitude of change observed by RT–PCR was not identical to the results of RNA-seq. The non-correspondence of magnitude reflects the differences in sensitivities of the RNA-seq and Taqman assays, which are two inherently different strategies.
Figure 2.
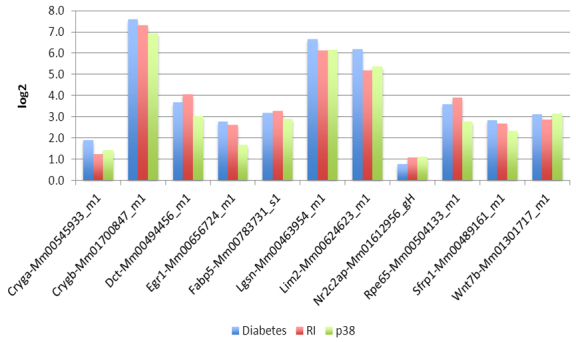
TaqMan real-time quantitative polymerase chain reaction (qPCR) validation was performed for selected transcripts. The transcripts were amplified as described in Methods section. Three technical replicates were performed for three biologic replicate samples of RNA isolated from nondiabetic animals, diabetic animals and diabetic animals treated with either the inhibitor of receptor for advanced glycation endproducts (RAGE) indicated as RI, or the inhibitor of p38 mitogen activated protein kinase (MAPK) designated as p38. The bars (left to right) represent samples corresponding to diabetic animals, diabetic animals treated with RAGE inhibitor, and diabetic animals treated with p38 MAPK inhibitor, respectively. Fold increase was calculated with respect to nondiabetic controls.
A few additional transcripts (Cdh3-cadherin, Fgf2-fibroblast growth factor, Lrat-lecithin retinol acyltransferase, Mnd1-meiotic nuclear divisions 1 homolog, Rgr-retinal G-protein coupled receptor, and Sema3c-semaphorin) were chosen for validation with SYBR green based real-time qPCR. All of these transcripts were significantly altered in diabetic mice (Figure 3). As stated for Taqman qPCR validation, the correspondence between RNA-seq and SYBR green qPCR was comparable but not identical.
Figure 3.
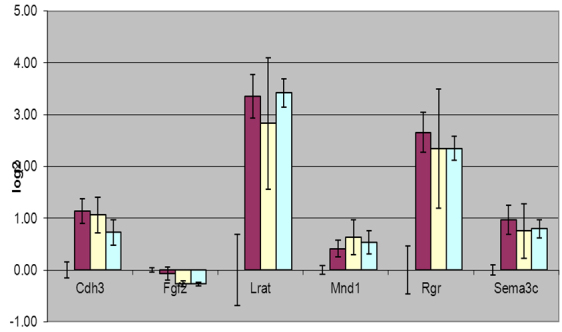
Cyanine (SYBR) green real-time quantitative polymerase chain reaction (qPCR) validation was performed for selected transcripts. The transcripts were amplified as described in Methods section. Three technical replicates were performed for three biologic replicate samples of RNA isolated from nondiabetic animals, diabetic animals and diabetic animals treated with either inhibitor of receptor for advanced glycation endproducts (RAGE) indicated as RI, or an inhibitor of p38 mitogen activated protein kinase (MAPK) designated as p38. The bars (left to right) represent samples corresponding to diabetic animals, diabetic animals treated with RAGE inhibitor, and diabetic animals treated with p38 MAPK inhibitor, respectively. The error bars represent standard deviation from the mean value (n=9). The levels of transcript in diabetic and treated animals were determined relative to the levels present in nondiabetic controls.
Transcripts, pathways and gene ontology (GO) categories are altered in retinas isolated from diabetic animals
The application of SAM and a false discovery rate of 25% for the data set revealed greater than 1.5 fold alterations in the levels of 173 transcripts. Alternative analysis with BWA-ANOVA, however, indicated alterations of greater than 1.5 fold in 298 transcripts. Among several GO categories, significantly altered transcripts revealed by either of the two methods were grouped together as structural constituents of eye lens, visual perception, sensory perception of light stimulus, eye development, structural molecule activity, microsomes, the vesicular fraction, and glucuronosyl transferase activity. Changes were also observed in transcripts involved in inflammatory pathway, apoptosis, microvasculature, Wnt signaling, photoreceptors, and neural cell function. The alterations in the levels of these transcripts are described below.
Crystallins, a major class of retinal genes, are altered in diabetic retinopathy
More than 20 variants of crystallins have been detected in the retina. Of these, 13 transcripts were increased by greater than 1.6 fold (Table 3). The levels of Cryaa, Cryba1, Cryba2, Cryba4, Crybb1, Crybb2, and Crygs in diabetic animals were elevated by >9.0 fold, whereas the abundance of Cryab, Crygb, Crygc, Crygd, and Crygn in diabetic animals varied between 1.6 and 6.6 fold compared to non-diabetic controls. These transcript changes were detected by both analysis methods.
Table 3. Alterations in the levels of crystallin transcripts in diabetic retinopathy and the reversal of their levels by RAGE inhibitor and p38 MAPK inhibitor.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| Crybb2 |
Crystallin, Beta B2 |
19.8 |
3.6 |
2.4 |
| Cryba1 |
Crystallin, Beta A1 |
19.3 |
3.7 |
1.9 |
| Cryba4 |
Crystallin, Beta A4 |
18.3 |
3.8 |
2.4 |
| Crygs |
Crystallin, Gamma S |
18.2 |
3.9 |
2.5 |
| Cryaa |
Crystallin, Alpha A |
17.9 |
3.5 |
2.4 |
| Cryba2 |
Crystallin, Beta A2 |
16.4 |
3.4 |
2.1 |
| Crybb1 |
Crystallin, Beta B1 |
15.1 |
3.9 |
2.8 |
| Crybb3 |
Crystallin, Beta B3 |
9.2 |
3.2 |
2.5 |
| Cryab |
Crystallin, Alpha B |
6.6 |
3.6 |
1.9 |
| Crygb |
Crystallin, Gamma B |
4.5 |
3.2 |
2.2 |
| Crygd |
Crystallin, Gamma D |
3.9 |
2.6 |
2.3 |
| Crygc |
Crystallin, Gamma C |
2.4 |
1.7 |
1.6 |
| Crygn | Crystallin, Gamma N | 1.6 | 1.5 | 1.2 |
*D=diabetic control, n=nondiabetic control, D+mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D+PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Alterations in Wnt signaling transcripts
Altered levels were observed for 14 transcripts that are either directly involved in Wnt signaling pathway or are targets of Wnt signaling (Table 4). While Sfrp1 and Wnt7b levels in diabetic animals increased by 3.9 and 2.9 fold, Wnt7a and Mfrp were upregulated by more than 2.0 fold. The increase in the levels of Wnt5b and axin, and decrease in Wisp 1 and Wnt2b transcripts were marginal. The levels of a few target genes of Wnt signaling such as FRAT2, BMP4, Cldn1, and Cldn2 were also altered.
Table 4. Alterations in the levels of transcripts involved in Wnt signaling pathway.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| Sfrp1 |
Secreted frizzled-related protein 1 |
3.9 |
2.9 |
1.9 |
| Wnt7b |
Wingless-related MMTV integration site 7B |
2.9 |
1.9 |
1.5 |
| Wnt7a |
Wingless-related MMTV integration site 7A |
2.1 |
1.5 |
1.5 |
| Mfrp |
Membrane-type frizzled-related protein |
2.1 |
1.9 |
1.5 |
| Cldn1 |
Claudin 1 |
1.6 |
1.4 |
1.4 |
| Cldn2 |
Claudin 2 |
1.6 |
1.4 |
1.6 |
| BMP4 |
Bone morphogenetic protein 4 |
1.5 |
1.6 |
1.5 |
| Id2 |
Inhibitor of DNA binding 2 |
1.4 |
1.6 |
1.4 |
| Axin1 |
Axin 1; axis inhibition protein 1 |
1.3 |
1.2 |
1.6 |
| FRAT2 |
Frequently rearranged in advanced T-cell lymphomas 2 |
1.2 |
1.4 |
1.8 |
| Wnt5b |
Wingless-related MMTV integration site 5B |
1.2 |
1.3 |
1.4 |
| Wisp1 |
WNT1 inducible signaling pathway protein 1 |
1.5 (down) |
1.6 (down) |
2.4 (down) |
| Wnt2b | Wingless related MMTV integration site 2b | 1.1 (down) | 1.1 (down) | 1.05 (down) |
*D=diabetic control, n=nondiabetic control, D+mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D+PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Transcripts involved in microvasculature and the inflammatory pathway
Endothelin and VEGF were among the vascular transcripts detected in the retina. The levels of Edn2 decreased by 1.4 fold, whereas an increase of 1.4 fold was observed in the levels of Edn3 (Table 5). The levels of other vasculature related genes were marginally altered in diabetic animals. Alterations were also observed in the abundance of inflammation associated transcripts, namely Ltbp1, Bmp4, Hspb1, CD44, C1qtnf5, Ifitm1, and Islr, which varied between 1.5 and 2.2 fold in diabetic animals (Table 6).
Table 5. Altered levels of microvasculature transcripts associated with diabetic retinopathy.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| Edn3 |
Endothelin 3 |
1.4 |
1.5 |
1.4 |
| VEGFB |
Vascular endothelial growth factor B |
1.1 |
1.3 (down) |
1.1 (down) |
| VEGFA |
Vascular endothelial growth factor A |
1.02 |
1.4 |
1.3 |
| Edn2 | Endothelin 2 | 1.4 (down) | 1.6 (down) | 2.0 (down) |
*D=diabetic control, n=nondiabetic control, D + mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D + PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Table 6. Relative levels of transcripts associated with inflammatory pathway in diabetic mice.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| C1qtnf5 |
C1q and tumor necrosis factor related protein 5 |
2.2 |
1.9 |
1.5 |
| CD44 |
CD44 antigen |
1.7 |
1.3 |
1.2 |
| Ltbp1 |
Latent transforming growth factor beta binding protein 1 |
1.6 |
1.4 |
1.4 |
| Islr |
Immunoglobulin superfamily containing leucine-rich repeat |
1.6 |
1.5 |
1.6 |
| Hspb1 |
Heat shock protein 1 |
1.6 |
1.6 |
1.2 |
| Ifitm1 |
Interferon induced transmembrane protein 1 |
1.5 |
1.4 |
1.3 |
| Bmp4 | Bone morphogenetic protein 4 | 1.5 | 1.6 | 1.5 |
*D=diabetic control, n=nondiabetic control, D+mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D+PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Transcripts involved in apoptosis
We analyzed a variety of pro-apoptotic and anti-apoptotic transcripts as well as other transcripts that influence the apoptosis pathways. A significant increase was observed in the levels of Dapl1 and Bid1, while the levels of Fads3, Fas, and Bag5 showed marginal alterations. An appreciable decrease was also noted in the levels of Trafip3, Tnfsf13, Tnfrsf18, and Casp7 (Table 7). It warrants mention that these transcripts were not shortlisted when the FDR and p-value cut-off were applied to the data analysis. Given the importance of apoptosis in diabetic retinopathy [36,37], these genes were specifically analyzed from the master list of sequenced tags and were found to have altered levels in the diabetic animals.
Table 7. Changes in the levels of transcripts involved in apoptosis pathway.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| Dapl1 |
Death associated protein-like 1 |
2.6 |
1.2 |
1.2 |
| Bid |
BH3 interacting domain death agonist |
1.6 |
1.5 |
1.3 |
| Fads3 |
Fatty acid desaturase 3 |
1.3 |
1.2 |
1.2 |
| Fas |
Fas (TNF receptor superfamily member 6) |
1.3 |
1.1 |
1.2 |
| Bag5 |
BCL2-associated athanogene 5 |
1.2 |
1.2 |
1.4 |
| Traf3ip3 |
TRAF3 interacting protein 3 |
1.5 (down) |
1.5 (down) |
2.5 (down) |
| Tnfsf13 |
Tumor Necrosis Factor superfamily member 13 |
1.4 (down) |
1.5 (down) |
1.6 (down) |
| Tnfrsf18 |
Tumor necrosis factor receptor superfamily, member 18 |
1.4 (down) |
1.7 (down) |
2.2 (down) |
| Casp7 |
Caspase 7 |
1.4 (down) |
1.5 (down) |
2.0 (down) |
| Bcl7c | B-cell CLL/lymphoma 7C | 1.1 (down) | 1.6 (down) | 1.3 (down) |
*D=diabetic control, n=nondiabetic control, D+mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D+PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Transcripts involved in neuronal functions and photoreceptors
The list of differentially expressed genes obtained by applying stringent threshold parameters did not contain any transcripts involved in neuronal development or signal transduction. However, a review of the transcript master list, without applying rigorous statistical significance, revealed changes in transcripts encoded by polycomb genes (Pcgf), acetyl choline receptor genes (Chrn), muscarinic cholinergic receptor genes (Chrm), potassium channels (Kcnq), glutamate receptor genes (Grin), solute carrier/transporter genes (Slc), and the fasciculation and elongation gene (Fez) (Table 8). Similar review revealed decreased levels of transcripts involved in the development and function of photoreceptors. These transcripts included cyclic nucleotide gated channel (Cngb3), arrestin (Arr), guanine nucleotide binding protein (Gnb3), and phosphodiesterase (Pde6h). Marginal decreases were also noticed in photoreceptor-specific transducin (Gnat2) and Crxos1. However, the levels of Nrl, Crx, and Nr2e3 remained relatively unchanged (Table 9).
Table 8. Altered levels of transcripts involved in neuronal functions.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| Pcgf2 |
Polycomb group ring finger 2 |
1.5 |
1.3 |
1.2 |
| Slc15a2 |
Solute carrier family 15 (H+/peptide transporter), member 2 |
1.3 |
1.1 |
1.1 |
| Kcnq4 |
Potassium voltage-gated channel, subfamily Q, member 4 |
1.2 |
1.1 |
1.1 (down) |
| Chrna2 |
Cholinergic receptor, nicotinic, alpha polypeptide 2 (neuronal) |
1.2 |
1.2 |
1.2 |
| Grinl1a |
Glutamate receptor, ionotropic, NMDA like 1A |
1.2 |
2 |
2 |
| Chrm4 |
Cholinergic receptor, muscarinic 4 |
1.04 |
1.2 |
1.4 |
| Slc6a5 |
Solute carrier family 6 (neurotransmitter transporter, glycine), member 5 |
1 |
1.39 (down) |
1.32 (down) |
| Kcnip4 |
Kv channel interacting protein 4 |
1.25 (down) |
1.13 (down) |
1.62 (down) |
| Kcnma1 |
Potassium large conductance calcium-activated channel, subfamily M, alpha member 1 |
1.24 (down) |
1.62 (down) |
1.57 (down) |
| Grin1 |
Glutamate receptor, ionotropic, NMDA1 (zeta 1) |
1.21 (down) |
1.79 (down) |
1.47 (down) |
| Chrnb3 |
Cholinergic receptor, nicotinic, beta polypeptide 3 |
1.08 (down) |
1.44 (down) |
1.35 (down) |
| Fez1 | Fasciculation and elongation protein zeta 1 (zygin I) | 1.03 (down) | 1.35 (down) | 1.29 (down) |
*D=diabetic control, n=nondiabetic control, D+mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D+PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Table 9. Altered levels of selected photoreceptor-specific transcripts.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| Cngb3 |
Cyclic nucleotide gated channel beta 3 |
1.3 (down) |
1.4 (down) |
1.4 (down) |
| Arr3 |
Arrestin 3, retinal |
1.3 (down) |
1.6 (down) |
1.8 (down) |
| Gnb3 |
Guanine nucleotide binding protein (G protein), beta 3 |
1.3 (down) |
1.7 (down) |
1.8 (down) |
| Pde6h |
Phosphodiesterase 6H, cGMP-specific, cone, gamma |
1.2 (down) |
1.8 (down) |
2.2 (down) |
| Pde6a |
Phosphodiesterase 6A, cGMP-specific, rod, alpha |
1.2 (down) |
1.4 (down) |
1.5 (down) |
| Gnat2 |
Guanine nucleotide binding protein, alpha transducing 2 |
1.1 (down) |
1.3 (down) |
1.2 (down) |
| Crxos1 |
Crx opposite strand transcript 1 |
1.1 (down) |
1.4 (down) |
1.7 (down) |
| Pde6c |
Phosphodiesterase 6C, cGMP specific, cone, alpha prime |
1.1 (down) |
1.7 (down) |
1.8 (down) |
| Nrl |
Neural retina leucine zipper gene |
1.02 (down) |
1.1 |
1.1 |
| Crx |
Cone-rod homeobox containing gene |
1.03 |
1.3 |
1.2 |
| Nr2e3 | Nuclear receptor subfamily 2, group E, member 3 | 1.01 | 1.11 (down) | 1.22 (down) |
*D=diabetic control, n=nondiabetic control, D+mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D+PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Altered expression of genes encoding glucuronosyl transferases in diabetic retinopathy
A significant increase was observed in transcripts for members of the UDP-glucuronosyl transferase gene family during diabetic retinopathy. The protein products of these genes are primarily responsible for the metabolism of xenobiotics. In particular, UGT1A5, UGT1A10, UGT1A1, UGT1A9, UGT1A2, UGT1A7C, UGT1A6A, and UGT1A6B transcripts were elevated by greater than 3.0 fold in diabetic animals (Table 10). These transcripts were detected in analyses performed by the ELAND-SAM method at the gene level with a FDR cutoff of 25%, but were absent in the list of transcripts generated by transcript isoform level BWA-ANOVA analysis at a p value cutoff of less than 0.05.
Table 10. Changes in the abundance of UDP glucuronosyl transferase transcripts in diabetic animals.
| |
|
Fold change* |
Fold change* |
Fold change* |
|---|---|---|---|---|
| Gene symbol | Gene name | D vs N | D + mRAGE-Fc vs N | D + PHA666859 vs N |
| Ugt1a5 |
UDP glucuronosyltransferase 1 family, polypeptide A5 |
3.3 |
2 |
1.5 |
| Ugt1a10 |
UDP glucuronosyltransferase 1 family, polypeptide A10 |
3.3 |
2 |
1.5 |
| Ugt1a1 |
UDP glucuronosyltransferase 1 family, polypeptide A1 |
3.3 |
2 |
1.5 |
| Ugt1a9 |
UDP glucuronosyltransferase 1 family, polypeptide A9 |
3.3 |
2 |
1.5 |
| Ugt1a2 |
UDP glucuronosyltransferase 1 family, polypeptide A2 |
3.3 |
2 |
1.5 |
| Ugt1a7c |
UDP glucuronosyltransferase 1 family, polypeptide A7C |
3.3 |
1.9 |
1.5 |
| Ugt1a6a |
UDP glucuronosyltransferase 1 family, polypeptide A6A |
3.3 |
2 |
1.5 |
| Ugt1a6b | UDP glucuronosyltransferase 1 family, polypeptide A6B | 3.1 | 2.1 | 1.5 |
*D=diabetic control, n=nondiabetic control, D+mRAGE-Fc=diabetic plus murine RAGE-Fc fusion protein, D+PHA666859=diabetic plus p38 MAPK inhibitor PHA666859.
Rhodopsin-retinol pathway altered in diabetic mice
Rhodopsin-retinol pathway transcripts showed increased levels for lecithin retinol acetyl transferasease, transthyretin, retinol binding protein, and Stra6 (stimulated by retinoic acid). However, decreases occurred in the levels of transcripts corresponding to retinoic acid metabolism enzyme Cyp26a1, retinol dehydrogenase, and rhodopsin phosphorylating GPCR kinase (Grk4).
Pathways related to altered metabolism in diabetic mice
Transcripts directly/indirectly related to glycemic pathway showed marginal increases in the case of glycerol-3-phosphate dehydrogenase and insulin secretion modulating Trpm4 protein, whereas transcripts levels decreased slightly for a glycogen interacting protein Trim7 and for insulin-like growth factor binding proteins Igfbp3 and Igfbp5.
Alternative splicing of genes in diabetic retinopathy
The RNA-seq method provides the identity of transcripts that have yielded an unambiguous sequence. Some of the sequence reads, which span exon boundaries, can be analyzed to predict alternatively spliced transcripts. We identified 55 genes that were alternatively spliced in the retina of diabetic animals (Table 11). The alternative splicing events in these genes were detected by selecting transcripts with a RPKM cutoff of 3.0 and using ASPIRE’s ΔI cutoff of 0.2, which indicates a 20% change in exon inclusion. This threshold of ΔI has been shown to yield accurate representations of alternatively spliced versions of transcripts [34].
Table 11. Alternatively spliced transcripts in diabetic animals.
| Gene symbol | Gene name | Gene symbol | Gene name |
|---|---|---|---|
| Abca8a |
ATP-binding cassette |
Agtpbp1 |
ATP/GTP binding protein |
| Ankrd39 |
ankyrin repeat domain |
Ank2 |
ankyrin 2, brain |
| Apip |
APAF1 interacting |
Dus1l |
dihydrouridine synthase |
| Bdh1 |
3-hydroxybutyrate dehydrogenase |
Eif4g3 |
eukaryotic translation initiation |
| Cltb |
clathrin |
Epb4.1l2 |
erythrocyte protein |
| Commd6 |
COMM domain |
Fam193b |
family with sequence similarity193 |
| Dclk2 |
doublecortin-like kinase |
Hook2 |
hook homolog 2 |
| Dedd2 |
death effector domain |
Pcmtd2 |
protein O-methyltransferase |
| Dync1li2 |
dynein |
Tpm3 |
tropomyosin 3, |
| Fam161a |
family with sequence similarity |
Arr3 |
arrestin 3, retinal |
| Fam184a |
family with sequence similarity184 |
Gprasp2 |
G protein-coupled receptor |
| Ggps1 |
geranylgeranyl diphosphate synth |
Gse1 |
genetic suppressor element |
| Gripap1 |
GRIP1 associated protein |
Nsmaf |
neutral sphingomyelinase |
| Impg2 |
interphotoreceptor matrix protein |
Sgip1 |
SH3-domain GRB2-like |
| Plekha6 |
pleckstrin homology domain |
Ubtf |
upstream transcription factor |
| Ppie |
peptidylprolyl isomerase E |
Unc79 |
unc-79 homolog |
| Ptprf |
protein tyrosine phosphatase |
Ccdc64 |
coiled-coil domain containing |
| Samhd1 |
SAM domain and HD domain |
Cugbp2 |
CUGBP, Elav-like |
| Scamp3 |
secretory carrier mem protein |
Dctn6 |
dynactin 6 |
| Smc5 |
structural maintenance of chrom |
Mak |
male germ cell-associated kinase |
| Sorbs1 |
sorbin and SH3 domain |
Mare |
nitrogen permease regulator-like 3 |
| Trpm1 |
transient receptor cation channel |
Scarb1 |
scavenger receptor class B, |
| Tsc2 |
tuberous sclerosis 2 |
Slc30a9 |
solute carrier family 30 (Zn) |
| Zcrb1 |
zinc finger CCHC-type |
Trnau1ap |
tRNA selenocysteine 1 protein |
| Zmiz2 | zinc finger, MIZ-type | Whrn | whirlin Gene |
It warrants mention that alternative splicing of transcripts is also prevalent in the retinas of normal animals. The 55 alternatively spliced transcripts therefore represent either novel spliced versions or spliced isoforms with altered abundance in diabetic animals, which suggests an association of alternative splicing with diabetic retinopathy. Noteworthy among these transcripts were those involved in neuronal cell survival, neural differentiation, apoptosis, endocytosis, intracellular transport, glucose metabolism and insulin impairment, G-protein coupled receptor signaling, and mRNA processing. Specifically, alternative splicing of arrestin (Arr), interphotoreceptor matrix proteoglycan (Impg2), and transient receptor membrane potential cation channel provide tentative evidence for the effects of diabetic retinopathy on the activity and integrity of photoreceptor cells in the retina.
Responsiveness to treatments and molecular side effects of therapeutic intervention with candidate drugs
We have characterized the molecular responsiveness of animals to treatments as positive, negative, or as side effects, based on a comparison with wild type mice. We define positive effects of drugs as a reversal of transcript levels toward normalcy, and negative effects as changes in transcript levels of treated animals that are greater than those seen in the diabetic animals when compared to wild type animals. If a transcript remains unchanged in retinas from diabetic animals and the therapy leads to a significant change in its abundance, this is classified as a side effect.
Treatments with inhibitors of RAGE and p38 MAPK inhibited diabetes-induced changes (positive effects) and also exacerbated other changes (negative effects; Table 3, Table 4, Table 5, Table 6, Table 7, Table 8, Table 9, and Table 10). Both inhibitors blocked the diabetes-induced changes in levels of all crystallin transcripts and the levels of UDP-glucuronosyl transferases. Both inhibitors worked well in preventing the changes in the levels of Wnt pathway transcripts or apoptosis transcripts, but the RAGE-inhibitor appeared to work positively on a greater number of transcripts, as was the case for transcripts of inflammatory pathways. Surprisingly, photoreceptor transcripts were adversely affected in animals treated with either of these drugs. Of the 837 transcripts that were altered by greater than 1.5 fold in diabetic mice or in any one of the treatments, 379 transcripts were further exacerbated by RAGE inhibitor. The treatment with p38 MAPK inhibitor, on the other hand, caused further adverse exacerbations in 535 transcripts. Whether these exacerbations of transcript levels by drugs were beneficial or detrimental is not known at present.
Similar analyses were also performed with the data related to alternative splicing (Figure 4). Comparison of diabetic versus nondiabetic, RAGE inhibitor-treated diabetic versus nondiabetic and p38 MAPK inhibitor-treated diabetic versus nondiabetic animals led to the identification of alternatively spliced transcripts specific to each group of animals (Table 11, Table 12, and Appendix 2). Some notable alternatively spliced transcripts in diabetic animals included doublecortin-like kinase Dclk3, death effector domain Dedd2, dynein Dync1li2, interphotoreceptor matrix protein Impg2, ankyrin 2, arrestin 3, coiled-coil domain containing Ccdc64, G-protein coupled receptor Gprasp2, Nsmaf, dynactin 6, and whirlin (Table 11). Among the transcripts responsive to RAGE inhibitor were photoreceptor specific gene arrestin, Nsmaf transcript involved in apoptosis, and endocytosis transcript Sgip1. The exacerbated levels of 76 alternatively spliced transcripts in p38 MAPK inhibitor-treated animals included the Wnt pathway transcript Axin1 and its target gene Cldn1, opsin transcript Opnm1w, Mapk8ip3, peroxisomal transcript Pex16, cilia transcript Cep164, and ocular regulator Pax6 (Table 12 and Appendix 2). A comparison of alternatively spliced transcripts revealed 12 transcripts common among nondiabetic animals, diabetic animals, diabetic animals treated with RAGE inhibitor, and diabetic animals treated with p38 MAPK inhibitor (Figure 4). Among others, these genes included G-protein coupled receptor associated sorting protein, kinesin family member 1A, nuclear receptor co-repressor, phospholipase A2, and zinc finger protein 444. Although the summary presented in Figure 4 does not allow us to decipher the mechanisms of these therapies, the results suggest that the changes in splicing patterns in response to therapy may reflect the positions of p38 MAP kinase and RAGE in the cell signaling pathways relative to each other.
Figure 4.
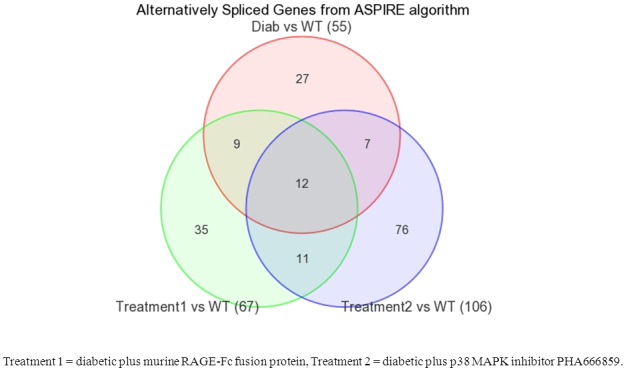
Unique and common splice forms of transcripts are present in various groups of animals. The splice variants from various samples were compared and the numbers of transcripts unique to a specific experimental pair or common to two or more experimental pairs are indicated.
Table 12. Treatment responsive alternatively spliced transcripts.
| Responsive to T1 and T2* | Unresponsive to T1 and T2* | Exacerbated by T2* | ||||
|---|---|---|---|---|---|---|
| Abca8a |
Ggps1 |
Efha2 |
Ablim1 |
Epb4.1l3 |
Os9 |
Sag |
| Ankrd39 |
Gripap1 |
Fam169a |
Adarb1 |
Fam177a |
P4ha1 |
Sfmbt1 |
| Apip |
Impg2 |
Gprasp1 |
Asph |
Fam57b |
Papola |
Slc16a3 |
| Bdh1 |
Plekha6 |
Hnrnph3 |
Axin1 |
Fbxo3 |
Pax6 |
Srp54a |
| Cltb |
Ppie |
Kif1a |
BC018101 |
Golga2 |
Pcdhga11 |
Srpk2 |
| Commd6 |
Ptprf |
Ncor1 |
BC037034 |
Golga4 |
Pcsk7 |
Srsf7 |
| Dclk2 |
Samhd1 |
Pla2g5 |
Bod1l |
Gpbp1 |
Pex16 |
Tacc2 |
| Dedd2 |
Scamp3 |
RP23–285G5.1 |
Brsk2 |
Gpr85 |
Pfkp |
Tlk2 |
| Dync1li2 |
Smc5 |
Slc25a19 |
Cacna1a |
Gtf2ird1 |
Pitpnm3 |
Trip12 |
| Fam161a |
Sorbs1 |
Strn |
Cep164 |
Heg1 |
Pkp4 |
Ypel3 |
| Fam184a |
Trpm1 |
Zfp444 |
Cldnd1 |
Immt |
Plekhb1 |
Zc3h11a |
| Zmiz2 |
Tsc2 |
|
Cnpy3 |
Ip6k2 |
Ppp2r3c |
Zdhhc2 |
| |
Zcrb1 |
|
Col4a3bp |
Iqsec3 |
Ppp6r2 |
Zfp207 |
| |
|
|
Crebl2 |
Itsn2 |
Pppde2 |
Zfp740 |
| |
|
|
Dlg4 |
Jmjd6 |
Ptcd3 |
Zfp788 |
| |
|
|
Dpp3 |
Mapk8ip3 |
Rab28 |
Zfp97 |
| |
|
|
Eno2 |
Mpp2 |
Ralgapa1 |
Zmynd8 |
| |
|
|
Nufip2 |
Msi1 |
Rufy3 |
Zscan29 |
| |
|
|
Opn1mw |
|
|
|
|
Responsive to T1* |
Responsive to T2* |
Exacerbated by T1 and T2* |
Exacerbated by T1* |
|||
| Arr3 |
Agtpbp1 |
Ccdc64 |
Akap8 |
Dph3 |
Mthfs |
Rho |
| Gprasp2 |
Ank2 |
Cugbp2 |
Amy2a2 |
Ehbp1 |
Nrcam |
Sfrs14 |
| Gse1 |
Dus1l |
Dctn6 |
Amy2a3 |
Epb4.1 |
Pan3 |
Smarcc2 |
| Nsmaf |
Eif4g3 |
Mak |
Amy2a4 |
Gm2382 |
Pdk1 |
Tmpo |
| Sgip1 |
Epb4.1l2 |
Mare |
Ap1b1 |
Gm672 |
Phactr4 |
Unc45a |
| Ubtf |
Fam193b |
Scarb1 |
Arhgap12 |
Gmps |
Ppfibp1 |
Zfp687 |
| Unc79 |
Hook2 |
Slc30a9 |
Atrx |
Ilf3 |
Prkcsh |
Zmym3 |
| |
Pcmtd2 |
Trnau1ap |
Cspp1 |
Maz |
Prom1 |
|
| Tpm3 | Whrn | D4Ertd22e | Mettl3 | R3hdm2 | ||
*Treatment 1=diabetic plus murine RAGE-Fc fusion protein, Treatment 2=diabetic plus p38 MAPK inhibitor PHA666859.
Discussion
This is the first report to describe the application of RNA-seq for comprehensive sequencing of transcripts in a diabetic retinopathy model and for evaluating the efficacy of candidate drugs. The RNA-seq methodology has allowed accurate and quantitative identification of molecular signatures. The quantitative RT–PCR data (by TaqMan or SYBR Green) validated the expression changes for all 15 transcripts that were examined. Interestingly, the rank order of differentially expressed transcripts (as a consequence of diabetes or treatment with drugs) differed between the two analysis methods used. The ELAND-SAM method used gene model annotations (RefGene) from UCSC, while the BWA-ANOVA method used the transcript model annotations (refflat) from UCSC. However, the transcripts revealed by both methods of analyses were verifiable by real-time PCR and thus the validity of both methods was supported. The validation of the RNA-seq platform for comprehensive transcriptome analysis has recently been described by Brooks et al. [24]. Given the growing use of next generation sequencing platforms, the development of additional analytical tools in the future should allow even better description of RNA-seq data.
As predicted, we observed changes in a variety of retinal transcripts due to diabetes. Some of these changes have been found to influence glucose metabolism or to alter the levels of hormone and hormone receptors in other tissues; these include glycerol-3-phosphate dehydrogenase, galanin receptor (Galr2) involved in G-protein coupled receptor signaling, transient receptor (Trpm4) involved in insulin secretion, insulin like growth factor binding proteins (Igfbp3 and Igfbp5), the kinesin family of proteins, and calpain. Since vascular cells occupy such a small portion of the retina, the majority of observed changes likely are due to changes in the retinal neuroglia. Consistent with this idea, the list of transcripts that were significantly altered in diabetes did not contain transcripts corresponding to retinal vasculature, but targeted analyses of specific transcripts (such as endothelin-2, endothelin-3 and VEGFB) nevertheless revealed diabetes-induced alterations of 1.1–1.4 fold in levels of these transcripts.
Inflammation has been previously demonstrated to play an important role in the pathogenesis of diabetic retinopathy in animals [38,39]. Previous microarray profiling in a rat diabetic retinopathy model indicated changes in the levels of a variety of transcripts involved in inflammatory pathways [19]. Our RNA-seq data showed many inflammation-associated genes with altered expression; these included C1q TNF-related gene 5 (C1qtnf5), latent TGF-β binding protein (Ltbp1), phospholipase (Pla2g2f), interleukin 17 receptor 7c (Il17rc), and C4b. The changes in relatively few transcripts of the inflammatory pathway by eight months of diabetes seen in our study, when compared to changes reported at shorter durations, might indicate that inflammation is most severe early during the etiology of diabetic retinopathy, and that its severity might diminish as the disease progresses.
A variety of transcripts were altered in late stage diabetes; our RNA-seq data identified the Wnt signaling pathway, crystallins, and various transcription factors. Significant alterations were observed in the abundance of transcripts corresponding to Wnt7b, secreted frizzled receptor related protein-1 (Sfrp1), dapper antagonist of β-catenin (Dact1), Wnt inducible secreted protein-1 (Wisp1), and membrane frizzled related protein (Mfrp). Modulation of the Wnt pathway has been reported as a likely target for intervention for diabetic retinopathy [40], and Wisp1 is known to upregulate the expression of the anti-apoptotic Bcl-X(L) gene [41]. In addition to the involvement of Wnt pathway proteins in other inflammatory diseases [42], alterations in Wnt signaling proteins have been shown in a rat diabetic retinopathy model [43].
We and others have previously observed a significant upregulation of different crystallin transcripts in diabetic retina [44]. The levels of fourteen crystallins increased significantly in diabetic mice. These proteins were initially characterized as structural components of the eye lens [45,46], but expression of crystallins has been shown in other tissues including the retina [47]. Crystallins that belong to the small heat shock protein family have a non-structural role [48], and retina-specific elevated levels of Cryaa, Cryba1, Crygs, Crybb2, Crygb, and Crygc have been reported in experimental uveitis [49]. Cryaa was shown to localize to photoreceptor cells in experimental uveitis, which likely represents a function in these cells to protect against apoptosis [49]. Our results clearly indicate a dramatic upregulation of various crystallins in diabetic retinopathy and suggest this to be a stress response, which may be similar to the reported effects of increased levels of Hspb1 transcript in retina [50]. Post-translational modifications have been reported to alter chaperone activity of crystallins [51–53], while glycation associated with aggregation is likely to compromise the protective effects of crystallins in diabetic animals. Transcriptional upregulation of crystallins may therefore possibly represent a stress response, but the translated proteins may be rendered dysfunctional by diabetes-induced and deleterious post-translational modifications and other crosslinking events.
The deleterious changes to the retina that occur in diabetes are likely to be associated with alterations in the levels of transcripts associated with apoptosis, survival, and metabolic injury. Consistent with this hypothesis, we have observed changes in a variety of transcripts associated with apoptosis pathways. The metabolic injury to the neural retina is suggested by the altered levels of transcripts that modulate oxidative stress in cells. Changes in transcripts such as cytochrome c oxidase, glutathione-S-transferase omega-1 and omega-2, microsomal glutathione S-transferase, and glutathione synthase indicate that cellular injury to the retina occurs due to oxidative stress. While cytochrome oxidase is a sink for molecular oxygen channeled through electron transport chain in mitochondria, glutathione synthase and glutathione-S-transferases are important components of redox reactions.
The comparative analysis of gene level expression data also revealed differential expression of UDP-glucuronosyl transferases in diabetic animals. The UDP-glucuronosyl transferases metabolize a variety of xenobiotics, but they are also important for reactions occurring in cellular metabolism, such as those involving steroid hormones. We speculate that these enzymes may possess some other signaling activities, in line with the reported observation that UDP-glucuronosyl transferase 2B17 depletion led to downregulation of the Mcl-1 gene [54]. If these types of signaling activities associated with UDP-glucuronosyl transferases are involved in the transcription of diabetic retinopathy-related genes, then targeted inhibition of specific UDP-glucuronosyl transferases might have therapeutic implications for diabetic retinopathy.
Numerous groups have been exploring ways to inhibit diabetic retinopathy by blocking specific metabolic pathways. Although none of these targeted approaches have been successful in the clinic to date, several candidate drugs are being evaluated for efficacy. The alterations in the levels of superoxide, nitric oxide, cyclooxygenase-2, VEGF, and NF-κβ in the early stages of diabetic retinopathy are consistent with the inflammation observed in retinal tissue. Our drug studies suggest that diabetes-induced alterations observed in inflammatory pathway transcripts are mediated in particular via p38 MAPK [40,55,56]. Another metabolic consequence of chronic hyperglycemia, in addition to inflammation, is the accumulation of advanced glycation end products (AGEs) [57]. These AGEs and other ligands are recognized by RAGE, and activated RAGE is associated with a variety of downstream consequences such as upregulation of VEGF and NF-κβ, and increased leucocyte adhesion in retinal microvascular endothelial cells, all of which are consistent with inflammation. Importantly, inhibitors of RAGE and p38 MAPK have been shown to inhibit the development of early stages of diabetic retinopathy in animals [3,25].
Since the phenotypic benefits of the two therapies are similar, assessing their efficacies against the molecular changes that they cause might provide important information. We have tested these effects to determine whether the drug prevents diabetes-induced changes in transcripts or further exacerbates diabetes-induced changes in transcript levels. Our data suggest that the RAGE inhibitor reversed the levels of a large number of transcripts whose levels were significantly altered in diabetic mice. Although the p38 MAPK inhibitor seemed comparably effective at inhibiting the development of diabetic retinopathy, it did not inhibit as many diabetes-induced changes in levels of transcripts, in contrast to what was seen with the RAGE inhibitor. One possible interpretation of this difference is that the transcripts normalized by inhibition of RAGE, but not p38 MAPK, are not critical for the development of the lesions that characterize early diabetic retinopathy. Interestingly, the p38 MAPK inhibitor caused further exacerbations in many more diabetes-induced changes in transcript levels than did the RAGE inhibitor. The intracellular protein p38 MAPK is downstream of RAGE [58] in one of the three pathways, such as PI3K, NF-kB, and MAPK that are initiated by AGE. Thus, one might expect a greater number of altered transcripts upon activation of RAGE, and reversal of transcript changes in animals treated with the inhibitor of RAGE.
An important and useful outcome of the RNA-seq approach was the identification of alternatively spliced transcripts during the development of diabetic retinopathy. Disease specific alternatively spliced transcript variants are shown to exist in various conditions [58,59], and our experimental studies show that alternatively spliced transcripts arise as diabetic retinopathy develops. Characterization of alternatively spliced transcripts that are specifically present or absent in a specific condition provides another opportunity for therapeutic intervention of diabetic retinopathy. Tocotrienol and EGCG are known to target splice sites and to modulate the abundance of alternatively spliced transcripts [60,61]. Exploration of the ability of these agents to inhibit diabetic retinopathy will be an interesting avenue of further research.
Reversal of the levels of alternatively spliced exons upon treatments also provides an additional validation of drug efficacy. Our novel finding of alternative splicing of transcripts in diabetes raises the possibility that these changes contribute to the pathogenesis of the retinopathy. From this perspective, the identification of photoreceptor-specific changes in arrestin, Impg2, and Trpm1 raises the further possibility of a contribution by photoreceptors to diabetic retinopathy. These possibilities remain to be tested experimentally.
Just as therapies were shown above to promote further exacerbation of some diabetes-induced changes in transcript levels, therapy-specific exacerbation of alternative splicing also occurs. These side effects were more prominent following treatment with the p38 MAPK inhibitor than with the RAGE inhibitor, but the significance of these differences is not known at present. It warrants mention that alternatively spliced transcripts have been identified in a variety of diseases [62], and that a specific spliced form of a gene such as COX-1 is one target of acetaminophen [59].
In conclusion, we have applied RNA-seq strategy and two analysis methods to determine the molecular signatures that involve representative transcripts of retinal vasculature, inflammatory pathway, Wnt signaling, apoptosis, crystallins, and UDP-glucuronosyl transferases. The importance of several of these abnormalities in the development of diabetic retinopathy has been identified previously, illustrating the power of the RNA-seq technique to identify important changes in disease. Of importance, however, is the identification of disease and treatment-specific alternative splicing by this strategy. The remarkable longevity of changes observed in transcript levels during the 8 months of diabetes in our study suggests that these changes may be necessary to promote and maintain the complications of diabetes. The inhibitors of RAGE and p38 MAPK reversed some of the diabetes-induced levels of various transcripts to normal levels, and exacerbated the changes in others. The significance and mechanisms underlying these alterations remain to be determined, but our results suggest RNA-seq as a desirable strategy for investigating the pathogenesis of diabetic retinopathy or other diseases, to distinguish between candidate drugs and to determine their mechanisms of action.
Acknowledgments
This work was supported by National Eye Institute IPA and OneSight Foundation (R.P.K.), NEI intramural program (A.S.), and NIH-EY00300 and the Medical Research Service of the Department of Veteran Affairs (T.S.K.). The drugs were donated by Galactica Pharmaceutical Inc., and Pfizer Research Laboratories.
Appendix 1. Comparison of transcript abundance changes obtained by two methods of analysis*.
To access the data, click or select the words “Appendix 1.” This will initiate the download of a compressed (pdf) archive that contains the file. *Method I (BWA-ANOVA) and method II (ELAND-SAM) have been described in the methods’ section. Fold changes are calculated as a ratio of transcripts in diabetic animals and nondiabetic controls.
Appendix 2. Treatment responsive alternatively spliced transcripts.
To access the data, click or select the words “Appendix 2.” This will initiate the download of a compressed (pdf) archive that contains the file. *Treatment 1=diabetic plus murine RAGE-Fc fusion protein, Treatment 2=diabetic plus p38 MAPK inhibitor PHA666859.
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global Prevalence of Diabetes. Diabetes Care. 2004;27:1047–53. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Kern TS, Engerman RL. Pharmacological inhibition of diabetic retinopathy: aminoguanidine and aspirin. Diabetes. 2001;50:1636–42. doi: 10.2337/diabetes.50.7.1636. [DOI] [PubMed] [Google Scholar]
- 3.Du Y, Tang J, Li G, Berti-Mattera L, Lee CA, Bartkowski D, Gale D, Monahan J, Niesman MR, Alton G, Kern TS. Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Invest Ophthalmol Vis Sci. 2010;51:2158–64. doi: 10.1167/iovs.09-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barile GR, Pachydaki SI, Tari SR, Lee SE, Donmoyer CM, Ma W, Rong LL, Buciarelli LG, Wendt T, Horig H, Hudson BI, Qu W, Weinberg AD, Yan SF, Schmidt AM. The RAGE axis in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:2916–24. doi: 10.1167/iovs.04-1409. [DOI] [PubMed] [Google Scholar]
- 5.Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–42. doi: 10.2337/diabetes.50.8.1938. [DOI] [PubMed] [Google Scholar]
- 6.Yatoh S, Mizutani M, Yokoo T, Kozawa T, Sone H, Toyoshima H, Suzuki S, Shimano H, Kawakami Y, Okuda Y, Yamada N. Antioxidants and an inhibitor of advanced glycation ameliorate death of retinal microvascular cells in diabetic retinopathy. Diabetes Metab Res Rev. 2006;22:38–45. doi: 10.1002/dmrr.562. [DOI] [PubMed] [Google Scholar]
- 7.Schulze A, Downward J. Navigating gene expression using microarrays–a technology review. Nat Cell Biol. 2001;3:E190–5. doi: 10.1038/35087138. [DOI] [PubMed] [Google Scholar]
- 8.Stears RL, Martinsky T, Schena M. Trends in microarray analysis. Nat Med. 2003;9:140–5. doi: 10.1038/nm0103-140. [DOI] [PubMed] [Google Scholar]
- 9.Kandpal R, Saviola B, Felton J. The era of 'omics unlimited. Biotechniques. 2009;46:351–2. doi: 10.2144/000113137. [DOI] [PubMed] [Google Scholar]
- 10.Díaz E, Yang YH, Ferreira T, Loh KC, Okazaki Y, Hayashizaki Y, Tessier-Lavigne M, Speed TP, Ngai J. Analysis of gene expression in the developing mouse retina. Proc Natl Acad Sci USA. 2003;100:5491–6. doi: 10.1073/pnas.0831080100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mu X, Zhao S, Pershad R, Hsieh TF, Scarpa A, Wang SW, White RA, Beremand PD, Thomas TL, Gan L, Klein WH. Gene expression in the developing mouse retina by EST sequencing and microarray analysis. Nucleic Acids Res. 2001;29:4983–93. doi: 10.1093/nar/29.24.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swaroop A, Zack DJ. Transcriptome analysis of the retina. Genome Biol. 2002;3:S1022. doi: 10.1186/gb-2002-3-8-reviews1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida S, Mears AJ, Friedman JS, Carter T, He S, Oh E, Jing Y, Farjo R, Fleury G, Barlow C, Hero AO, Swaroop A. Expression profiling of the developing and mature Nrl−/− mouse retina: Identification of retinal disease candidates and transcriptional regulatory targets of Nrl. Hum Mol Genet. 2004;13:1487–503. doi: 10.1093/hmg/ddh160. [DOI] [PubMed] [Google Scholar]
- 14.Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh EC, Jing Y, Linares JL, Brooks M, Zareparsi S, Mears AJ, Hero A, Glaser T, Swaroop A. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc Natl Acad Sci USA. 2006;103:3890–5. doi: 10.1073/pnas.0508214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue W, Cojocaru RI, Dudley VJ, Brooks M, Swaroop A, Sarthy VP. Ciliary neurotrophic factor induces genes associated with inflammation and gliosis in the retina: a gene profiling study of flow-sorted, Müller cells. PLoS ONE. 2011;6:e20326. doi: 10.1371/journal.pone.0020326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida S, Yamaji Y, Yoshida A, Kuwahara R, Yamamoto K, Kubata T, Ishibashi T. Novel triple missense mutations of GUCY2D gene in Japanese family with cone-rod dystrophy: possible use of genotyping microarray. Mol Vis. 2006;12:1558–64. [PubMed] [Google Scholar]
- 17.Liu J, Wang J, Huang Q, Higdon J, Magdaleno S, Curran T, Zuo J. Gene expression profiles of mouse retinas during the second and third postnatal weeks. Brain Res. 2006;1098:113–25. doi: 10.1016/j.brainres.2006.04.086. [DOI] [PubMed] [Google Scholar]
- 18.Corbo JC, Myers CA, Lawrence KA, Jadhav AP, Cepko CL. A typology of photoreceptor gene expression patterns in the mouse. Proc Natl Acad Sci USA. 2007;104:12069–74. doi: 10.1073/pnas.0705465104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brucklacher RM, Patel KM, VanGuilder HD, Bixler GV, Barber AJ, Antonetti DA, Lin CM, LaNoue KF, Gardner TW, Bronson SK, Freeman WM. Whole genome assessment of the retinal response to diabetes reveals a progressive neurovascular inflammatory response. BMC Med Genomics. 2008;1:26. doi: 10.1186/1755-8794-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeman WM, Bixler GV, Brucklacher RM, Walsh E, Kimball SR, Jefferson LS, Bronson SK. Transcriptomic comparison of the retina in two mouse models of diabetes. J Ocul Biol Dis Infor. 2009;2:202–13. doi: 10.1007/s12177-009-9045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–7. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.t Hoen PA, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36:e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks MJ, Rajasimha HK, Roger JE, Swaroop A. Next generation sequencing facilitates quantitative analysis of wild type and Nrl−/− retinal transcriptomes. Mol Vis. 2011;17:3034–54. [PMC free article] [PubMed] [Google Scholar]
- 25.Li G, Tang J, Du Y, Lee CH, Kern TS. Beneficial effects of RAGE-Ig fusion protein on early diabetic retinopathy and tactile allodynia. Mol Vis. 2011;17:3156–65. [PMC free article] [PubMed] [Google Scholar]
- 26.Rio DC, Ares M, Jr, Hannon GJ, Nilsen TW. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc. 2010;2010:t5439. doi: 10.1101/pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- 27.Applied Biosystems A. Tech Notes. 2002;9:5. [Google Scholar]
- 28.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 32.Emig D, Salomonis N, Baumbach J, Lengauer T, Conklin BR, Albrecht M. AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res. 2010;38:W755–62. doi: 10.1093/nar/gkq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, Darnell RB. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005;37:844–52. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 35.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc, B. 1995;57:289–300. [Google Scholar]
- 36.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS, King GL. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15:1298–306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, Poulaki V, Semkova I, Kociok N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–28. [PMC free article] [PubMed] [Google Scholar]
- 38.Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–58. doi: 10.1016/j.preteyeres.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–2. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 40.Chen Y, Hu Y, Zhou T, Zhou KK, Mott R, Wu M, Boulton M, Lyons TJ, Gao G, Ma JX. Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am J Pathol. 2009;175:2676–85. doi: 10.2353/ajpath.2009.080945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su F, Overholtzer M, Besser D, Levine AJ. WISP-1 attenuates p53-mediated apoptosis in response to DNA damage through activation of the Akt kinase. Genes Dev. 2002;16:46–57. doi: 10.1101/gad.942902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You XJ, Bryant PJ, Jurnak F, Holcombe RF. Expression of Wnt pathway components frizzled and disheveled in colon cancer arising in patients with inflammatory bowel disease. Oncol Rep. 2007;18:691–4. [PubMed] [Google Scholar]
- 43.Zhou T, Zhou KK, Lee K, Gao G, Lyons TJ, Kowluru R, Ma JX. The role of lipid peroxidation products and oxidative stress in activation of the canonical wingless-type MMTV integration site (WNT) pathway in a rat model of diabetic retinopathy. Diabetologia. 2011;54:459–68. doi: 10.1007/s00125-010-1943-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xi J, Farjo R, Yoshida S, Kern TS, Swaroop A, Andley UP. A comprehensive analysis of the expression of crystallins in mouse retina. Mol Vis. 2003;9:410–9. [PubMed] [Google Scholar]
- 45.Tomarev SI, Piatigorsky J. Lens crystallins of invertebrates–diversity and recruitment from detoxification enzymes and novel proteins. Eur J Biochem. 1996;235:449–65. doi: 10.1111/j.1432-1033.1996.00449.x. [DOI] [PubMed] [Google Scholar]
- 46.Fort PE, Freeman WM, Losiewicz MK, Singh RS, Gardner TW. The retinal proteome in experimental diabetic retinopathy: up-regulation of crystallins and reversal by systemic and periocular insulin. Mol Cell Proteomics. 2009;8:767–79. doi: 10.1074/mcp.M800326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xi J, Farjo R, Yoshida S, Kern TS, Swaroop A, Andley UP. A comprehensive analysis of the expression of crystallins in mouse retina. Mol Vis. 2003;9:410–9. [PubMed] [Google Scholar]
- 48.Piatigorsky J. Crystallin genes: specialization by changes in gene regulation may precede gene duplication. J Struct Funct Genomics. 2003;3:131–7. [PubMed] [Google Scholar]
- 49.Rao NA, Saraswathy S, Wu GS, Katselis GS, Wawrousek EF, Bhat S. Elevated retina-specific expression of the small heat shock protein, alphaA-crystallin, is associated with photoreceptor protection in experimental uveitis. Invest Ophthalmol Vis Sci. 2008;49:1161–71. doi: 10.1167/iovs.07-1259. [DOI] [PubMed] [Google Scholar]
- 50.O'Reilly AM, Currie RW, Clarke DB. HspB1 (Hsp 27) expression and neuroprotection in the retina. Mol Neurobiol. 2010;42:124–32. doi: 10.1007/s12035-010-8143-3. [DOI] [PubMed] [Google Scholar]
- 51.Kumar PA, Kumar MS, Reddy GB. Effect of glycation on alpha-crystallin structure and chaperone-like function. Biochem J. 2007;408:251–8. doi: 10.1042/BJ20070989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puttaiah S, Biswas A, Staniszewska M, Nagaraj RH. Methylglyoxal inhibits glycation-mediated loss in chaperone function and synthesis of pentosidine in alpha-crystallin. Exp Eye Res. 2007;84:914–21. doi: 10.1016/j.exer.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 53.Losiewicz MK, Fort PE. Diabetes Impairs the Neuroprotective Properties of Retinal Alpha-crystallins. Invest Ophthalmol Vis Sci. 2011;52:5034–42. doi: 10.1167/iovs.10-6931. [DOI] [PubMed] [Google Scholar]
- 54.Hirata H, Hinoda Y, Zaman MS, Chen Y, Ueno K, Majid S, Tripsas C, Rubin M, Chen LM, Dahiya R. Function of UDP-glucuronosyltransferase 2B17 (UGT2B17) is involved in endometrial cancer. Carcinogenesis. 2010;31:1620–6. doi: 10.1093/carcin/bgq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84. doi: 10.1007/s00281-008-0111-x. [DOI] [PubMed] [Google Scholar]
- 57.Lin L, Park S, Lakatta EG. RAGE signaling in inflammation and arterial aging. Front Biosci. 2009;14:1403–13. doi: 10.2741/3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim E, Goren A, Ast G. Alternative splicing and disease. RNA Biol. 2008;5:17–9. doi: 10.4161/rna.5.1.5944. [DOI] [PubMed] [Google Scholar]
- 59.Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22:535–46. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 60.Anderson SL, Qiu J, Rubin BY. Tocotrienols induce IKBKAP expression: a possible therapy for familial dysautonomia. Biochem Biophys Res Commun. 2003;306:303–9. doi: 10.1016/s0006-291x(03)00971-9. [DOI] [PubMed] [Google Scholar]
- 61.Kim MH. Protein phosphatase 1 activation and alternative splicing of Bcl-X and Mcl-1 by EGCG + ibuprofen. J Cell Biochem. 2008;104:1491–9. doi: 10.1002/jcb.21725. [DOI] [PubMed] [Google Scholar]
- 62.Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochim Biophys Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]