

Abstract
Reactive oxygen species (ROS), generated endogenously during respiration or exogenously by genotoxic agents, induce oxidized bases and single-strand breaks (SSBs) in DNA that are repaired via the base excision/SSB repair (BER/SSBR) pathway in both the nucleus and mitochondria. Tightly regulated BER/SSBR with multiple sub-pathways is highly complex, and is linked to the replication and transcription. The repair-initiating DNA glycosylases (DGs) or AP-endonuclease (APE1), control the sub-pathway by stably interacting with downstream proteins usually via their common interacting domain (CID). A nonconserved CID with disordered structure usually located at one of the termini, includes the sequences for covalent modifications and/or organelle targeting. While the DGs are individually dispensable, the SSBR-initiating APE1 and polynucleotide kinase 3′ phosphatase (PNKP), are essential. BER/SSBR of mammalian nuclear and mitochondrial genomes share the same early enzymes. Accumulation of oxidative damage in nuclear and mitochondrial genomes has been implicated in aging and various neurological disorders. While defects in BER/SSBR proteins have been linked to hereditary neurodegenerative diseases, our recent studies implicated transition metal-induced inhibition of NEIL family DGs in sporadic diseases. This review focuses on the recent advances in repair of oxidatively damages in mammalian genomes and their linkage to aging and neurological disorders.
Keywords: DNA base excision repair, DNA glycosylases, single-strand break repair, protein-protein and protein-DNA interactions, aging, neurodegenerative disorders, reactive oxygen species
1. Introduction
The genomes of all organisms are constantly exposed to damage whose efficient repair is critical for maintaining genome integrity and cell survival. Oxidative damage represents the major class of insult to aerobic organisms due to continuous generation of reactive oxygen species (ROS) in vivo as by-product of mitochondrial (mt) oxidative phosphorylation and a variety of external insults including environmental toxins, chemotherapeutic drugs, UV light, ionizing radiation and inflammatory response (Lindahl, 1993; Mitra et al., 2001). ROS including O2•- (superoxide radical), OH• (hydroxyl radical) and H2O2 (hydrogen peroxide), generate a plethora of oxidized DNA bases, oxidized abasic (AP) sites, base deamination products, oxidized sugar fragments and DNA strand breaks (SSBs) (Dizdaroglu et al., 2002; Hegde et al., 2008a). It is generally estimated that ~105 DNA lesions are produced in a mammalian cell genome each day from spontaneous decay, replication errors and cellular metabolism alone (Hoeijmakers, 2009) among which ~104 lesions are oxidized bases and single-strand breaks (SSBs) (Lindahl, 1993). This is likely to be a gross underestimate because of continuous repair, thus the actual damage level may be much higher, underscoring the importance of their efficient repair for maintaining genome integrity. Many oxidized lesions and abasic (AP) sites could be transcribed or replicated by replicative or translesion DNA synthesis polymerases and are generally mutagenic in addition to being cytotoxic. For example, C→ 5-OHU lesion causes G-C → A-T transition; G→8-oxoG lesion causes G-C → T-A transversion (Shibutani et al., 1991). Furthermore, SSBs generated directly by reaction of deoxyribose with oxygen radicals or as intermediates during repair of oxidized bases contain various nonligatable termini including 3′ phosphoglycolate, 3′ phosphoglycoladehyde, 3′ phosphate (3′ P) and/or 5′ OH, 5′ phosphosugar derivative such as deoxyribonolactone (Hegde et al., 2008a; Mitra et al., 2001). Persistent oxidative genome damage or their defective or inefficient repair has been etiologically implicated in several human diseases in particular cancer, and inherited and acquired neurological disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease, amyotrophic lateral sclerosis (ALS) and also in aging (Acevedo-Torres et al., 2009; Aguirre et al., 2005; Alam et al., 1997; Bogdanov et al., 2000; Dorszewska et al., 2007; Lyras et al., 1997; Polidori et al., 1999; Yasuhara et al., 2007). In this review we discuss the recent advances in our understanding of oxidized genome damage repair and its role in preventing aging and neurological disorders.
2. Basic mechanism of DNA base excision/single-strand break repair (BER/SSBR)
The evolutionarily conserved BER pathway is responsible for repairing alkylated and oxidized DNA bases, as well as AP sites, while SSBs with various nonligatable termini are repaired via the SSBR pathway. BER was first identified in E. coli, and until a few years ago, believed to be the simplest and most characterized among various DNA repair pathways (Friedberg et al., 2006). However, recent studies suggest BER/SSBR, particularly in higher organisms, to be far more complex, involving a network of repair sub-pathways utilizing various noncanonical proteins. The basic BER reaction comprises four minimal steps: (i) base lesion recognition and excision by a DNA glycosylase (DG), and cleavage of the resulting AP site in a concerted reaction by the DG itself (for bifunctional DG) or by APE1 (for monofunctional DGs), (ii) cleaning of 3′ blocked termini at the strand break by APE1 and/or polynucleotide kinase 3′ phosphatase (PNKP) and 5′ blocking phosphodeoxyribose by DNA polymerase β (Polβ), (iii) gap filling by a DNA polymerase, and (iv) nick sealing by DNA ligases to complete the repair. While alkylated bases and uracil are excised by a monofunctional DG, all oxidized base-specific DGs are bifunctional with an intrinsic AP lyase activity (Mitra et al., 2001). Five oxidized base-specific DGs have been characterized in human cells so far, belonging to two families: Nth and Nei, named after their prototypes in E. coli. The first discovered 8-oxoguanine DNA glycosylase (OGG1), and NTH1 belong to the Nth family (McCullough et al., 1999). More recently discovered (by us and others) NEIL1 and NEIL2 belong to the Nei group (Bandaru et al., 2002; Hazra et al., 2002a; Hazra et al., 2002b; Takao et al., 2002). Recently, a fifth Nei family member NEIL3 was identified but its functions have not been completely characterized (Liu et al., 2010). MYH, a DG and ortholog of E. coli MutY, excises the normal base A from the 8-oxoG•A pair in DNA (Lu and Chang, 1988). Concerted lesion excision and strand cleavage activity of oxidized base specific DGs result in an SSB with two types of blocked 3′terminus: OGG1 and NTH1 carry out AP β lyase reaction and generate 3′ phospho-α,β-unsaturated aldehyde (3′PUA, also referred to as 3′ dRP) and 5′ P groups, while NEIL1 and NEIL2 catalyze βδ elimination to generate 3′ P and 5′ P termini (Wiederhold et al., 2004). In the next step, the 3′ dRP and 3′ P are removed by APE1 and PNKP, respectively. Furthermore, AP sites generated by monofunctional DGs in human cells are primarily processed by APE1 generating strand breaks with 3′OH and 5′ deoxyribosephosphate (5′ dRP) end. In mammalian cells, DNA polymerase β (Polβ) with intrinsic 5′ dRP lyase activity removes the dRP residue to generate ligatable 5′ P terminus (Sobol et al., 2000) and also incorporates the missing nucleotide (nt) at the 3′OH terminus. The resulting nick is sealed by a DNA ligase to complete single nucleotide incorporation (SN)-BER. However, an oxidized 5′ dRP residue is resistant to the lyase activity of Polβ. In this case 2 to 8 upstream nts are displaced by the DNA polymerase generating a 5′ overhang flap that is removed by flap endonuclease 1 (FEN-1), an essential enzyme involved in the removal of Okazaki fragment primers during lagging strand DNA replication. A second flap endonuclease, DNA2, has recently been discovered in both nucleus and mitochondria of human cells that acts in concert with FEN-1 in processing long flap structures (Duxin et al., 2009; Zheng et al., 2008). Gap filling synthesis then produces a longer repair patch. This alternative repair pathway with repair patch of 2 to 8 nts was named long-patch (LP)-BER. Thus, the nature of the 5′ terminus may dictate the choice between SN-vs. LP-BER pathways. BER in the nucleus utilizes two major DNA polymerases, Polβ and replicative Polδ (and possibly Polε); the former normally carries out SN-BER in non-dividing cells, while latter along with other components of DNA replication machinery including the sliding clamp PCNA, clamp loader replication factor-C (RF-C) participate in LP-BER (Dou et al., 2008; Hegde et al., 2008b; Prasad et al., 2000). However, Polβ has been shown to participate in LP-BER in vitro in co-ordination with FEN-1 (Liu et al., 2004). The involvement of DNA replication proteins with LP-BER suggests that LP-BER could be the preferred pathway during DNA replication, irrespective of the 5′ terminal group. The final step of nick sealing during nu BER/SSBR utilizes two DNA ligases: DNA ligase IIIα (LigIIIα) or DNA Ligase 1 (LigI). LigI’s primary role is in DNA replication and LigIII’s in BER, presumably in non-replicating cells where SN-BER is predominant. However LigI could also be involved in LP-BER in replicating cells which utilizes other replication-associated proteins. (Ellenberger and Tomkinson, 2008). Furthermore, LigIII has nu and mt isoforms but only the nu isoform is stabilized by XRCC1, the scaffold for assembling the BER complex (Caldecott et al., 1994), The mt isoform of LigIII, essential for mt DNA replication and repair, is not in a complex with XRCC1 (Lakshmipathy and Campbell, 2000). Recent studies demonstrated that LigIII is essential for mt DNA integrity but dispensable for nuclear DNA repair where LigI takes over LigIII functions in the latter’s absence (Gao et al., 2011; Simsek et al., 2011).
2.1. Diverse end-processing for SSBR
In addition to the common gap-filling and nick sealing in the BER/SSBR pathways, SSBR additionally involves several end-processing enzymes to remove multiple blocked termini generated by ROS (Hegde et al., 2008a). The most common block at ROS induced SSB is 3′ ′phosphoglycolate or 3′ phosphoglycolaldehyde, which is removed by APE1. Tyrosyl DNA phosphodiesterase 1 (TDP1), another 3′ end-processing enzyme, cleaves Top1(Tyr)-crosslinked to 3′ P at the strand break generated by abortive topoisomerase 1 (Top1) reaction. The resulting 3′ P is then removed by PNKP (El-Khamisy et al., 2005; Pouliot et al., 1999; Yang et al., 1996), which also phosphorylates the 5′OH generated at an SSB. A unique type of 5′ blocking groups is formed as intermediates during abortive DNA ligation, namely, adenylate linked via 5′•5′ bond to the 5′P terminus at an SSB. Aprataxin releases 5′AMP to restore the 5′ P terminus (Ahel et al., 2006; Caldecott, 2007). Some blocked 3′ termini could also be removed by ERCC1/XPF nuclease, a functional homologue of the yeast Rad1/Rad10 complex proposed to conduct this role in the budding yeast (Guzder et al., 2004). Thus, end cleaning is perhaps the most diverse enzymatic step in BER/SSBR, due to the variety of termini generated. The various end processing enzymes and their substrate are listed in Table 1.
Table 1.
Major end processing enzymes for BER/SSBR in mammalian cells and their reaction mechanism. Blocking groups are represented in red.
| End-processing enzymes | Substrate | → | Product |
|---|---|---|---|
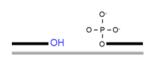
| (i) 5′ end processing enzymes PNKP (5′ kinase) |
→ |  |
|
| Polβ (5′ dRP lyase) |  |
→ |  |
| Aprataxin (5′adenylate•5′P cleavage) |
 |
→ |  |
| (ii) 3′ end processing enzymes APE1 (3′dRP lyase) (also removes 3′phosphoglycoladehyde) |
 |
→ | 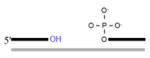 |
| PNKP (3′phosphatase) |  |
→ |  |
| TDP1 (removes Topl(Tyr)-linked 3′ termini to form 3′P which is then removed by PNKP) |
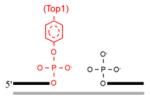 |
→ |  |
 |
|||
3. Diversity of early BER reaction
3.1. Substrate specificity and dispensability of individual DGs
Damage recognition in highly condensed chromatin is a critical step particularly for the oxidized base lesions whose structures may not be significantly different from the parent base and which often retain near normal base pairing properties with only minor perturbations in DNA helix (Hegde et al., 2010a). The mechanism of base excision by a DG involves its extrahelical flipping of the base into the catalytic pocket of the enzyme (Parker et al., 2007; Slupphaug et al., 1996). Thus, DG’s specificity results from the fit of the substrate lesion into the binding pocket where the binding is stabilized by various types of interactions. However, because a number of oxidized bases (>20) are repaired by only four (or five) DGs in the mammalian cells, the DGs usually possess broad, overlapping substrate range. For example, while OGG1’s primary substrate is 8-oxoG, it also excises ring-opened purine, formamidoguanine (FapyG). The NEILs initially shown to excise oxidized pyrimidines, were later observed to efficiently excise purine-derived lesions including 8-oxoG and hydantoins (Krishnamurthy et al., 2008). NTH1 is primarily responsible for the repair of oxidized pyrimidines including 5-OHU, 5-OHC, 5-formylC etc. It is likely that plasticity of DGs’ catalytic pockets allows induced fit of diverse substrates. This could also explain the low turnover, a hallmark of most DGs. Furthermore, the overlapping substrate specificity of DGs is consistent with the general observation that their individual requirement is not essential as deficiency of an individual DG is not lethal. Neither OGG1 or NTH1 null mice nor embryonic fibroblasts derived therefrom has a significant phenotype (Klungland et al., 1999; Minowa et al., 2000; Osterod et al., 2001). Although an increase in cancer incidence was observed in OGG1-null mice after exposure to bromate (Arai et al., 2002), the lack of immediate effect was unexpected, particularly because the lack of downstream BER proteins, such as APE1, XRCC1 or Polβ, induces embryonic lethality (Gu et al., 1994). Recent studies have shown that in spite of broad substrate specificity, DGs have distinct preferential and back-up functions (Hegde et al., 2008a). DGs and their preferred substrates are represented in Table 2.
Table 2.
Mammalian oxidized base-specific DNA glycosylases and their preferred substrates. Oxidative modifications are indicated in red.
| OGG1 | NTH1 | NEIL1 | NEIL2 |
|---|---|---|---|
(i) 8-oxoG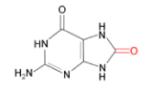
|
(i) Thymine glycol
|
(i) 5-OHU (and 5-OHC)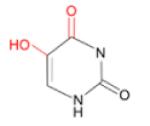
|
(i) Guanidinohydantoin
|
(ii) FapyG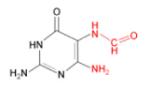
|
(ii) 5-OHU
|
(ii) FapyA (and FapyG)
|
(ii) 5-OHU
|
(iii) 5-OHC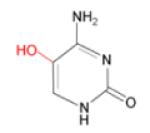
|
(iii) 8-oxoG
|
(iii) Dihydrouracil (DHU)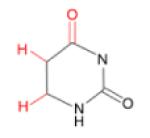
|
|
(iv) 5-formy1U
|
(iv) Thymine glycol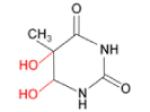
|
3.2. Repair in replicating and non-dividing cells: unique functions of NEILs
While OGG1 and NTH1 are active only with the duplex DNA, we showed that NEILs are more active with single-stranded (ss) DNA structures like those present in a bubble or a fork, than with the duplex DNA, which led us to hypothesize that NEILs preferentially function in the repair during DNA replication or transcription (Dou et al., 2003). We subsequently characterized the role of NEIL2 in preferential repair of transcribed genes, where NEIL2 functionally interacts with RNA polymerase II and hnRNP-U, a component of the transcription machinery (Banerjee et al., 2011). Unlike NEIL2, NEIL1 is induced in S phase cells and functionally interacts with DNA replication proteins including PCNA, replication protein A (RPA) FEN-1 and Werner’s helicase (WRN), based on which we propose that NEIL1 is preferentially involved in replication-coordinated BER (Dou et al., 2008; Hegde et al., 2008b; Theriot et al., 2010). Thus BER involves a network of distinct cell cycle- and genome region-specific repair sub-pathways and utilizes several noncanonical proteins (Banerjee et al., 2011; Das et al., 2007b). While the sequence fidelity of only the transcribed strand in functional genes is critical for maintaining information fidelity in non-replicating, terminally differentiated cells like neurons, integrity of both strands of genes should be essential for maintaining genome integrity in replicating cells. Furthermore, oxidized bases generated due to oxidation of the dNTP pool could be incorporated into DNA and induce mutations. Mutation due to incorporation of A opposite 8-oxoG in the template is prevented by MYH, an 8-oxoG/A specific glycosylase that excises A from the progeny strand (Michaels and Miller, 1992). We proposed that removal of A in the template opposite 8-oxoG in the nascent strand by MYH would be mutagenic, and suggested a distinct repair process involving excision of 8-oxoG from the nascent strand instead (Hazra et al., 1998). Similarly, a U:A mispair generated during replication via incorporation of a U opposite an A, was shown to be repaired post-replicatively by UNG2, the nuclear isoform of uracil-DNA glycosylase (UDG) (Otterlei et al., 1999). Both UNG2 and MYH associate with PCNA at the replication foci, where these glycosylases are recruited to excise U and A in the nascent chain respectively to preserve genomic fidelity (Kavli et al., 2002). Thus for oxidatively damaged bases such as 8-oxoG which are generated in situ or incorporated into DNA from the deoxynucleotide pool, we postulate distinct post-replicative vs. pre-replicative repair pathways in replicating genomes. Coordination between DNA replication and repair ensures that the DNA replication-associated proteins are co-opted during the S-phase to carry out repair synthesis following excision of an oxidized base in the template (Dou et al., 2008; Parlanti et al., 2007).
4. BERosome-mediated repair of oxidized bases: a new paradigm
4.1. Stable interaction of DGs and APE1 with downstream repair proteins for sub-pathway choice
The general view of BER as a series of sequential steps with individual repair enzymes carrying out reactions independently of one another was initially proposed (Izumi et al., 2003). However, recent studies have shown that early BER/SSBR enzymes (e.g., NEIL1 or APE1) stably interact with downstream components including DNA ligase, via their common interacting domain (Das et al., 2007a; Dou et al., 2008; Hegde et al., 2010a; Hegde et al., 2008b; Mitra, 2009; Theriot et al., 2010; Wiederhold et al., 2004). NEIL1 and NEIL2 immunoprecipitates (IP) from human cells contain the SN-BER proteins PNKP, Polβ, LigIIIα and XRCC1, with whom the NEILs also interact in a pairwise fashion. Recently we have also shown NEIL1’s interaction with LP-BER specific DNA replication proteins including PCNA, FEN-1 (Dou et al., 2008; Hegde et al., 2008b). Similarly, APE1 IP contains Polβ, LigIIIα and XRCC1, and more importantly, is proficient in complete repair activity in vitro for repairing tetrahydrofuran (AP site mimic)-containing duplex oligo substrate [(Szczesny et al., 2008); Mantha et al., unpublished data]. This has led us to propose a new paradigm in BER of collaboration of multiple proteins in coordinated fashion involving dynamic protein-protein interactions for enhancing repair efficiency. Recent studies provide evidence for a role of XRCC1 in the organization of BER into distinct multiprotein complexes of different sizes (Hanssen-Bauer et al., 2011), in support of our model. However, the concept of model of large preformed complex(es) raises the issue of stoichiometry and steric interference. Thus detailed mapping of interaction interfaces among these proteins is vital for unraveling the nature of these complexes. We propose that interaction of DGs with the distal repair proteins (like DNA ligases) is involved in sub-pathway selection.
4.2. Common, disordered interaction interface of early BER proteins
Mammalian DGs possess unique structural features compared to their homologs in lower organisms; namely, an unfolded extension or tail which participates in subcellular translocation and more importantly in protein-protein interactions [reviewed in (Hegde et al., 2010a)]. The sequence alignment of human (h) NTH1 with Nths from E. coli, archea and other lower organisms shows that the mammalian NTH1 has an N-terminal extension, absent in E. coli or archea. This N-terminal segment (residues 1-95) contains putative nu and mt localization signals (Sarker et al., 1998). It was also shown that hNTH1 has reduced activity compared to E. coli Nth (Ikeda et al., 1998; Thayer et al., 1995) likely due to the inhibitory role of the N-terminal tail by reducing the rate of product release without affecting base excision or AP lyase activities (Liu and Roy, 2002). The crystal structure of catalytically active deletion construct of hNEIL1 (lacking 56 C-terminal residues) and PONDR (Predictor Of Naturally Disordered Regions in proteins) modeling indicated that hNEIL1 has an unfolded C-terminal extension (~100 residues) which is absent in E. coli Nei (Doublie et al., 2004; Hegde et al., 2010a). We have previously shown that NEIL1 interacts with downstream SN-BER proteins, including Polβ, LigIIIα and XRCC1 via its unfolded C-terminal extension (between residues 289-349) (Wiederhold et al., 2004). We similarly identified nonconserved N-terminal disordered segment (~65 residues) in hAPE1 that is absent in its counterpart in E. coli, involved in protein-protein interactions (Chattopadhyay et al., 2008; Hegde et al., 2010a; Vascotto et al., 2009). The terminal disordered segments in NEIL1 and APE1 and their interacting partners are shown in Fig. 1. It is intriguing how NEIL1 or APE1 could simultaneously bind multiple proteins with high affinity and specificity via a small common interaction interface. Recent studies have indicated that such a phenomenon is not uncommon for mammalian “hub” proteins, whose binding segments invariably have disordered structure (Hegde et al., 2010a). The flexibility of the disordered domain could facilitate interaction with diverse partners via induced fit mechanism.
Fig.1.

Disordered extension in human (and other mammalian) NEIL1 or APE1 that is absent in the E. coli prototype, and is involved in interaction(s) with downstream repair proteins or other noncanonical proteins as indicated.
4.3. Covalent modifications of BER proteins
The functions of DNA repair proteins are often regulated via their posttranslational modifications including phosphorylation by kinases such as ATM, ATR and DNA-PK, acetylation by p300 and CBP, mono- or poly-ADP ribosylation, mono- and poly-ubiquitylation, sumoylation, methylation among others. Such modifications that regulate stability, interactions, intracellular distribution have been identified in many BER/SSBR proteins [reviewed in (Seet et al., 2006)]. Among the DGs, acetylation of NEIL2 and OGG1 have characterized (Bhakat et al., 2004; Bhakat et al., 2006). We showed that about 20% OGG1 in unstressed HeLa cells is acetylated at Lys338 and 341 which enhances OGG1 turnover by decreasing its affinity for the product (Bhakat et al., 2006). Moreover, oxidative stress increases the level of acetylated OGG1, most likely as a result of ROS-induced activation of p300. We have speculated that OGG1 acetylation provides a mechanism for rapid repair response without requiring de novo synthesis of the enzyme when enhanced repair is promptly needed for increased lesion load in cell genomes after exposure to oxidative stress. This is further supported by our observation that the repair of 8-oxoG is correlated with the level of acetylated OGG1. We have also shown acetylation of NEIL2 predominantly at Lys49 and 153 (Bhakat et al., 2004). Moreover, acetylation of Lys49 but not Lys153 strongly inhibits NEIL2 activity. Lys49 in NEIL2 is conserved and its mutation to Arg or other amino acids inactivates the enzyme. We proposed that acetylation at Lys49 acts as a regulatory switch for NEIL2’s activity, while Lys153 acetylation regulates its interactions (Bhakat et al., 2004).
APE1 was shown to be acetylated, phosphorylated as well as ubiquitylated in vitro and in cells (Busso et al., 2009; Izumi et al., 2003; Yacoub et al., 1997). Hsieh et al. provided first in vivo evidence for PKC-mediated phosphorylation of APE1 (Hsieh et al., 2001). APE1 can be phosphorylated in vitro by casein kinase I and II (CK1 and CK2) at various sites including consensus sequences for CKI, CKII and protein kinase C (PKC) (Bhakat et al., 2009; Yacoub et al., 1997). Interestingly, phosphorylation of APE1 by CKII abolished DNA repair activity in vitro, while by CKI or PKC had no such effect (Yacoub et al., 1997). Furthermore, phosphorylation of APE1 by CKII enhances trans-activation of the AP-1 transcription factor with no effect on its DNA repair activity (Fritz and Kaina, 1999). Recently another phosphorylation (Thr232) site in APE1 has been shown to affect its AP enodonuclease activity and has been implicated in PD and AD (Huang et al., 2010). We showed APE1 acetylation at Lys6 and Lys7 by the p300 both in vivo and in vitro (Izumi et al., 2003). The in vitro studies with recombinant WT APE1 or K6R/K7R or K6L/K7L mutants and p300, identified the acetyl-acceptor residues as Lys6 and Lys7 (Izumi et al., 2003), but mass spectroscopic analysis of in vitro acetylated APE1 (AcAPE1) could not detect diacetylated APE1 that could be explained by the possible steric effects of acetyl groups attached to -amino groups during second acetylation (Izumi et al., 2003). Thus, our data indicate that either Lys6 or Lys7 but not both can be acetylated in the same APE1 molecule. It is interesting to note that while Lys6 and Lys7 are conserved in most mammalian APE1 including human, mouse, and bovine, Lys7 is not conserved in rat and Chinese hamster suggesting that acetylation of Lys6 is more critical for APE1’s function. In addition, we showed that APE1 acetylation stimulates the formation of nCaRE-B complex which also contains hnRNP-L and HDAC1 (Izumi et al., 2003). Thus, it appears likely that acetylation-mediated conformational change in the disordered N-terminal segment (~40 residues), which is dispensable for APE1’s endonuclease activity, modulates protein-protein interactions. APE1 stably interacts with Y-box-binding protein 1 (YB-1) and acetylation further enhances its binding with YB-1 both in vivo and in vitro (Chattopadhyay et al., 2006) leading to the activation of multi drug resistance (MDR1) gene. Recently, we have established the molecular basis of APE1’s acetylation-dependent regulatory function in inducing MDR1-mediated drug resistance (Sengupta et al., 2010). The physiological importance of APE1 acetylation became more evident with our findings that the levels of acetylated APE1 is increased in response to variety of cellular stress or changes in intracellular Ca2+ or bacterial infection to gastric epithelial cells (Bhattacharyya et al., 2009; Sengupta et al., 2010). The S-nitrosation of APE1 has been also shown to occur in vivo. Treatment of S-nitroglutathione, an S-nitrosating agent, stimulated nu export of APE1 through S-nitrosation of Cys93 and Cys310 in a CRM1-independnent manner (Caldecott, 2007). Furthermore, a recent study demonstrated APE1’s ubiquitylation, which could act as a signal for APE1’s degradation by maintaining its subcellular localization (Busso et al., 2009). These studies clearly show that posttranslational modifications of key BER/SSBR proteins play an important role in regulation of its specific activity, modulate interactions with other repair proteins and altering their subcellular distribution.
Alteration of APE1’s expression has been implicated in several neurodegenerative diseases including Alzheimer’s (AD) and amyotrophic lateral sclerosis (ALS) (Davydov et al., 2003; Tan et al., 1998) and hyperoxia (100% oxygen) was shown to stimulate APE1 expression in the hippocampus and basal forebrain of young rats, but not in aged rats (Edwards et al., 1998; Quach et al., 2005). However, how altered APE1 levels or whether alteration of its modification is etiologically linked to these diseases is not known. Thus despite evidences for a critical role of posttranslational modifications of BER/SSBR proteins in regulation of their repair functions, and association of these proteins (depletion or functional deficiency) with aging and neurodegenerative diseases, as discussed later in this review, there is inadequate information on the status and role of posttranslational modifications linked to these end points. Future investigations should focus on these issues which we believe would be critical in developing novel therapeutic perspectives.
5. Repair of mitochondrial genome
ATP synthesis is often considered to be the major function of mitochondria, although its role in programmed cell death, lipid metabolism, biogenesis of steroids, calcium homeostasis, are also essential for normal cellular functions (Kakkar and Singh, 2007). Thus, maintaining mt homeostasis as well as the integrity of its genome may be critical for cellular survival. A single mammalian cell contains up to several thousands copies of duplex, circular 16.5 kb mt genome localized within 80-700 mitochondria, depending on the cell type (Miller et al., 2003). The mtDNA encodes 13 essential subunits of the respiratory chain, 22 tRNA and 2 rRNAs, all of which are critical for maintaining oxidative phosphorylation (OXPHOS; Anderson et al., 1981). OXPHOS consumes about 85% of oxygen utilized by the cell and early measurement indicated that ~5% of consumed oxygen escape OXPHOS to form ROS under physiological conditions (Turrens, 2003). Therefore, the mitochondria are considered as the major site for continuous ROS generation (Cadenas and Davies, 2000). Due to close proximity to the site of ROS generation, nonchromatinized state and lack of protective histones in the mt genome, the mutation rate of human mtDNA is 20-100-fold higher than that in the nu DNA (Pesole et al., 1999). Continuous ROS exposure to the mt genome induces multiple types of oxidized bases, AP sites, and SSBs containing 3′ and 5′ blocking groups in mtDNA, which cause mutations and impair mt as well as overall cell functions (Cooke et al., 2003). Although the mt genome is also susceptible to UV damage in contrast to the nu genome, no repair of UV induced pyrimidine photoproducts has been observed in mtDNA from mouse L cells, human KB or HeLa cells. It was proposed that the lack of NER activity in the mitochondria warrants efficient mtDNA replication in order to dilute out pyrimidine photoproduct-containing mtDNA molecules (Clayton et al., 1974). On the other hand, purified human mitochondria were shown to possess mismatch repair activity in vitro (Mason et al., 2003); however, whether MSH2, the critical mismatch repair recognition protein, is present in the mitochondria is not settled (Berneburg et al., 2006; Larsen et al., 2005). YB1 was proposed as a mt counterpart of MSH2 (de Souza-Pinto et al., 2009). Similarly, the mitochondria isolated from Chinese hamster ovary cells were shown to be capable of repairing double strand breaks, and human mt extracts were able to catalyze homologous recombination of different DNA substrates (LeDoux et al., 1992). The presence of critical homologous recombination proteins in human mitochondria were recently reported (Sage et al., 2010). In contrast to the uncertainty of NER, mismatch repair and homologous recombination in the mt genome, the repair of alkylated and oxidized base lesions via the BER pathway was unequivocally established (Croteau and Bohr, 1997; Driggers et al., 1993). The first clear evidence for BER in the mitochondria came from the ability of mt extracts from rat liver to repair uracil (Miller et al., 2003; Stierum et al., 1999b). Complete repair uracil occurred solely via SN-BER and repair synthesis was carried out by DNA polymerase gamma (Polγ; Fig. 2A). Since additional mt BER enzymes were characterized including OGG1 (Nishioka et al., 1999), uracil DNA glycosylase (UDG) (Anderson and Friedberg, 1980), thyminglycol glycosylase (TDG) (Stierum et al., 1999a; Takao et al., 1998), APE1 (Chattopadhyay et al., 2006; Tell et al., 2001), Polγ (Bolden et al., 1977; Hubscher et al., 1979) and DNA ligase III (Lakshmipathy and Campbell, 1999). Recent reports documented the presence of TDP1 and aprataxin in mitochondria underscoring the importance of end-processing activity in mt genome maintenance (Das et al., 2010; Sykora et al., 2011). It should be mentioned that among the 16 known eukaryotic DNA polymerases, Polγ is the only one detected in mammalian mitochondria and should hence be involved in both DNA repair and replication (Graziewicz et al., 2006). Similarly, the sole mt DNA ligase III, a product of alternative translational initiation site of the ligase III gene, is involved in both mt DNA replication and repair (Lakshmipathy and Campbell, 1999).
Fig. 2.

Models of mitochondrial BER pathways. A) Early model of uracil-initiated single-nucleotide BER proposed by (Stierum et al., 1999b). B) Repair of uracil via multi-nucleotide incorporation in LP-BER (Akbari et al., 2008). C) Multi-nucleotide BER of 2-deoxyribonolactone (dL) (Liu et al., 2008a; Zheng et al., 2008). D) Repair of tetrahydrofuran (THF) via multi-nucleotide BER [(Szczesny et al., 2008); Tann et al unpublished data].
Recently, we (Szczesny et al., 2008) and others (Akbari et al., 2008; Liu et al., 2008b) independently reported the presence of LP-BER activity in mt extracts from mouse and human cell lines. While the broad conclusions were similar, these studies did not agree on the identity of the mt 5′ exonuclease involved in 5′end processing. Our group (Szczesny et al., 2008) and Akbari et al., (Akbari et al., 2008) concluded that mt LP-BER does not require FEN-1 involved in nu LP-BER. Repair of uracil by the mt extract was shown to involve incorporation of several-nucleotides in concert with the 5′ flap processing but the involved enzyme was not identified (Akbari et al., 2008; Fig. 2B). We detected the 5′ exo/endonuclease activity which generated very short DNA fragments in mammalian mitochondria, distinct from the product of FEN-1 activity (Szczesny et al., 2008). Moreover, our results support the model of mt LP-BER where polγ incorporates nucleotides in vitro both upstream and downstream to the damage possibly due to robust 3′ exonuclease (proofreading) activity of Polγ (Longley et al., 1998). We speculated that due to proofreading activity of Polγ, repair of all oxidative lesions in mtDNA occurs via incorporation of multiple nucleotides (Szczesny et al., 2008; Fig. 2D). In contrast, Liu et al., have shown that repair of 2-deoxyribonolactone (dL), a common AP site oxidation product, was dependent on FEN-1 activity although the presence of an additional flap-removing enzyme was also suggested (Liu et al., 2008b). Subsequently DNA2, a helicase/nuclease, which plays a versatile role in processing of nu DNA intermediates in yeast genome during replication and repair, was shown to be present in the human mitochondria, and was proposed to process 5′ flap intermediates in DNA synergistically with FEN-1 during mt replication and repair (Zheng et al., 2008; Fig. 2C). Initially, unlike yeast DNA2 that is present both in nucleus and mitochondria, human DNA2 was detected only in mitochondria but subsequent studies shown its nu localization as well (Duxin et al., 2009). HDNA2 depletion led to a modest decrease in mt replication intermediates and inefficient repair of mt genome damage but surprisingly resulted in the appearance of aneuploid cells together with formation of internuclear chromatin bridges, indicating human DNA2’s important role in maintaining stability of the nu genome (Duxin et al., 2009). Thus, establishing the precise role of hDNA2 in maintaining integrity of nu and mt genomes needs further investigation. In addition, another mt-specific 5′ exo/endonuclease, EXOG, was identified as a paralog of Endonuclease G, with nuclease activity towards ssDNA; however its role in the maintenance of mt genome and possible role in mt BER was not investigated until recently (Cymerman et al., 2008; Kieper et al., 2010; Fig. 2D). Interestingly, EXOG generates mainly a di-nucleotide excision product, similar to what we had observed in repair with mt extract (Szczesny et al., 2008). We recently demonstrated that EXOG, which is unique to the mitochondria unlike FEN1 or DNA2, has been implicated in the removal of the 5′-blocking residue and is required for repairing endogenous SSBs in the mt genome (Tann et al., 2011). Further, EXOG depletion induces persistent SSBs in the mtDNA, enhances ROS levels, and causes apoptosis in normal cells but not in mt genome-deficient (rho0) cells, suggesting that persistent SSBs in the mt genome alone could provide the initial trigger for apoptotic signaling in mammalian cells.
We should stress here that a big challenge in identification of new mt proteins including DNA repair proteins is to provide unequivocal evidence for their mt localization when it is extremely difficult to purify mitochondria completely devoid of nu, cytosolic or endoplasmic reticulum (ER) contamination. There is growing evidence that DNA repair proteins are not only present in the nucleus and mitochondria but also in cytoplasm and ER (Szczesny and Mitra, 2005).
6. BER/SSBR defects in central nervous system (CNS) in aging and neurodegenerative diseases
The CNS includes the brain and spinal cord consisting of multiple cell types including neurons and glial cells (astrocytes, oligodendrocytes and microglia). A distinct feature of neurons is that replication-associated DNA repair cannot occur in these terminally differentiated, postmitotic cells (Hanawalt, 2008). Thus, the major challenge for neuronal function is genome repair associated with transcription (Nouspikel and Hanawalt, 2002). The genomes of neurons are particularly susceptible to oxidative damage because of the abundance of ROS generated due to high O2 consumption and relatively weak anti-oxidant defense system (Liu et al., 2002).
Because the brain is generally protected from environmental genotoxins by the blood-brain barrier, endogenous ROS-induced genome damage is the most critical threat to neuronal cells (Chen et al., 2002; Liu et al., 1996). BER/SSBR activities have been characterized in the brain cells (Chen et al., 2002; Englander et al., 2002; Liu et al., 1996). Among the DGs, OGG1 expression decreased during rat brain development (transition from non-differentiated to differentiated neurons), while NEIL1 and NEIL2 levels increased (Wilson and McNeill, 2007). This is consistent with high transcriptional activity in the brain (Lu et al., 2004) and likely involvement of the NEILs in transcription-associated repair (Banerjee et al., 2011; Dou et al., 2003; Dou et al., 2008; Hegde et al., 2008a; Hegde et al., 2008b). Among other BER proteins, NTH1 showed comparable expression and activity in brain, testes, liver and kidney, but OGG1 activity was low in the brain relative to other tissues. PNKP and APE1 are generally highly expressed in the brain. Polβ is ubiquitously expressed in different brain regions, and appears to be the major DNA polymerase in neurons (Wilson, 1998) whose activity decreases with age (Rao et al., 2001). Further, DNA replication/repair proteins FEN-1, Polδ/ε, PCNA, and Lig I levels are low in adult neurons, while XRCC1 and Lig IIIα are highly expressed in the brain compared to other tissue types (Wilson and McNeill, 2007). These studies suggest that the neurons are deficient in LP-BER activity with SN-BER being the predominant mode of repair. However, a comparison among tissues/cell types for protein expression may not provide insight into the repair capacity per se, and the published studies only confirm the presence of BER/SSBR proteins in the neurons. Furthermore, the lack of a detailed picture of genome repair in brain could be attributed to the fact that human brain genome damage/repair can only be studied in postmortem samples and difficulty in examining these processes in individual cell type-specific manner, which would be critical because of likely differences in damage tolerance in these cell types.
6.1. Aging
The oxidative stress theory of aging proposes that accumulation of oxidative DNA damage over the life span of an organism leads to gradual decline of cellular functions and eventual death (Bohr, 2002). This model is supported by several circumstantial evidences including the observation that lower free radical production and/or antioxidant treatment protects against age-related deterioration, and cognitive decline (Lemon et al., 2003). Further, deficit or decrease in the repair of oxidative DNA damage appears to correlate with premature aging and age-related diseases (Bohr et al., 2007). It appears likely that overall genome repair, specifically the balance between DNA damage and its repair is a major determinant of the longevity and cell viability. A specific defect in processing 5′dRP residue at the strand break in Sir2 (SIRT6 homolog)-deficient mice displayed age-related degenerative phenotype (Mostoslavsky et al., 2006). The activities of DGs OGG1, NTH1 and uracil DNA glycosylase (UNG) in brain mitochondria decrease significantly with age (Gredilla et al., 2010).
6.2. Inherited neurological disorders
Some 200 neurological disorders with diverse etiologies have been reported in humans, many of which have been linked to inherited defect in various DNA repair pathways. Mutations or altered expression of BER [e.g., OGG1, XRCC1; (Coppede et al., 2010a; Dogru-Abbasoglu et al., 2007; Qian et al., 2010)], SSBR [e.g., TDP1, aprataxin, PNKP; (Ahel et al., 2006; El-Khamisy et al., 2007; Shen et al., 2010)] and DSBR (e.g., ATM, NBS1) proteins have been observed in humans predisposed to various hereditary neurodegenerative diseases [see review and references therein (Caldecott, 2008)]. Deficiencies in OGG1 or APE1, combined with exposure to oxidative stress could lead to neurodegenerative symptoms. XRCC1 was linked to autosomal recessive spinocerebellar ataxias with various neurological symptoms (Paulson and Miller, 2005; Taroni and DiDonato, 2004). Further, defects in end-processing proteins, aprataxin and TDP1, have been shown to be associated with ataxia with oculomotor apraxia 1 and spinocerebellar ataxia with axonal neuropathy, respectively (El-Khamisy et al., 2007; Hirano et al., 2007; Rass et al., 2007). A recent study linked mutations in PNKP to autosomal recessive disease characterized by severe neurological abnormalities including microcephaly, early-onset, intractable seizures and developmental delay [denoted MCSZ; (Shen et al., 2010)]. While compelling evidences link SSBR deficiency to neurodegenerative disorders (Katyal and McKinnon, 2008), similar association with early BER proteins (DGs, APE1) has not been established. A common OGG1 Ser326Cys polymorphic variant with reduced activity and linked to increased cancer risk was weakly associated with HD and ALS risk (Coppede et al., 2007a), but not with AD or PD (Coppede et al., 2010a; Coppede et al., 2007b; Coppede and Migliore, 2009). Asp148Glu polymorphism in APE1 with lower repair activity was associated with increased ALS risk (Hayward et al., 1999). Association between the XRCC1 Arg399Gln polymorphism and ALS risk was also reported (Coppede et al., 2010b). Taken together, these studies suggest association of BER/SSBR gene SNP variants with predisposition to neurodegenerative diseases.
6.3 Reduced BER/SSBR activity in sporadic neurodegenerative diseases - inhibition of repair by transition metals
Decreased capacity to repair oxidized DNA damage was observed in brain cell extracts in sporadic neurodegenerative diseases which constitute >80 % of disease incidences, with unknown etiology (Weissman et al., 2007; Wilson and McNeill, 2007). Significant BER deficiency was observed in brains of AD patients which is due to limited DNA base damage excision by DGs and reduced repair synthesis by Polβ (Weissman et al., 2007). Similarly, decreased mt Polγ activity was observed in neurons affected with neurodegenerative diseases (Adlam et al., 2005). Reduction of total BER capacity in sporadic diseases has not been correlated with the expression of BER enzymes in brain tissues which suggested involvement of additional mechanisms in BER defects.
Excessive accumulation of transition metals, particularly Cu and Fe, have been implicated in AD, PD and also other human neurodegenerative diseases like Huntington’s disease and Wilson’s disease (Dawson et al., 1993; Hegde et al., 2004; Rao, 1999). The toxicity issue of metals which are essential micronutrients for normal physiological processes is very complex because these metals are typically sequestered in vivo in catalytically inactive form by metal storage proteins such as ferritin, transferrin, or cerruloplasmin (Dorszewska et al., 2007). However, in PD brains, increased iron is often accompanied with decreased ferritin synthesis, resulting in free iron overload (Alam et al., 1997). Furthermore, redox-cycling iron and copper could generate ROS which oxidize cellular components including DNA. We have recently shown that Fe/Cu significantly inhibit the activity of NEIL1 and NEIL2, but not OGG1 at physiological levels both in neuronal cells and in vitro (Hegde et al., 2010b; Hegde et al., 2011). The metals affected both base excision and AP lyase activities of NEILs and inhibited NEIL1’s interaction with downstream repair proteins like Polβ, and FEN-1, further inhibiting the overall repair. Fe(II) and Cu(II) stably bind to the NEILs with high affinity, affecting their secondary structure. The lack of OGG1 inhibition under similar conditions suggested binding specificity and eliminates the metal ions’ direct binding to DNA. These results showed for first time that Fe/Cu overload associated with neurodegenerative diseases could act as a double-edged sword by both increasing oxidative genome damage and inhibiting its repair. Previously, divalent metal forms of Fe(II) and Cu(II) were found to be elevated specifically in early phase of neurodegenerative disorders and Fe(III) accumulates in the later phase presumably due to dyshomeostasis of metals (Hegde et al., 2009; Hegde et al., 2011; Rao, 1999). Thus our results showing that the NEIL-initiated BER is preferentially compromised by Fe(II) and Cu(II) raises the possibility that a similar situation could occur in the early phase of neurodegenerative disorders. Many other BER/SSBR proteins are also inhibited by transition metals. Whiteside et al., showed that Cd and Cu inhibit both phosphatase and kinase activities of PNKP with human cell extracts and recombinant protein (Whiteside et al., 2010). Heavy metals could delay SSB rejoining in mammalian cells (De Boeck et al., 1998; Lynn et al., 1997). It was shown earlier that APE1 endonuclease activities are inhibited by heavy metals lead, iron, and cadmium (McNeill et al., 2004). Another study revealed that elevated Fe levels caused reduction in FEN-1 and Lig III activities due to interference of repair protein binding to their DNA substrates (Li et al., 2009). Thus metal mediated genotoxicity could involve induction of oxidative genome damage via free radical generation, direct DNA binding; and inhibition of DNA repair via inhibition of key BER/SSBR enzymes, and affecting protein-protein interactions among the repair proteins. Thus reduced or defective oxidized genome damage repair due to inactivating mutations in the repair enzymes, as well as their inhibition by transition metals leading to an imbalance in genome damages and their repair has been proposed to play a vital role in the pathogenesis of neurodegenerative diseases.
Interestingly, metal chelators (e.g., EDTA, desferrioxamine) could reverse Fe(II) mediated inhibition of NEILs but a reducing agent (TCEP or DTT) was required for the reversal of Cu-mediated inhibition (Hegde et al., 2010b; Hegde et al., 2011). Based on these observations, we proposed distinct mechanisms for Fe and Cu mediated inhibition: Cu inhibition involves oxidation of cysteine residues in NEIL1. Further, curcumin, a natural spice component with both metal chelation and reducing activities, was able to reverse the metal induced inhibition of NEILs both in vitro and in neuronal cells (Hegde et al., 2010b).
7. Conclusions
Repair of oxidative genome damage that include base lesions, AP sites and their oxidation products (Greenberg et al., 2004), and SSBs via BER/SSBR pathway both in the nucleus, and mitochondria is critical for maintaining genomic integrity and cell survival particularly in aerobic organisms that are continuously exposed to endogenous ROS. Multiple BER/SSBR pathways have evolved in mammalian cells to ensure efficient repair of a variety of base lesions and SSBs with multiple, ROS-induced termini. These repair sub-pathways have preferential as well as back up functions, thus providing versatility particularly at the first step of repair. The importance of BER/SSBR in prevention of neurodegenerative diseases was initially questioned by the lack of linkage between accumulation of oxidized bases, AP sites and SSBs and human diseases. Recently, defective BER/SSBR machinery is being increasingly linked to these diseases. An overall reduction in DNA repair activity has been associated with aging in human brain suggesting it to be a normal aspect of the aging process. Furthermore, our recent studies showing specific inhibition of NEIL-initiated BER by transition metals suggested that similar scenario could prevail in neurodegenerative diseases. Future studies should focus on the interplay of BER/SSBER components with specific pathologies associated with these diseases and how repair of neurons’ genomes in specific brain regions are affected in various CNS disorders.
Highlights.
This review focuses on the broad repair mechanisms for oxidative damage in mammalian nuclear and mitochondrial genomes.
Multiple repair sub-pathways involving distinct repair complexes.
Role of disordered terminal extensions and posttranslational modifications in the early BER proteins are summarized.
The impact of BER/SSBR deficiency in central nervous system (CNS) on aging and neurodegenerative diseases is discussed.
Acknowledgements
The research in the authors’ laboratory is supported by USPHS grants, R01 CA81063, R01 CA53791, P01 CA92586, P01 AG10514, P30 ES06676 (SM), R01 CA102271 (TKH) and American Parkinson’s Disease Association’s postdoctoral fellowship (MLH). Because of the limited focus of the article, many appropriate references could not be included, for which the authors apologize. We thank Drs. KSJ Rao (INDICASAT-AIP, Panama) and Istvan Boldogh (Department of Microbiology and Immunology, UTMB) for helpful discussions.
Abbreviations
- DG
DNA glycosylase
- AP
abasic
- APE1
AP endonuclease 1
- PNKP
polynucleotide 5′kinase/3′phosphatase
- ROS
reactive oxygen species
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- Pol
DNA polymerase
- Lig
DNA ligase
- FEN-1
flap endonuclease 1
- BER
base excision repair
- SSB
single-strand break
- SSBR
SSB repair
- mt
mitochondrial
- nu
nuclear
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acevedo-Torres K, Berrios L, Rosario N, Dufault V, Skatchkov S, Eaton MJ, Torres-Ramos CA, Ayala-Torres S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair (Amst) 2009;8:126–36. doi: 10.1016/j.dnarep.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005;19:1088–95. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- Aguirre N, Beal MF, Matson WR, Bogdanov MB. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic Res. 2005;39:383–8. doi: 10.1080/10715760400027979. [DOI] [PubMed] [Google Scholar]
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–6. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- Akbari M, Visnes T, Krokan HE, Otterlei M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amst) 2008;7:605–16. doi: 10.1016/j.dnarep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- Anderson CT, Friedberg EC. The presence of nuclear and mitochondrial uracil-DNA glycosylase in extracts of human KB cells. Nucleic Acids Res. 1980;8:875–88. [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Arai T, Kelly VP, Minowa O, Noda T, Nishimura S. High accumulation of oxidative DNA damage, 8-hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis. 2002;23:2005–10. doi: 10.1093/carcin/23.12.2005. [DOI] [PubMed] [Google Scholar]
- Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002;1:517–29. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, Boldogh I, Sarkar PS, Mitra S, Hazra TK. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J Biol Chem. 2011 doi: 10.1074/jbc.M110.198796. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berneburg M, Kamenisch Y, Krutmann J, Rocken M. ‘To repair or not to repair - no longer a question’: repair of mitochondrial DNA shielding against age and cancer. Exp Dermatol. 2006;15:1005–15. doi: 10.1111/j.1600-0625.2006.00508.x. [DOI] [PubMed] [Google Scholar]
- Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–9. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009;11:621–38. doi: 10.1089/ars.2008.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol. 2006;26:1654–65. doi: 10.1128/MCB.26.5.1654-1665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Chattopadhyay R, Burnette BR, Cross JV, Mitra S, Ernst PB, Bhakat KK, Crowe SE. Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology. 2009;136:2258–69. doi: 10.1053/j.gastro.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Brown RH, Matson W, Smart R, Hayden D, O’Donnell H, Beal M. Flint, Cudkowicz M. Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med. 2000;29:652–8. doi: 10.1016/s0891-5849(00)00349-x. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–12. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- Bohr VA, Ottersen OP, Tonjum T. Genome instability and DNA repair in brain, ageing and neurological disease. Neuroscience. 2007;145:1183–6. doi: 10.1016/j.neuroscience.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Bolden A, Noy GP, Weissbach A. DNA polymerase of mitochondria is a gamma-polymerase. J Biol Chem. 1977;252:3351–6. [PubMed] [Google Scholar]
- Busso CS, Iwakuma T, Izumi T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene. 2009;28:1616–25. doi: 10.1038/onc.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–30. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Caldecott KW. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Repair (Amst) 2007;6:443–53. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–31. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay R, Das S, Maiti AK, Boldogh I, Xie J, Hazra TK, Kohno K, Mitra S, Bhakat KK. Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol Cell Biol. 2008;28:7066–80. doi: 10.1128/MCB.00244-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–76. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Cao G, Hastings T, Feng Y, Pei W, O’Horo C, Chen J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J Neurochem. 2002;81:1273–84. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- Clayton DA, Doda JN, Friedberg EC. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc Natl Acad Sci U S A. 1974;71:2777–81. doi: 10.1073/pnas.71.7.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. Faseb J. 2003;17:1195–214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- Coppede F, Ceravolo R, Migheli F, Fanucchi F, Frosini D, Siciliano G, Bonuccelli U, Migliore L. The hOGG1 Ser326Cys polymorphism is not associated with sporadic Parkinson’s disease. Neurosci Lett. 2010a;473:248–51. doi: 10.1016/j.neulet.2010.02.059. [DOI] [PubMed] [Google Scholar]
- Coppede F, Mancuso M, Gerfo A. Lo, Carlesi C, Piazza S, Rocchi A, Petrozzi L, Nesti C, Micheli D, Bacci A, Migliore L, Murri L, Siciliano G. Association of the hOGG1 Ser326Cys polymorphism with sporadic amyotrophic lateral sclerosis. Neurosci Lett. 2007a;420:163–8. doi: 10.1016/j.neulet.2007.04.067. [DOI] [PubMed] [Google Scholar]
- Coppede F, Mancuso M, Gerfo A. Lo, Manca ML, Petrozzi L, Migliore L, Siciliano G, Murri L. A Ser326Cys polymorphism in the DNA repair gene hOGG1 is not associated with sporadic Alzheimer’s disease. Neurosci Lett. 2007b;414:282–5. doi: 10.1016/j.neulet.2006.12.035. [DOI] [PubMed] [Google Scholar]
- Coppede F, Migheli F, Gerfo A. Lo, Fabbrizi MR, Carlesi C, Mancuso M, Corti S, Mezzina N, del Bo R, Comi GP, Siciliano G, Migliore L. Association study between XRCC1 gene polymorphisms and sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010b;11:122–4. doi: 10.3109/17482960903220297. [DOI] [PubMed] [Google Scholar]
- Coppede F, Migliore L. DNA damage and repair in Alzheimer’s disease. Curr Alzheimer Res. 2009;6:36–47. doi: 10.2174/156720509787313970. [DOI] [PubMed] [Google Scholar]
- Croteau DL, Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem. 1997;272:25409–12. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- Cymerman IA, Chung I, Beckmann BM, Bujnicki JM, Meiss G. EXOG, a novel paralog of Endonuclease G in higher eukaryotes. Nucleic Acids Res. 2008;36:1369–79. doi: 10.1093/nar/gkm1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Boldogh I, Lee JW, Harrigan JA, Hegde ML, Piotrowski J, de Souza Pinto N, Ramos W, Greenberg MM, Hazra TK, Mitra S, Bohr VA. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J Biol Chem. 2007a;282:26591–602. doi: 10.1074/jbc.M703343200. [DOI] [PubMed] [Google Scholar]
- Das BB, Dexheimer TS, Maddali K, Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc Natl Acad Sci U S A. 2010;107:19790–5. doi: 10.1073/pnas.1009814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Chattopadhyay R, Bhakat KK, Boldogh I, Kohno K, Prasad R, Wilson SH, Hazra TK. Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J Biol Chem. 2007b;282:28474–84. doi: 10.1074/jbc.M704672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov V, Hansen LA, Shackelford DA. Is DNA repair compromised in Alzheimer’s disease? Neurobiol Aging. 2003;24:953–68. doi: 10.1016/s0197-4580(02)00229-4. [DOI] [PubMed] [Google Scholar]
- Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264:C961–7. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- De Boeck M, Lison D, Kirsch-Volders M. Evaluation of the in vitro direct and indirect genotoxic effects of cobalt compounds using the alkaline comet assay. Influence of interdonor and interexperimental variability. Carcinogenesis. 1998;19:2021–9. doi: 10.1093/carcin/19.11.2021. [DOI] [PubMed] [Google Scholar]
- de Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D, Lebel M, Stevnsner TV, Rasmussen LJ, Bohr VA. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst) 2009;8:704–19. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radic Biol Med. 2002;32:1102–15. doi: 10.1016/s0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- Dogru-Abbasoglu S, Aykac-Toker G, Hanagasi HA, Gurvit H, Emre M, Uysal M. The Arg194Trp polymorphism in DNA repair gene XRCC1 and the risk for sporadic late-onset Alzheimer’s disease. Neurol Sci. 2007;28:31–4. doi: 10.1007/s10072-007-0744-x. [DOI] [PubMed] [Google Scholar]
- Dorszewska J, Florczak J, Rozycka A, Kempisty B, Jaroszewska-Kolecka J, Chojnacka K, Trzeciak WH, Kozubski W. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer’s and Parkinson’s diseases. Acta Neurobiol Exp (Wars) 2007;67:113–29. doi: 10.55782/ane-2007-1639. [DOI] [PubMed] [Google Scholar]
- Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–84. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- Dou H, Theriot CA, Das A, Hegde ML, Matsumoto Y, Boldogh I, Hazra TK, Bhakat KK, Mitra S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J Biol Chem. 2008;283:3130–40. doi: 10.1074/jbc.M709186200. [DOI] [PubMed] [Google Scholar]
- Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci U S A. 2004;101:10284–9. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers WJ, LeDoux SP, Wilson GL. Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. J Biol Chem. 1993;268:22042–5. [PubMed] [Google Scholar]
- Duxin JP, Dao B, Martinsson P, Rajala N, Guittat L, Campbell JL, Spelbrink JN, Stewart SA. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–82. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, Rassin DK, Izumi T, Mitra S, Perez-Polo JR. APE/Ref-1 responses to oxidative stress in aged rats. Journal of neuroscience research. 1998;54:635–8. doi: 10.1002/(SICI)1097-4547(19981201)54:5<635::AID-JNR8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Hartsuiker E, Caldecott KW. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst) 2007;6:1485–95. doi: 10.1016/j.dnarep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–13. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–38. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander EW, Hu Z, Sharma A, Lee HM, Wu ZH, Greeley GH. Rat MYH, a glycosylase for repair of oxidatively damaged DNA, has brain-specific isoforms that localize to neuronal mitochondria. J Neurochem. 2002;83:1471–80. doi: 10.1046/j.1471-4159.2002.01259.x. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2nd Edition ASM Press; Washington: 2006. [Google Scholar]
- Fritz G, Kaina B. Phosphorylation of the DNA repair protein APE/REF-1 by CKII affects redox regulation of AP-1. Oncogene. 1999;18:1033–40. doi: 10.1038/sj.onc.1202394. [DOI] [PubMed] [Google Scholar]
- Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, McKinnon PJ. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. 2011;471:240–4. doi: 10.1038/nature09773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- Gredilla R, Garm C, Holm R, Bohr VA, Stevnsner T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol Aging. 2010;31:993–1002. doi: 10.1016/j.neurobiolaging.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MM, Weledji YN, Kim J, Bales BC. Repair of oxidized abasic sites by exonuclease III, endonuclease IV, and endonuclease III. Biochemistry. 2004;43:8178–83. doi: 10.1021/bi0496236. [DOI] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–6. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- Guzder SN, Torres-Ramos C, Johnson RE, Haracska L, Prakash L, Prakash S. Requirement of yeast Rad1-Rad10 nuclease for the removal of 3′-blocked termini from DNA strand breaks induced by reactive oxygen species. Genes Dev. 2004;18:2283–91. doi: 10.1101/gad.1232804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC. Emerging links between premature ageing and defective DNA repair. Mech Ageing Dev. 2008;129:503–5. doi: 10.1016/j.mad.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen-Bauer A, Solvang-Garten K, Sundheim O, Pena-Diaz J, Andersen S, Slupphaug G, Krokan HE, Wilson DM, 3rd, Akbari M, Otterlei M. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ Mol Mutagen. 2011;52:623–35. doi: 10.1002/em.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward C, Colville S, Swingler RJ, Brock DJ. Molecular genetic analysis of the APEX nuclease gene in amyotrophic lateral sclerosis. Neurology. 1999;52:1899–901. doi: 10.1212/wnl.52.9.1899. [DOI] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002a;99:3523–8. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Maidt L, Floyd RA, Mitra S. The presence of two distinct 8-oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucleic Acids Res. 1998;26:5116–22. doi: 10.1093/nar/26.22.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002b;277:30417–20. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, Srinivas P, Sambamurti K, Rao KJ, Scancar J, Messori L, Zecca L, Zatta P. Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis. 2009;17:457–68. doi: 10.3233/JAD-2009-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008a;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Hazra TK, Mitra S. Functions of disordered regions in mammalian early base excision repair proteins. Cell Mol Life Sci. 2010a;67:3573–87. doi: 10.1007/s00018-010-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Hegde PM, Holthauzen LM, Hazra TK, Rao KS, Mitra S. Specific Inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: potential etiological linkage to neurodegenerative diseases. J Biol Chem. 2010b;285:28812–25. doi: 10.1074/jbc.M110.126664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Hegde PM, Rao K.S. Jagannatha, Mitra S. Oxidative Genome Damage and its Repair in Neurodegenerative Diseases: Function of Transition Metals as a Double-Edged Sword. J Alzheimers Dis. 2011 doi: 10.3233/JAD-2011-110281. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Shanmugavelu P, Vengamma B, Rao TS, Menon RB, Rao RV, Rao KS. Serum trace element levels and the complexity of inter-element relations in patients with Parkinson’s disease. J Trace Elem Med Biol. 2004;18:163–71. doi: 10.1016/j.jtemb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Hegde ML, Theriot CA, Das A, Hegde PM, Guo Z, Gary RK, Hazra TK, Shen B, Mitra S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J Biol Chem. 2008b;283:27028–37. doi: 10.1074/jbc.M802712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, Northrop JL, Salih MA, Inoue K, Armstrong DL, Champoux JJ, Takashima H, Boerkoel CF. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? Embo J. 2007;26:4732–43. doi: 10.1038/sj.emboj.7601885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Hsieh MM, Hegde V, Kelley MR, Deutsch WA. Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic Acids Res. 2001;29:3116–22. doi: 10.1093/nar/29.14.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E, Qu D, Zhang Y, Venderova K, Haque ME, Rousseaux MW, Slack RS, Woulfe JM, Park DS. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat Cell Biol. 2010;12:563–71. doi: 10.1038/ncb2058. [DOI] [PubMed] [Google Scholar]
- Hubscher U, Kuenzle CC, Spadari S. Functional roles of DNA polymerases beta and gamma. Proc Natl Acad Sci U S A. 1979;76:2316–20. doi: 10.1073/pnas.76.5.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Biswas T, Roy R, Izumi T, Boldogh I, Kurosky A, Sarker AH, Seki S, Mitra S. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J Biol Chem. 1998;273:21585–93. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- Izumi T, Wiederhold LR, Roy G, Roy R, Jaiswal A, Bhakat KK, Mitra S, Hazra TK. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193:43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- Kakkar P, Singh BK. Mitochondria: a hub of redox activities and cellular distress control. Mol Cell Biochem. 2007;305:235–53. doi: 10.1007/s11010-007-9520-8. [DOI] [PubMed] [Google Scholar]
- Katyal S, McKinnon PJ. DNA strand breaks, neurodegeneration and aging in the brain. Mech Ageing Dev. 2008;129:483–91. doi: 10.1016/j.mad.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:39926–36. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- Kieper J, Lauber C, Gimadutdinow O, Urbanska A, Cymerman I, Ghosh M, Szczesny B, Meiss G. Production and characterization of recombinant protein preparations of Endonuclease G-homologs from yeast, C. elegans and humans. Protein Expr Purif. 2010;73:99–106. doi: 10.1016/j.pep.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci U S A. 1999;96:13300–5. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy N, Zhao X, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–46. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmipathy U, Campbell C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol Cell Biol. 1999;19:3869–76. doi: 10.1128/mcb.19.5.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmipathy U, Campbell C. Mitochondrial DNA ligase III function is independent of Xrcc1. Nucleic Acids Res. 2000;28:3880–6. doi: 10.1093/nar/28.20.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- LeDoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis. 1992;13:1967–73. doi: 10.1093/carcin/13.11.1967. [DOI] [PubMed] [Google Scholar]
- Lemon JA, Boreham DR, Rollo CD. A dietary supplement abolishes age-related cognitive decline in transgenic mice expressing elevated free radical processes. Experimental biology and medicine. 2003;228:800–10. doi: 10.1177/15353702-0322807-05. [DOI] [PubMed] [Google Scholar]
- Li H, Swiercz R, Englander EW. Elevated metals compromise repair of oxidative DNA damage via the base excision repair pathway: implications of pathologic iron overload in the brain on integrity of neuronal DNA. J Neurochem. 2009;110:1774–83. doi: 10.1111/j.1471-4159.2009.06271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha - lipoic acid. Proc Natl Acad Sci U S A. 2002;99:2356–61. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:4925–30. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Qian L, Sung JS, de Souza-Pinto NC, Zheng L, Bogenhagen DF, Bohr VA, Wilson DM, 3rd, Shen B, Demple B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol Cell Biol. 2008a;28:4975–87. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Qian L, Sung JS, de Souza-Pinto NC, Zheng L, Bogenhagen DF, Bohr VA, Wilson DM, 3rd, Shen B, Demple B. Removal of Oxidative DNA Damage via FEN1-Dependent Long-Patch Base Excision Repair in Human Cell Mitochondria. Mol Cell Biol. 2008b doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair, and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J Neurosci. 1996;16:6795–806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Roy R. Truncation of amino-terminal tail stimulates activity of human endonuclease III (hNTH1) J Mol Biol. 2002;321:265–76. doi: 10.1016/s0022-2836(02)00623-x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- Longley MJ, Ropp PA, Lim SE, Copeland WC. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry (Mosc) 1998;37:10529–39. doi: 10.1021/bi980772w. [DOI] [PubMed] [Google Scholar]
- Lu AL, Chang DY. Repair of single base-pair transversion mismatches of Escherichia coli in vitro: correction of certain A/G mismatches is independent of dam methylation and host mutHLS gene functions. Genetics. 1988;118:593–600. doi: 10.1093/genetics/118.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–91. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Lynn S, Lai HT, Kao SM, Lai J, Jan KY. Cadmium inhibits DNA strand break rejoining in methyl methanesulfonate-treated CHO-K1 cells. Toxicol Appl Pharmacol. 1997;144:171–6. doi: 10.1006/taap.1997.8116. [DOI] [PubMed] [Google Scholar]
- Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J Neurochem. 1997;68:2061–9. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- Mason PA, Matheson EC, Hall AG, Lightowlers RN. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003;31:1052–8. doi: 10.1093/nar/gkg167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu Rev Biochem. 1999;68:255–85. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- McNeill DR, Narayana A, Wong HK, Wilson DM., 3rd Inhibition of Ape1 nuclease activity by lead, iron, and cadmium. Environ Health Perspect. 2004;112:799–804. doi: 10.1289/ehp.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J Bacteriol. 1992;174:6321–5. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31:e61. doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa O, Arai T, Hirano M, Monden Y, Nakai S, Fukuda M, Itoh M, Takano H, Hippou Y, Aburatani H, Masumura K, Nohmi T, Nishimura S, Noda T. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc Natl Acad Sci U S A. 2000;97:4156–61. doi: 10.1073/pnas.050404497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Boldogh I, Izumi T, Hazra TK. Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ Mol Mutagen. 2001;38:180–90. doi: 10.1002/em.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Hegde ML, Theriot CA, Das A, Hegde PM, Hazra TK. Complexity in repair of oxidative genome damage and its regulation. Proceedings of Princess Takamatsu Symposium; Tokyo, Japan. 2009. [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–29. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, Nakabeppu Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell. 1999;10:1637–52. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC. DNA repair in terminally differentiated cells. DNA Repair (Amst) 2002;1:59–75. doi: 10.1016/s1568-7864(01)00005-2. [DOI] [PubMed] [Google Scholar]
- Osterod M, Hollenbach S, Hengstler JG, Barnes DE, Lindahl T, Epe B. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis. 2001;22:1459–63. doi: 10.1093/carcin/22.9.1459. [DOI] [PubMed] [Google Scholar]
- Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE. Post-replicative base excision repair in replication foci. Embo J. 1999;18:3834–44. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–7. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlanti E, Locatelli G, Maga G, Dogliotti E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007;35:1569–77. doi: 10.1093/nar/gkl1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL, Miller VM. Breaks in coordination: DNA repair in inherited ataxia. Neuron. 2005;46:845–8. doi: 10.1016/j.neuron.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Pesole G, Gissi C, De Chirico A, Saccone C. Nucleotide substitution rate of mammalian mitochondrial genomes. J Mol Evol. 1999;48:427–34. doi: 10.1007/pl00006487. [DOI] [PubMed] [Google Scholar]
- Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci Lett. 1999;272:53–6. doi: 10.1016/s0304-3940(99)00578-9. [DOI] [PubMed] [Google Scholar]
- Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–5. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000;275:4460–6. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- Qian Y, Chen W, Wu J, Tao T, Bi L, Xu W, Qi H, Wang Y, Guo L. Association of polymorphism of DNA repair gene XRCC1 with sporadic late-onset Alzheimer’s disease and age of onset in elderly Han Chinese. J Neurol Sci. 2010;295:62–5. doi: 10.1016/j.jns.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Quach N, Chan T, Lu TA, Schreiber SS, Tan Z. Induction of DNA repair proteins, Ref-1 and XRCC1, in adult rat brain following kainic acid-induced seizures. Brain Res. 2005;1042:236–40. doi: 10.1016/j.brainres.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Rao KS, Annapurna VV, Raji NS. DNA polymerase-beta may be the main player for defective DNA repair in aging rat neurons. Annals of the New York Academy of Sciences. 2001;928:113–20. doi: 10.1111/j.1749-6632.2001.tb05641.x. [DOI] [PubMed] [Google Scholar]
- Rao KSJ, Rao RV, Shanmugavelu P, Menon RB. Trace elements in Alzheimer’s disease brain: A new hypothesis. Alz Rep. 1999;2:241–246. [Google Scholar]
- Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- Sage JM, Gildemeister OS, Knight KL. Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J Biol Chem. 2010;285:18984–90. doi: 10.1074/jbc.M109.099846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker AH, Ikeda S, Nakano H, Terato H, Ide H, Imai K, Akiyama K, Tsutsui K, Bo Z, Kubo K, Yamamoto K, Yasui A, Yoshida MC, Seki S. Cloning and characterization of a mouse homologue (mNthl1) of Escherichia coli endonuclease III. J Mol Biol. 1998;282:761–74. doi: 10.1006/jmbi.1998.2042. [DOI] [PubMed] [Google Scholar]
- Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol. 2006;7:473–83. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Mantha AK, Mitra S, Bhakat KK. Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene. 2010;30:482–93. doi: 10.1038/onc.2010.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, Bodell A, Barry B, Gleason D, Allen K, Ganesh VS, Chang BS, Grix A, Hill RS, Topcu M, Caldecott KW, Barkovich AJ, Walsh CA. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet. 2010;42:245–9. doi: 10.1038/ng.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–4. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- Simsek D, Furda A, Gao Y, Artus J, Brunet E, Hadjantonakis AK, Van Houten B, Shuman S, McKinnon PJ, Jasin M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. 2011;471:245–8. doi: 10.1038/nature09794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–10. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- Stierum RH, Croteau DL, Bohr VA. Purification and characterization of a mitochondrial thymine glycol endonuclease from rat liver. J Biol Chem. 1999a;274:7128–36. doi: 10.1074/jbc.274.11.7128. [DOI] [PubMed] [Google Scholar]
- Stierum RH, Dianov GL, Bohr VA. Single-nucleotide patch base excision repair of uracil in DNA by mitochondrial protein extracts. Nucleic Acids Res. 1999b;27:3712–9. doi: 10.1093/nar/27.18.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora P, Croteau DL, Bohr VA, Wilson DM., 3rd Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:7437–42. doi: 10.1073/pnas.1100084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Mitra S. Effect of aging on intracellular distribution of abasic (AP) endonuclease 1 in the mouse liver. Mech Ageing Dev. 2005;126:1071–8. doi: 10.1016/j.mad.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Szczesny B, Tann AW, Longley MJ, Copeland WC, Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J Biol Chem. 2008;283:26349–56. doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao M, Aburatani H, Kobayashi K, Yasui A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998;26:2917–22. doi: 10.1093/nar/26.12.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao M, Kanno S, Shiromoto T, Hasegawa R, Ide H, Ikeda S, Sarker AH, Seki S, Xing JZ, Le XC, Weinfeld M, Kobayashi K, Miyazaki J, Muijtjens M, Hoeijmakers JH, van der Horst G, Yasui A. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. Embo J. 2002;21:3486–93. doi: 10.1093/emboj/cdf350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Sun N, Schreiber SS. Immunohistochemical localization of redox factor-1 (Ref-1) in Alzheimer’s hippocampus. Neuroreport. 1998;9:2749–52. doi: 10.1097/00001756-199808240-00012. [DOI] [PubMed] [Google Scholar]
- Tann AW, Boldogh I, Meiss G, Qian W, Van Houten B, Mitra S, Szczesny B. Apoptosis Induced by Persistent Single-strand Breaks in Mitochondrial Genome: CRITICAL ROLE OF EXOG (5′-EXO/ENDONUCLEASE) IN THEIR REPAIR. J Biol Chem. 2011;286:31975–83. doi: 10.1074/jbc.M110.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taroni F, DiDonato S. Pathways to motor incoordination: the inherited ataxias. Nature reviews. Neuroscience. 2004;5:641–55. doi: 10.1038/nrn1474. [DOI] [PubMed] [Google Scholar]
- Tell G, Crivellato E, Pines A, Paron I, Pucillo C, Manzini G, Bandiera A, Kelley MR, Di Loreto C, Damante G. Mitochondrial localization of APE/Ref-1 in thyroid cells. Mutat Res. 2001;485:143–52. doi: 10.1016/s0921-8777(00)00068-9. [DOI] [PubMed] [Google Scholar]
- Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. Embo J. 1995;14:4108–20. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CA, Hegde ML, Hazra TK, Mitra S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair (Amst) 2010;9:643–52. doi: 10.1016/j.dnarep.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A, Radicella JP, Kelley MR, D’Ambrosio C, Scaloni A, Quadrifoglio F, Tell G. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol. 2009;29:1834–54. doi: 10.1128/MCB.01337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–55. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside JR, Box CL, McMillan TJ, Allinson SL. Cadmium and copper inhibit both DNA repair activities of polynucleotide kinase. DNA Repair (Amst) 2010;9:83–9. doi: 10.1016/j.dnarep.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–20. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Wilson DM, 3rd, McNeill DR. Base excision repair and the central nervous system. Neuroscience. 2007;145:1187–200. doi: 10.1016/j.neuroscience.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Wilson SH. Mammalian base excision repair and DNA polymerase beta. Mutat Res. 1998;407:203–15. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- Yacoub A, Kelley MR, Deutsch WA. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res. 1997;57:5457–9. [PubMed] [Google Scholar]
- Yang SW, Burgin AB, Jr., Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci U S A. 1996;93:11534–9. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara T, Hara K, Sethi KD, Morgan JC, Borlongan CV. Increased 8-OHdG levels in the urine, serum, and substantia nigra of hemiparkinsonian rats. Brain Res. 2007;1133:49–52. doi: 10.1016/j.brainres.2006.11.072. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zhou M, Guo Z, Lu H, Qian L, Dai H, Qiu J, Yakubovskaya E, Bogenhagen DF, Demple B, Shen B. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–36. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]