

Abstract
This article reviews the pathogenic mechanism of non-steroidal anti-inflammatory drug (NSAID)-induced gastric damage, focusing on the relation between cyclooxygenase (COX) inhibition and various functional events. NSAIDs, such as indomethacin, at a dose that inhibits prostaglandin (PG) production, enhance gastric motility, resulting in an increase in mucosal permeability, neutrophil infiltration and oxyradical production, and eventually producing gastric lesions. These lesions are prevented by pretreatment with PGE2 and antisecretory drugs, and also via an atropine-sensitive mechanism, not related to antisecretory action. Although neither rofecoxib (a selective COX-2 inhibitor) nor SC-560 (a selective COX-1 inhibitor) alone damages the stomach, the combined administration of these drugs provokes gastric lesions. SC-560, but not rofecoxib, decreases prostaglandin E2 (PGE2) production and causes gastric hypermotility and an increase in mucosal permeability. COX-2 mRNA is expressed in the stomach after administration of indomethacin and SC-560 but not rofecoxib. The up-regulation of indomethacin-induced COX-2 expression is prevented by atropine at a dose that inhibits gastric hypermotility. In addition, selective COX-2 inhibitors have deleterious influences on the stomach when COX-2 is overexpressed under various conditions, including adrenalectomy, arthritis, and Helicobacter pylori-infection. In summary, gastric hypermotility plays a primary role in the pathogenesis of NSAID-induced gastric damage, and the response, causally related with PG deficiency due to COX-1 inhibition, occurs prior to other pathogenic events such as increased mucosal permeability; and the ulcerogenic properties of NSAIDs require the inhibition of both COX-1 and COX-2, the inhibition of COX-1 upregulates COX-2 expression in association with gastric hypermotility, and PGs produced by COX-2 counteract the deleterious effect of COX-1 inhibition.
Keywords: Non-steroidal anti-inflammatory drug, Gastric damage, Pathogenesis, Gastric motility, Neutrophil
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs) are frequently used to treat inflammatory pain. A major limitation to their use, however, is the adverse reaction they cause to the gastrointestinal (GI) tract, including the formation of gastric lesions, the potentiation of ulcerogenic responses to stress, and the impairment of gastric ulcer healing[1-4]. Concerning the mechanism of NSAID-induced gastric damage, prostaglandin (PG) deficiency is of prime importance to the gastric ulcerogenic response to NSAIDs, yet it has proven to be more complicated than expected and involves multiple, closely interacting elements, including hypermotility, neutrophils, free radicals, and so on[1,5-13].
The PG deficiency caused by NSAIDs is due to the inhibition of cyclooxygenase (COX). COX exists in two isozymes, COX-1 and COX-2; the former is constitutively expressed in various tissues, including the stomach, while the latter appears to be expressed in most tissues in response to growth factors and cytokines[14,15]. This tissue specificity of the COX isozymes has led to the idea that COX-1 is critical for housekeeping actions in the GI mucosa, whereas COX-2 functions under pathological conditions such as inflammation. Indeed, it has been reported that the gastric ulcerogenic properties of NSAIDs are due to the inhibition of COX-1, but not COX-2[16]. However, studies using selective COX-1 and COX-2 inhibitors demonstrated that the GI ulcerogenic effects of NSAIDs are not accounted for solely by inhibition of COX-1, but require inhibition of COX-2 as well[17-21]. It has also been shown that inhibition of COX-1 upregulated COX-2 expression in the GI mucosa, and PGs produced by COX-2 may help maintain the mucosal integrity when there is a deficiency of PGs due to COX-1 inhibition[18,20,21]. This idea was supported by the finding that the selective COX-2 inhibitor by itself damaged the gastric mucosa when the expression of COX-2 was upregulated in the stomach of rats subjected to adrenalectomy (glucocorticoid deficiency) or induction of adjuvant arthritis or Helicobacter pylori (H. pylori) infection[22-24].
In this article, we reviewed the pathogenesis of NSAID-induced gastric damage, mainly based on our own publications, including the roles of functional events, particularly, gastric hypermotility, as well as the influences of arthritis and H. pylori infection, and discussed the relation between COX-1 or COX-2 inhibition and pathogenic elements such as gastric motility and neutrophil infiltration.
GENERAL ASPECTS OF NSAID-INDUCED GASTRIC DAMAGE
Relation to PG deficiency
There is no doubt that a deficiency of endogenous PG is a background factor in NSAID-induced gastric ulceration. Indeed, when various NSAIDs, such as indomethacin (30 mg/kg), flurbiprofen (20 mg/kg), naproxen (40 mg/kg), dicrofenac (40 mg/kg) and aspirin (200 mg/kg), were administered to rats subcutaneously, all of these agents, except aspirin, produced damage in the stomach at doses that significantly decreased the mucosal prostaglandin E2 (PGE2) concentration[18,19] (Figure 1). Characteristically, the damage was observed along the long axis of the stomach and consisted mostly of hemorrhagic lesions, with a few non-hemorrhagic lesions. Interestingly, aspirin given subcutaneously did not produce any damage, despite inhibiting PG production as effectively as other NSAIDs[18,25]. Notwithstanding, it is assumed that PG deficiency is causally related to the gastric ulcerogenic action of NSAIDs, but this factor alone is not sufficient for the development of gastric lesions. The reason why parenterally administered aspirin does not cause gastric damage will be discussed in another section of this article.
Figure 1.

Gastric ulcerogenic responses (A) and changes in mucosal prostaglandin E2 content (B) induced by various non-steroidal anti-inflammatory drugs in rat stomach. The animals were given indomethacin (30 mg/kg), aspirin (200 mg/kg), naproxen (40 mg/kg), flurbiprofen (20 mg/kg) and diclofenac (40 mg/kg) s.c., and killed 4 h later. Data are presented as the mean ± SE in 5 rats. aP < 0.05 vs control (data from ref. 18 after modification).
Effect of various drugs
The development of gastric lesions in response to indomethacin was inhibited by prior administration of PGE2. These lesions were also prevented by antisecretory drugs such as cimetidine, omeprazole and atropine[6,26,27], confirming the importance of luminal acid in the pathogenesis of these lesions. Of interest, since atropine was effective even when 150 mmol of HCl was applied to the lumen, it is assumed that this protective action is not associated with the antisecretory effect and initiated by factors other than inhibition of acid secretion (Figure 2). Neither cimetidine nor omeprazole was effective against indomethacin-induced gastric damage in the presence of exogenous acid. In addition, anti-neutrophil antiserum also reduced the severity of these lesions, but much less effectively than other agents[28]. Pretreatment with both atropine and dmPGE2 significantly inhibited the development of gastric lesions at all time points during a 4 h-test period following administration of indomethacin. By contrast, the anti-neutrophil antiserum did not affect the onset but significantly reduced the severity of lesions at 4 h after indomethacin treatment. It is assumed that the gastric ulcerogenic response to indomethacin is prevented by supplementation with PGE2 and inhibition of acid secretion as well as an atropine-sensitive mechanism, not related to the antisecretory action. Neutrophils do not play a role in the onset of these lesions but may be involved in the later extension of the damage. Suematsu et al[29] recently reported that the severity of gastric lesions produced by indomethacin was worse in mice lacking heat shock factor 1 (HSF1), a transcription factor for HSP genes, than in control mice, while these lesions were ameliorated in transgenic mice expressing HSP70. They suggested that expression of HSP70 ameliorates indomethacin-induced gastric damage by affecting mucosal apoptosis, probably via the activation of Bax.
Figure 2.

Effects of atropine, cimetidine and omeprazole on gastric lesions produced by indomethacin in rats. The animals were given indomethacin (30 mg/kg, s.c.) and killed 4 h later. Omeprazole (30 mg/kg), cimetidine (100 mg/kg) and atropine (3 mg/kg, s.c.) were given 1 h before indomethacin. In some cases, the animals were given 1 mL of 100 mmol HCl p.o. immediately after the administration of indomethacin. Data are presented as the mean ± SE for 5-8 rats. aP < 0.05 vs vehicle (data from refs. 6, 26 and 27 after modification).
Functional alterations involved in pathogenesis
Gastric hypermotility: Mersereau et al[7] first emphasized the importance of stomach hypermotility and mucosal foldings in the genesis of gastric lesions in response to phenylbutazone. As expected, all NSAIDs, except aspirin, increased gastric motility at ulcerogenic doses, leading to the development of gastric lesions[19] (Figure 3). Gastric hypermotility causes microvascular disturbances, especially at specific sites on mucosal foldings, leading to various events including neutrophil-endothelial interaction. Garrick et al[30] reported that high-amplitude contractions during cold-restraint stress resulted in a temporal restriction of mucosal blood flow and lowered the mucosal resistance to injury. The gastric damage induced by indomethacin occurred linearly along the long axis of the stomach, and microscopically, was seen at the top or the bottom of mucosal foldings, the sites most influenced by mucosal compression due to contraction of the stomach, where mucosal blood flow is restricted, leading to microvascular disturbances (Figure 4A and B). The inhibition of gastric motility may lead to a flattening of the mucosal foldings and a decrease in microvascular disturbances, resulting in prevention of the fold-related band-like lesions, as observed after the administration of indomethacin[6,10,26,31]. A role for muscle elements in the pathogenic mechanism of gastric ulceration has been demonstrated[6,10,31-33]. Yamaguchi et al[32]monitored gastric mucosal hemodynamics and motility simultaneously and found oscillatory changes in the hemodynamics during gastric hypermotility induced by water-immersion stress. We also found that indomethacin caused oscillatory changes in mucosal blood flow associated with hypermotility of the stomach, and such blood flow changes were prevented when the hypermotility was inhibited by atropine[10] (Figure 4C). It is assumed that indomethacin induces the sequential events in the early stage of lesion formation in the stomach during hypermotility; the microcirculatory disturbances due to abnormal compression of the gastric wall, followed by increased vascular permeability, leading to cellular damage[10,33]. Anyway, the indomethacin-induced gastric hypermotility was inhibited by both atropine and PGE2 but not by either omeprazole or the anti-neutrophil antiserum[6,10]. Since atropine prevented indomethacin-induced gastric damage, even in the presence of exogenous acid[26], the inhibitory effect on gastric hypermotility may account for the protective action of this agent. In addition, indomethacin caused oxyradical production and lipid peroxidation in the gastric mucosa, probably resulting from the ischemic-reperfusion changes due to rhythmic hypercontraction of the stomach[10]. Certainly, these changes were prevented by atropine, again confirming an importance of gastric hypermotility. At present, the exact mechanism by which NSAIDs cause gastric hypermotility remains unknown. However, it is assumed that indomethacin-induced gastric hypermotility is mediated by a vagal-cholinergic mechanism, involving a glycoprivic response[6,31].
Figure 3.

Representative recordings showing the effects of various non-steroidal anti-inflammatory drug on gastric motility in rats. Indomethacin (35 g/kg), aspirin (200 mg/kg), naproxen (40 mg/kg), flurbiprofen (20 mg/kg) or diclofenac (40 mg/kg) was given s.c. after basal motility had stabilized (data from ref.18 after modification).
Figure 4.

Macro- and microscopical observations of gastric lesions induced by indomethacin in rats (A and B) and simultaneous recordings of gastric motility and mucosal blood flow in the rat before and after administration of indomethacin (C). A, B: The animals were given indomethacin (25 mg/kg, s.c.), and the stomachs were excised 4 h later. Note that the lesions were located, in most cases, on the upper part of the mucosal folds (arrow) and in some cases at the base of the folds (arrows); C: Indomethacin (25 mg/kg, s.c.) was given, while atropine (1 mg/kg, s.c.) was given 1 h after indomethacin treatment. Note that during hypermotility states the mucosal blood flow repeated a decrease and an increase, respectively, corresponding to contraction and relaxation of the stomach wall (data from refs. 8 and 33 after modification). GMBF: Gastric mucosal blood flow.
Neutrophils: Neutrophils have been implicated in the damage associated with NSAIDs[35]. These cells are recruited to a site of injury by chemotaxins and participate in amplifying the inflammatory response. Many studies including ours have shown that indomethacin-induced gastric damage could be prevented by an anti-neutrophil antiserum or monoclonal antibody against the CD18 adhesion molecule[13,34,35]. However, there have been few studies showing the less importance of neutrophils in NSAID-induced gastric damage[36,37]. Trevethick et al[36] reported that neutrophil infiltration does not contribute to the ulcerogenic effects of indomethacin in the rat gastric mucosa. Similarly, Melange et al[37] showed that neutropenia does not prevent indomethacin-induced gastrointestinal damage in rats. Santucci et al[38] even showed that granulocyte colony stimulating factor, though it markedly increased myeloperoxidase (MPO) activity, significantly prevented gastric lesions from forming, suggesting no relationship between MPO activity and the ulcerogenic response to indomethacin. A study by Morise et al[34] also showed that indomethacin provoked the development of gastric lesions even in CD18, intercellular adhesion molecule 1, or P-selectin-deficient mice, the degree of severity being about 70% of that in wild-type mice. We reported that the anti-neutrophil antiserum caused a significant inhibition of indomethacin- induced gastric damage, yet the degree of inhibition was much less than that shown by atropine or dmPGE2[28] (Figure 5). Furthermore, it was shown that the anti-neutrophil antiserum did not prevent the onset of damage until 3 h after indomethacin treatment and significantly reduced the severity of damage 4 h later. These results suggest that the neutralization of neutrophils itself is not sufficient to prevent the onset of damage but reduces the overall expression of gastric lesions in response to indomethacin. Anthony et al[32] examined the sequence of histological changes in the rat stomach after indomethacin treatment and identified an early phase of injury that involves mucosal contraction and vascular fibrin deposition but does not involve neutrophil infiltration. Thus, the neutrophil infiltration may be secondary to the events associated with gastric hypermotility, and not a primary event preceding the onset of gastric damage. Indeed, the increase in MPO activity as well as formation of lesions induced by indomethacin was prevented when the enhanced gastric motility was inhibited by atropine[28].
Figure 5.

Time-course of changes in gastric lesions following administration of indomethacin (30 mg/kg, s.c.) in rats, with or without pretreatment. Atropine (1 mg/kg) was given s.c. 30 min before indomethacin, while dmPGE2 (10 μg/kg) or anti-neutrophil antiserum (ANS, 0.2 mL/rat) was given i.v. 10 min and 1 h, respectively, before indomethacin. Data are presented as the mean ± SE in 4-6 rats. aP < 0.05 vs control group given normal serum (data from ref. 28 after modification).
PROSTAGLANDIN E RECEPTOR SUBTYPE INVOLVED IN PGE2-INDUCED PROTECTION
Although exogenous PGs, especially PGE2, prevent NSAID-induced gastric damage, how they do so remains unknown. We examined the effect of various prostanoids, subtype-specific prostaglandin E (EP) agonists, on the development of gastric lesions in response to indomethacin and determined which functional alteration is most closely associated with this action[39]. Such an approach would be helpful to understanding of which event(s) may be critically important to the pathogenic mechanism of NSAID-induced gastric damage.
Gastric ulcerogenic response
PGE2 exhibited a potent inhibitory effect on indomethacin-induced gastric damage. This effect was mimicked by other prostanoids such as 17-phenyl PGE2 (EP1 agonist) and sulprostone (EP1/EP3 agonist)[39] (Figure 6). Neither butaprost (EP2 agonist), ONO-NT-012 (EP3 agonist), nor 11-deoxy PGE1 (EP3/EP4 agonist), was effective in reducing the severity of these lesions, indicating that the activation of the EP2, EP3, and EP4 receptors does not provide gastric protection against indomethacin[39,40]. These results strongly suggest that the protective effect of PGE2 against indomethacin- induced gastric damage is brought about by activation of the EP1 receptor. This idea is supported by the finding that the protective action of PGE2 against indomethacin was totally mitigated by prior administration of ONO-AE-829, a selective EP1 receptor antagonist. In addition, indomethacin caused gastric damage similarly in both wild-type and knockout mice lacking EP1 or EP3 receptors, yet the protective action of PGE2 was observed in wild-type and EP3-receptor knockout mice but not in mice lacking EP1 receptors. Given the above findings, it is assumed that PGE2 prevents indomethacin-induced gastric ulceration through the activation of EP1 receptors.
Figure 6.
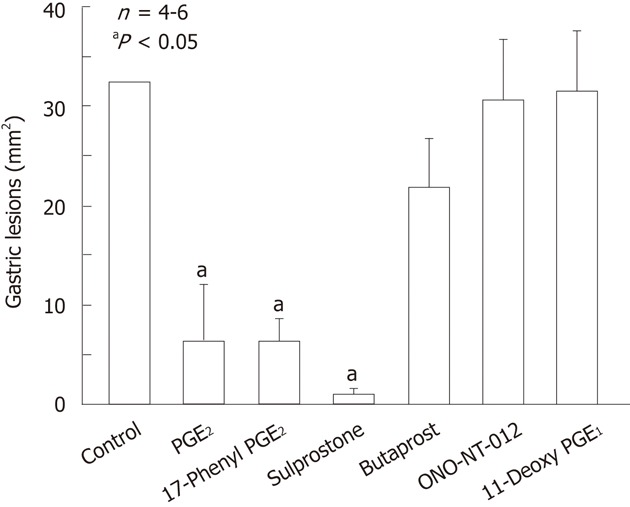
Effects of various prostaglandin E agonists on gastric lesions generated by indomethacin in rats. The animals were given indomethacin (30 mg/kg) s.c. and killed 4 h later. Prostaglandin E2 (PGE2, 0.3 mg/kg), 17-phenyl PGE2 (0.3 mg/kg; EP1 agonist), sulprostone (0.3 mg/kg; EP1/EP3 agonist), butaprost (10 mg/kg; EP2 agonist), ONO-NT-012 (10 mg/kg; EP3 agonist) and 11-deoxy PGE1 (3 mg/kg; EP3/EP4 agonist) were given i.v. 10 min before indomethacin. Data are presented as the mean ± SE in 4-6 rats. aP < 0.05 vs control (data from ref. 39 after modification).
Gastric functional alterations
The prostanoids exhibiting a preference for the EP1 receptors inhibited gastric hypermotility and damage in response to indomethacin (Figure 7). These effects were antagonized by ONO-AE-829, an EP1 antagonist, strongly suggesting that the antigastric motility effect of PGE is paralleled by a reduction in gross mucosal injury of the stomach with the use of indomethacin. Both butaprost and ONO-NT-012 reportedly increased gastric mucosal blood flow[41], yet these drugs did not provide any protection against indomethacin-induced gastric damage, suggesting that the protective action is not functionally associated with the increased mucosal blood flow. Certainly, since inhibition of gastric motility may lead to an attenuation of microvascular disturbances due to contraction of the stomach, prostanoids acting through EP1 receptors may help maintain mucosal blood flow after the administration of indomethacin. It is assumed that the actions of PGE2 to prevent indomethacin-induced gastric damage are functionally associated with the inhibition of gastric hypermotility. The mechanism by which PGE2 inhibits gastric motility through EP1 receptors remains unknown. Milenov et al[42] reported that PGE2 relaxed the circular muscle but contracted the longitudinal muscle of the canine stomach. Narumiya and his group reported the distribution of mRNA of the EP receptors along the gastrointestinal tract[43,44]. They found that strong signals for EP1 transcripts occurred in the smooth muscle cells in the muscularis mucosa throughout the tract. Since EP1 receptors are coupled to phosphatidyl inositol turnover[45], it is assumed that contraction of longitudinal smooth muscle by PGE2 is associated with an increase of cytosolic calcium. Contraction of circular smooth muscle leads to the appearance of mucosal folds, which have been implicated in the pathogenesis of ulcers including indomethacin-generated gastric lesions[6-8,10,13]. At present, the mechanism by which PGE2 relaxes circular smooth muscle through activation of EP1 receptors still remains unclear.
Figure 7.
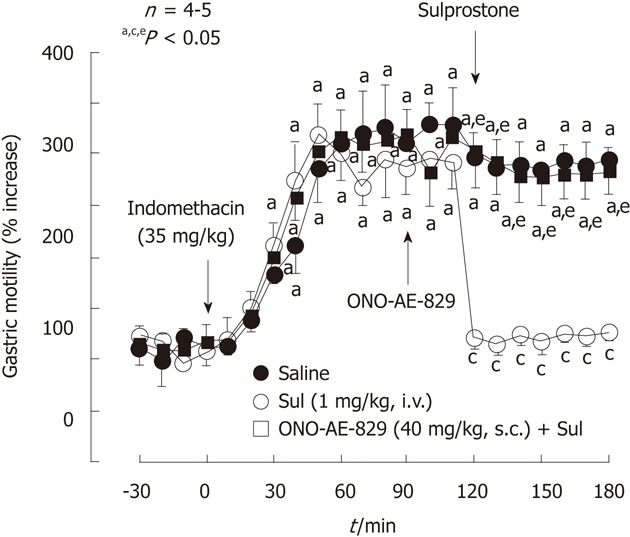
Effect of sulprostone on the increased gastric motility caused by indomethacin in rats. Indomethacin (35 mg/kg) was given s.c.. Sulprostone (Sul, 1 mg/kg) was given i.v. as a single injection 2 h after indomethacin, while ONO-AE-829 (40 mg/kg) was given s.c. 30 min before the administration of sulprostone. Data are presented as the mean ± SE of values determined every 10 min in 4-5 rats. Significant difference at P < 0.05; afrom basal values in the corresponding group; cfrom saline group; efrom indomethacin plus sulprostone (data from ref. 39 after modification).
It is known that PGE2 has an inhibitory effect on neutrophil functions, including chemotaxis[46]. We confirmed that PGE2 exhibited an inhibitory effect on the migration of neutrophils caused by formyl-methionyl-leucyl-phenylalanine in vitro[39]. The same inhibitory action was shown by both butaprost and 11-deoxy PGE1, but not by 17-phenyl PGE2, sulprostone, or ONO-NT-012, clearly indicating that the anti-neutrophil chemotaxis action of PGE2 is mediated by activation of EP2 and EP4 receptors (Figure 8). Thus, it is assumed that the inhibition of neutrophil migration by itself is not sufficient to reduce the overall expression of gastric lesions in response to indomethacin. Since the increase in MPO activity as well as ulceration induced by indomethacin was prevented when the enhanced gastric motility was inhibited by atropine[13,28,47], it is likely that the neutrophil infiltration is secondary to the event associated with gastric hypermotility following indomethacin treatment. As mentioned before, Melange et al[37] even showed that NSAID-induced gastric injury is neutrophil-independent in the neutropenic rats. These results strongly suggest that the protective effect of PGE2 is functionally associated with the inhibition of gastric motility, but not neutrophil infiltration.
Figure 8.

Effects of atropine and various prostaglandin E agonists on the neutrophil chemotaxis stimulated by formyl-methionyl-leucyl-phenylalanine. Neutrophils were pretreated for 45 min with atropine and various prostaglandin E agonists such as PGE2, 17-phenyl PGE2, butaprost, ONO-NT-012 and 11-deoxy PGE1 at the indicated concentrations, and then the cells were stimulated by incubation with formyl-methionyl-leucyl-phenylalanine (fMLP, 1x10-7 mol) for another 45 min. Data are expressed as a percentage of the stimulated values (control) observed in the presence of fMLP and represent the mean ± SE from 4 experiments. Significant difference at P < 0.05; afrom normal; cfrom control (data from ref. 39 after modification).
ROLE OF COX INHIBITION IN NSAID-INDUCED GASTRIC DAMAGE
Ulcerogenic properties of various COX inhibitors
COX, the enzyme responsible for PG production, exists in two isozymes, the constitutively expressed COX-1 and the inducible COX-2[14,15]. NSAIDs inhibit the activity of both COX-1 and COX-2, yet it is believed that the inhibition of COX-1 is critical for their ulcerogenic properties in the stomach. However, Wallace et al[17] reported that inhibition of both COX-1 and COX-2 is required for the induction of gastric lesions. This finding was confirmed in our experiment using the selective COX-1 inhibitor SC-560 and the COX-2 inhibitor rofecoxib[19,20,40]. As shown in Figure 9, indomethacin at 30 mg/kg produced gastric lesions with a marked decrease in mucosal PGE2 content. As expected, the selective COX-2 inhibitor rofecoxib did not induce any damage at 30 mg/kg, with no effect on mucosal PGE2 content. Likewise, the COX-1 inhibitor SC-560 did not cause gastric damage even at 30 mg/kg, despite inhibiting PGE2 production, as effectively as indomethacin. However, these agents given together provoked damage in the stomach. In this case, when SC-560 at 10 mg/kg was given together with various doses of rofecoxib, the severity of the damage increased depending on the dose of the selective COX-2 inhibitor (Figure 10). Similarly, when rofecoxib at 10 mg/kg was given together with SC-560, the damage increased in a manner dependent on the dose of SC-560. These results do not support the paradigm that COX-1 but not COX-2 plays a “housekeeping” role in the stomach, and strongly suggest that inhibition of both COX-1 and COX-2 is required for the occurrence of NSAID-induced gastric injury. Langenbach et al[47] reported that the indomethacin-induced gastric lesions were inhibited in animals lacking the COX-1 enzyme, casting a doubt on the role of PG/COX-1 in the pathogenesis. However, since the inhibition of COX-1 induces the expression of COX-2[19,48], it is possible that the PGs produced by COX-2 compensate for the PG deficiency in COX-1 knockout animals.
Figure 9.

Gastric ulcerogenic responses to various cyclooxygenase inhibitors and their effects on prostaglandin E2 content in rat stomachs. A: Gastric ulcerogenic responses induced by various cyclooxygenase (COX) inhibitors in rat stomach. The animals were given indomethacin (nonselective COX inhibitor; 10 and 30 mg/kg), SC-560 (selective COX-1 inhibitor; 10 and 30 mg/kg), or rofecoxib (selective COX-2 inhibitor; 10 and 30 mg/kg) p.o. and killed 8 h later; B: Effects of various COX inhibitors on gastric mucosal prostaglandin E2 (PGE2) content in rats. The animals were given indomethacin (10 mg/kg), SC-560 (10 mg/kg), or rofecoxib (10 mg/kg) p.o. and killed 2 h later. Data are presented as the mean ± SE in 5-6 rats. aP < 0.05 vs control (data from refs. 18 and 19 after modification).
Figure 10.
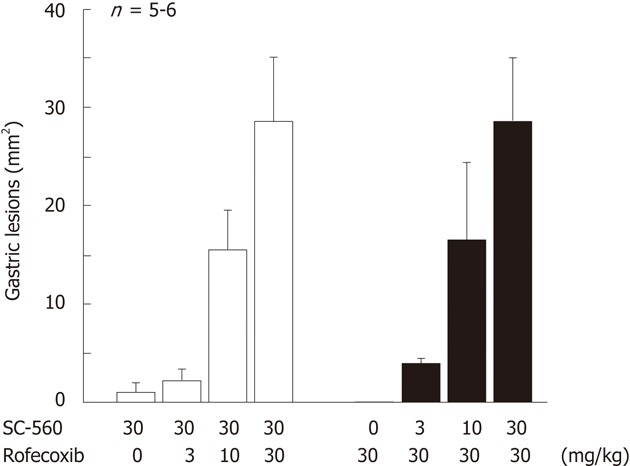
Gastric ulcerogenic response induced by combined administration of SC-560 and rofecoxib in rats. The animals were administered SC-560 (3-30 mg/kg) and rofecoxib (3-30 mg/kg) p.o. either alone or in combination, and killed 8 h later. Data are presented as the means ± SE in 5-6 rats (data from ref. 18 after modification).
COX inhibition and various pathogenic events
The pathogenic mechanism of NSAID-induced gastric damage involves multiple functional alterations, including gastric hypermotility, microcirculatory disturbance, neutrophil activation, and microvascular permeability[5-13]. A marked increase in gastric motility was observed after the administration of SC-560 but not rofecoxib, although the duration of the hypermotility was short as compared with that induced by nonselective COX inhibitors, suggesting that gastric hypermotility induced by NSAIDs is associated with a PG deficiency caused by COX-1 inhibition[18]. Likewise, SC-560 but not rofecoxib increased microvascular permeability in the stomach, similar to indomethacin. These results for SC-560 are reasonable, because indomethacin at an ulcerogenic dose is known to cause microcirculatory disturbances resulting from abnormal mucosal compression of the stomach wall due to gastric hypermotility[10]. On the other hand, Wallace et al[17] reported that SC-560, but not celecoxib, decreased the gastric mucosal blood flow, suggesting a role for PGs derived from COX-1 in the maintenance of mucosal blood flow. They also showed that the selective COX-2 inhibitor celecoxib elicited neutrophil adherence in mesenteric venules, as potently as indomethacin, whereas the selective COX-1 inhibitor SC-560 did not. However, we observed that neither SC-560 nor rofecoxib alone affected MPO activity in the gastric mucosa, yet these two agents together apparently increased MPO activity to the levels comparable to those induced by indomethacin[19]. This event might be hampered by PGs derived from COX-2, probably at later stages following gastric hypermotility, since microcirculatory disturbances are known to enhance the adhesion of neutrophils to endothelial cells[13,34]. These results strongly suggest that the inhibition of both COX-1 and COX-2 is required for enhancement of neutrophil migration in the gastric mucosa and that neutrophils may be involved in the damage process later on, but do not play a role in the onset of gastric damage induced by NSAIDs.
Upregulation of COX-2 expression
The most important event is that the expression of COX-2 mRNA was induced in the gastric mucosa after administration of NSAIDs[18,19] (Figure 11A). The upregulation of COX-2 expression was similarly observed in the rat stomach after administration of SC-560 but not rofecoxib, suggesting a causal relationship between COX-1 inhibition and COX-2 expression (Figure 11B). We also reported the upregulation of COX-2 expression in the small intestine following administration of both indomethacin and SC-560[20,48,49]. It is assumed that inhibition of COX-1 induces a PG deficiency but upregulates the expression of COX-2, which contributes to a restoration of PG production. Indeed, the mucosal PGE2 content of the stomach was markedly decreased by SC-560, yet values recovered significantly 8 h after the administration in a rofecoxib-sensitive manner[18] (Figure 12A). Thus, the upregulation of COX-2 expression following inhibition of COX-1 may represent a compensatory response to inhibition of PG biosynthesis and contribute to maintenance of the mucosal integrity of the stomach. This speculation is supported by the fact that combined treatment with SC-560 and rofecoxib did provoke gross damage in the stomach, and that such damage was prevented by administration of PGE2 4 h after the use of COX inhibitors[19]. The exact mechanism by which the expression of COX-2 is induced by inhibition of COX-1 remains unknown. Since the expression of COX-2 induced by indomethacin was attenuated by atropine at the dose that inhibited the gastric hypermotility[6,21,48], it is possible that the upregulation of COX-2 expression is due to vascular injury caused by abnormal mucosal compression of the stomach wall during gastric hypermotility (Figure 12B). Indeed, atropine significantly inhibited the recovery of PGE2 levels following administration of SC-560, similar to rofecoxib[48]. Alternatively, because NSAIDs release tumor necrosis factor α (TNF-α)[38,50], the upregulation of COX-2 expression observed under COX-1 inhibition is mediated by TNF-α. Omeprazole had no effect on the expression of COX-2 induced by indomethacin, suggesting no role for luminal acid in this phenomenon[48].
Figure 11.
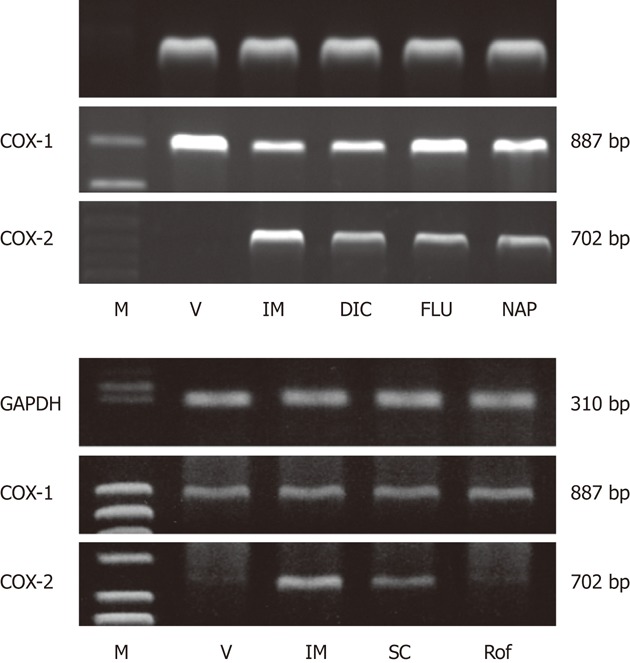
Gene expression of cyclooxygenase-1 and cyclooxygenase-2 in rat gastric mucosa after administration of various non-steroidal anti-inflammatory drugs (A) or various cyclooxygenase inhibitors (B). The animals were given indomethacin (IM, 30 mg/kg), naproxen (NAP, 40 mg/kg), flurbiprofen (FLU, 20 mg/kg), dicrofenac (DIC, 40 mg/kg), SC-560 (SC, 30 mg/kg), or rofecoxib (Rof, 30 mg/kg) p.o., and the expression of cyclooxygenase (COX)-1 and COX-2 mRNA was examined by reverse transcription polymerase chain reaction 4 h later. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; M: Marker, V: Vehicle (data from refs. 18 and 19 after modification).
Figure 12.
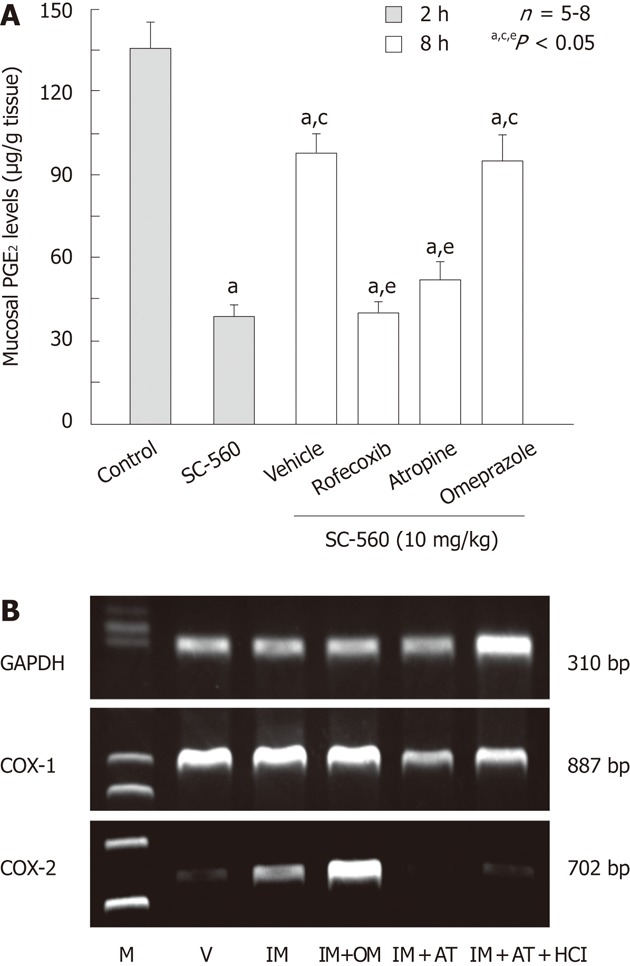
Effects of various drugs on prostaglandin E2 production and cyclooxygenase-2 expression in rat gastric mucosa. A: Effects of various drugs on prostaglandin E2 (PGE2) levels in the rat gastric mucosa at 8 h after administration of SC-560. The animals were administered SC-560 (10 mg/kg) p.o., and killed 2 or 8 h later. Rofecoxib (30 mg/kg) was given p.o. together with SC-560 while omeprazole (OM, 30 mg/kg) or atropine (AT, 3 mg/kg) was given s.c. 1 h before the administration of SC-560. Data are presented as the mean ± SE in 6 rats. Significant difference at P < 0.05; afrom control; cfrom SC-560 (2 h); efrom vehicle; B: Effect of OM and AT on cyclooxygenase (COX)-2 expression after administration of indomethacin (IM) in rat stomach. The animals were administered IM (30 mg/kg) p.o., and killed 4 h later. OM (30 mg/kg) or AT (3 mg/kg) was given s.c. 1 h before indomethacin. Some animals were given 1 mL of 100 mmol HCl immediately after administration of IM. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; M: Marker, V: Vehicle (data from ref. 48 after modification).
POTENTIATION OF NSAID-INDUCED GASTRIC DAMAGE
Adrenalectomy
Takeuchi et al[9] demonstrated that indomethacin-induced gastric damage was markedly aggravated in adrenalectomized rats and the dose required to produce lesions was decreased in these rats. Filaretova et al[22] confirmed the aggravation of NSAID-induced gastric ulceration in adrenalectomized rats (glucocorticoid-deficient conditions) and further investigated the influence of adrenalectomy on the expression of COX-2 in the stomach as well as the ulcerogenic effect of celecoxib (a selective COX-2 inhibitor) in these rat stomachs. It was found that adrenalectomy decreased plasma corticosterone levels and markedly aggravated indomethacin-induced gastric lesions. This aggravation was significantly prevented by corticosterone replacement, suggesting that glucocorticoid deficiency is the reason for the aggravation of indomethacin-induced gastric injury in adrenalectomized rats. Moreover, in adrenalectomized rats, celecoxib provoked gross damage that was prevented by corticosterone pretreatment. Mucosal PGE2 content was increased 3-fold after adrenalectomy, and this response was prevented by both celecoxib and corticosterone. COX-2 mRNA expression was up-regulated in the stomach of adrenalectomized rats, but suppressed by corticosterone replacement. It is assumed that adrenalectomy, probably via a glucocorticoid deficiency, increases PGE2 production in the stomach due to COX-2 expression, and the selective COX-2 inhibitor produces gastric lesions by suppressing this additional PG production in adrenalectomized rats. These findings also support the idea that COX-2 as well as COX-1 play a role in maintaining gastric mucosal integrity under glucocorticoid-deficient conditions.
Adjuvant arthritis
Patients with rheumatoid arthritis (RA) are reportedly more susceptible to NSAID-induced gastropathy than other NSAID users[51,52]. This observation has been validated in arthritic rat models induced by injecting Freund’s complete adjuvant into the planter region of a hindfoot, where the gastric ulcerogenic response to indomethacin was markedly aggravated in comparison with normal animals[23,53,54]. Since the aggravation of these lesions in arthritic rats was dependent on the degree of arthritic change, it is assumed that there is a cause-effect relationship between the systemic inflammation and the increased gastric mucosal susceptibility to indomethacin. As several studies including ours showed increased serum gastrin levels and acid secretion in arthritic rats[53,55], it is speculated that the increased gastric ulcerogenic response is partly attributable to hyperacidity in the stomach. However, because the aggravation of these lesions was similarly observed in arthritic rats, even in the presence of exogenous acid to mask endogenous hyperacidic conditions[53], it is unlikely that the increased mucosal susceptibility to indomethacin in arthritic rats is associated with the increase of acid secretion. Interestingly, the aggravation of indomethacin-induced gastric damage in arthritic rats was prevented by prior administration of NG-nitro-L-arginine methyl ester, a nonselective nitric oxide synthase (NOS) inhibitor, and aminoguanidine, a selective inducible NOS (iNOS) inhibitor, as well as dexamethasone, an inhibitor of iNOS mRNA transcription, although they did not affect the severity of the lesions observed in normal rats[53]. Moreover, the distinct expression of iNOS mRNA was observed in the stomach of arthritic rats, accompanied with an increase in NO production. These findings suggest that the increased ulcerogenic response to NSAIDs in arthritic rats is associated at least partly with endogenous NO, mainly produced by iNOS. It is possible that the increased susceptibility of arthritic rat stomachs to NSAIDs might be explained by production of peroxynitrite, resulting from the interaction of NO/iNOS with superoxide radicals[56].
As mentioned, selective COX-2 inhibitors such as rofecoxib and celecoxib, even at a higher dose (100 mg/kg), did not damage the normal rat stomach[18]. However, they produced gross lesions in the stomach of arthritic rats[23] (Figure 13A). Moreover, PG generation in the arthritic rat stomach was significantly enhanced with a concomitant increase of COX-2 expression (Figure 13B). Certainly, the mucosal PG content was reduced by indomethacin in both normal and arthritic rat stomachs. In contrast, the COX-2 inhibitor rofecoxib did not affect PG generation in normal rats but significantly decreased PG content in the stomach of arthritic rats, suggesting that COX-2 activity caused the increase in PG production in arthritic rat stomachs. These findings suggest that COX-2 plays an important role in maintaining the integrity of the gastric mucosa in arthritic rats. It is possible that the increased COX-2 expression level in the stomach occur in association with inflammation or stress caused by pain. Since SC-560, a selective COX-1 inhibitor, worsened stress-induced gastric lesions[57], SC-560 may produce hemorrhagic lesions in the stomach by potentiating the ulcerogenic response to arthritis-related stress. Further study is certainly required to verify this point.
Figure 13.
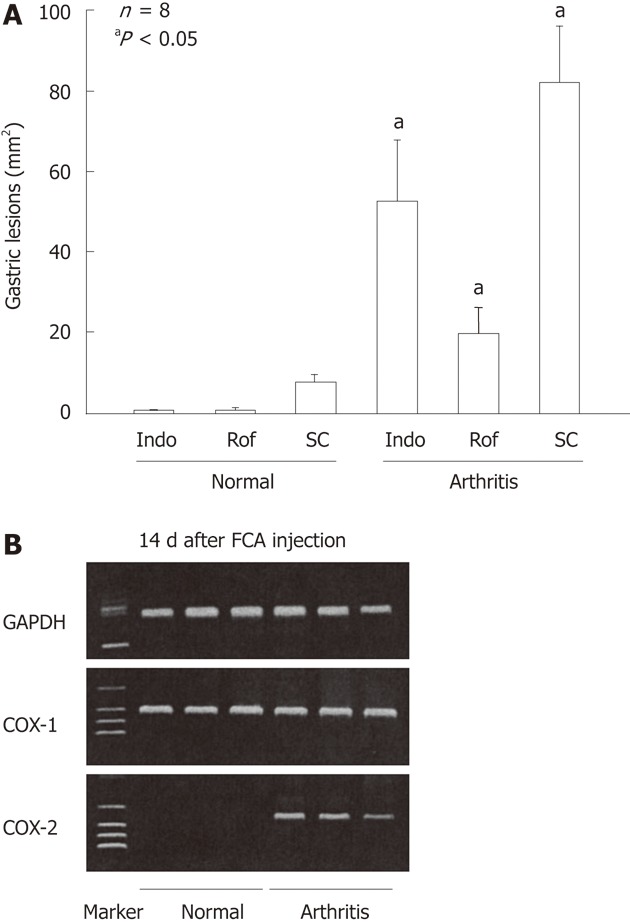
Gastric ulcerogenic effect of indomethacin, rofecoxib and SC-560 and the expression of cyclooxygenase-1 and cyclooxygenase-2 mRNA in the gastric mucosa of normal and arthritic rats. A: Arthritis was induced by injecting Freund’s complete adjuvant (FCA) into the plantar region of the right hindfoot, and the experiments were performed 14 d after the injection. Indomethacin (Indo) (3 mg/kg), rofecoxib (Rof) (30 mg/kg), or SC-560 (SC) (30 mg/kg) were administered p.o., and the animals were killed 4 h later. Data are presented as the mean ± SE in 4-8 animals, aP < 0.05 vs the corresponding group in normal rats; B: COX-2 mRNA was not detected in the normal rats, but clearly observed in the arthritic rats on day 14 after the FCA injection, whereas COX-1 mRNA was observed in the stomach of both normal and arthritic rats. Lane 1, marker; lanes 2-4, normal rats; lanes 5-7, arthritic rats (data from ref. 23 after modification). GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
H. pylori infection
Takahashi et al[24] examined the expression of COX proteins and production of PGE2 in the gastric mucosa during H. pylori infection. The level of COX-1 remained nearly constant during the infection. In contrast, the COX-2 protein was not found in normal mucosa or in H. pylori -infected mucosa at 2 wk, but was markedly elevated 4 wk after the infection, with a significant rise in PGE2 production. To investigate the role of COX-2 in H. pylori-induced gastritis, they also examined the effects of NSAIDs on PGE2 production and gastric pathology caused by H. pylori. NS-398 (a COX-2-selective inhibitor) at 10 mg/kg or indomethacin at 2 mg/kg was administered for 4 wk to normal and H. pylori-infected animals. NS-398 failed to inhibit PGE2 production in normal mucosa but significantly reduced the H. pylori-increased PGE2 production. In contrast, indomethacin potently inhibited PGE2 production in both normal and H. pylori-infected mucosa. Hemorrhagic erosions, neutrophil infiltration, lymphoid follicles, and epithelium damage were induced by H. pylori infection. NS-398 and indomethacin aggravated these pathological changes, but did not increase viable H. pylori numbers. Overall, these results indicate that both COX-2 and COX-1 might play anti-inflammatory roles in H. pylori-induced gastritis. Similar findings were also obtained by Tanigawa et al[58] who showed that PGE2 derived from either COX-1 or COX-2 is involved in the regulation of gastric mucosal inflammation and contributes to the maintenance of mucosal integrity during H. pylori infection via inhibition of TNF-α expression.
BIPHASIC EFFECT OF ASPIRIN
Conventional NSAIDs cause gastric damage with a concomitant decrease in mucosal PGE2 production, irrespective of the route of administration[18,19]. However, since aspirin is not ulcerogenic in the stomach, despite that it reduces mucosal PGE2 production as effectively as other NSAIDs, it is likely that a depletion of endogenous PGs by itself is not sufficient for gastric lesions to form and other factors are required for the onset of gastric damage. However, when administered orally, aspirin damages the stomach, similar to other NSAIDs. Several studies have proposed a role for neutrophils or TNF-α in the pathogenesis of NSAID-induced gastric damage[34,35,50]. These events are considered to occur in relation to a decrease in PG biosynthesis in the gastric mucosa due to suppression of COX activity. However, aspirin given parenterally inhibited PGE2 production in the stomach, yet did not cause any damage in the mucosa[19]. Furthermore, salicylate reportedly inhibited TNF-α production by suppressing nuclear factor kappa B[59]. Considering all these points, we assumed that the topical irritant action of oral aspirin is most crucial in causing gastric mucosal damage.
As mentioned earlier, aspirin does not damage the stomach but shows a dose-dependent inhibition of indomethacin-induced gastric injury[24] (Figure 14A). This result is consistent with the finding by Robert et al[60], who showed for the first time that aspirin provided protection against gastric damage in response to various noxious agents including indomethacin. Following the subcutaneous administration of aspirin (200 mg/kg) in rats, plasma levels of salicylate increased with time, reaching almost a plateau within 30 min, and remained elevated for more than 4 h. A small amount of aspirin was detected in the blood for the first 15 min, but it had disappeared almost totally 30 min later. As expected, since salicylate, the major metabolite of aspirin, also prevented indomethacin-induced gastric damage, it is possible that the protective action of aspirin is mediated by salicylate. Interestingly, aspirin and salicylate did not increase basal gastric motility but suppressed the enhanced gastric motility following indomethacin treatment, suggesting again a relationship between the inhibition of gastric hypermotility and prevention of gastric damage (Figure 14B)[25]. At present, the exact mechanism by which salicylate (aspirin) suppresses the gastric hypermotility induced by indomethacin remains unknown.
Figure 14.
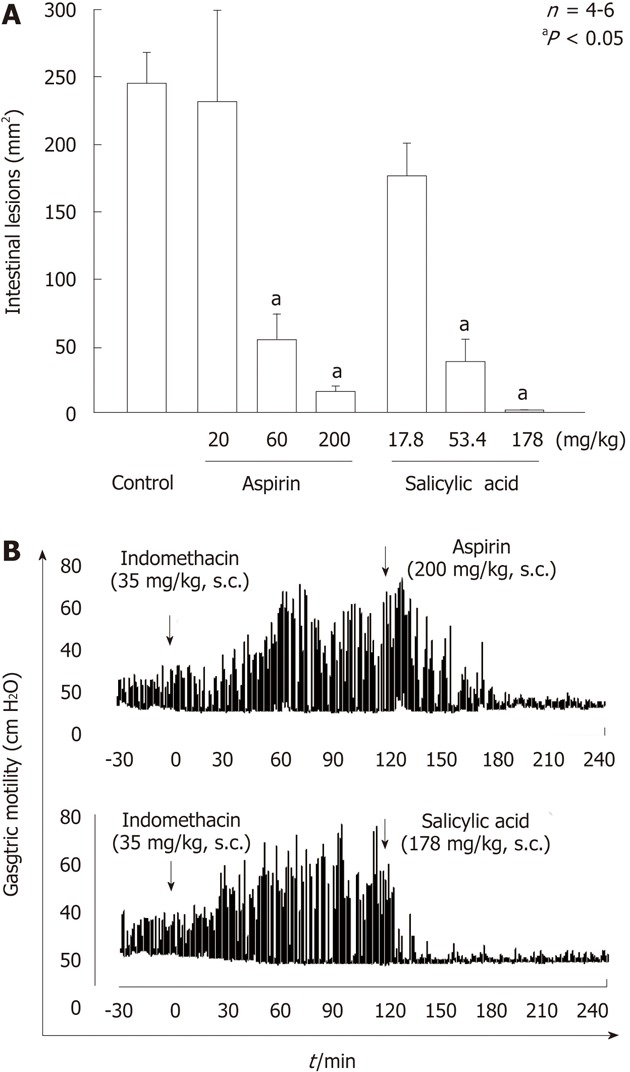
Effects of aspirin and salicylic acid on gastric lesions (A) and gastric hypermotility (B) caused by indomethacin in rats. A: The animals were given indomethacin (35 mg/kg) s.c., and killed 24 h later. Aspirin (20-200 mg/kg) or salicylic acid (17.8-178 mg/kg) was given s.c. 30 min before indomethacin. Data are presented as the mean ± SE in 4-6 rats. aP < 0.05 vs control; B: Animals were given indomethacin (30 mg/kg) s.c. and subsequently aspirin (200 mg/kg) or salicylic acid (178 mg/kg) s.c. 2 h later. Note that both aspirin and salicylic acid markedly inhibited the intestinal hypermotility induced by indomethacin, with the effect of salicylic acid appearing much earlier than that of aspirin (data from ref. 25 after modification).
Unlike other NSAIDs, COX-2’s acetylation by aspirin switches eicosanoid biosynthesis from PGE2 to lipoxin A4, which exerts protective effects in the stomach. Co-administration of aspirin and a selective COX-2 inhibitor, such as celecoxib or rofecoxib, resulted in substantially more severe gastric injury than that produced with either agent alone[61,62]. We also observed that the gastric ulcerogenic response to aspirin was significantly worsened by co-administration of rofecoxib but not SC-560[63]. These results confirmed the importance of COX-2’s inhibition in this phenomenon related to the suppression of lipoxin A4’s production.
SUMMARY AND FUTURE PROSPECTS
The gastric ulcerogenic properties of NSAIDs are not accounted for solely by the inhibition of COX-1 and require the inhibition of both COX-1 and COX-2[17-19]. This idea is supported by the finding that neither the selective COX-1 nor COX-2 inhibitor alone caused gross damage in the stomach, but the combined administration of these two inhibitors provoked the development of gastric lesions. Indomethacin caused an increase of gastric motility, microvascular permeability and MPO activity following administration of indomethacin[6,10,26,28,34,64,65] and showed that the former two events were due to COX-1 inhibition, but the increase of MPO activity occurred only when both COX-1 and COX-2 were inhibited[19]. On the other hand, NSAIDs up-regulate the expression of COX-2, and the PGs produced by COX-2 may suppress the neutrophil-endothelial interaction caused by the vascular disturbances due to COX-1 inhibition. These sequential events related to COX-1 and/or COX-2 inhibition explain why gastric damage occurs only when both COX-1 and COX-2 are inhibited (Figure 15). It should also be noted that selective COX-2 inhibitors by themselves damage the gastric mucosa when an overexpression of COX-2 occurs in the stomach under conditions of adrenalectomy, arthritis, or H. pylori infection[22-24]. Independent of the type of NSAIDs, the users of NSAIDs should be aware of these side effects if they are infected with H. pylori or have a glucocorticoid deficiency or arthritic condition. Interestingly, aspirin acts to protect against indomethacin-induced gastric damage, although this agent given p.o. damages the stomach due to its direct irritative action. The failure of aspirin to induce gastric injury may be explained, at least partly, by a protective action of salicylic acid, the metabolite of aspirin, and this action is also functionally associated with inhibition of gastric hypermotility in response to indomethacin.
Figure 15.
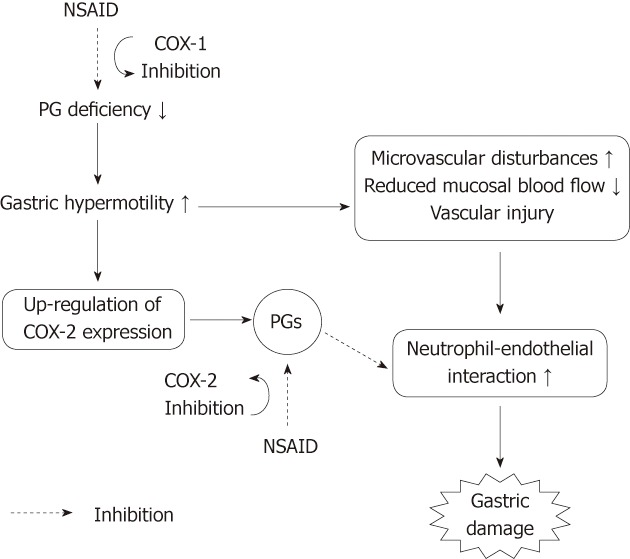
Working hypothesis on the roles of COX-1 and COX-2 in the pathogenic mechanism of non-steroidal anti-inflammatory drug-induced gastric damage. Non-steroidal anti-inflammatory drugs (NSAIDs) cause gastric hypermotility, followed by microvascular disturbances and neutrophil activation, leading to gastric damage. Gastric hypermotility and subsequent vascular disturbances are associated with a prostaglandin (PG) deficiency caused by COX-1 inhibition. The inhibition of COX-1 up-regulates COX-2 expression, and PGs produced by COX-2 may suppress the neutrophil-endothelial interaction caused by microvascular disturbances due to COX-1 inhibition.
There is no doubt that gastric hypermotility plays a primary role in the pathogenesis of NSAID-induced damage in the stomach[3-7]. This response, causally related with PG depletion due to COX-1 inhibition, occurs prior to other pathogenic events involved in NSAID-induced gastric damage, such as microvascular disturbances and neutrophil infiltration as well as COX-2 expression[6,10,19,28,63]. However, the mechanism underlying NSAID-induced gastric hypermotility remains unknown. Since the gastric hypermotility induced by indomethacin was inhibited by atropine and vagotomy as well as intravenous glucose infusion[6,7,66], it is assumed that the response occurs in association with PG deficiency caused by COX-1 inhibition and is mediated by the vagal-cholinergic pathway through central glucose receptors. The upregulation of NSAID-induced COX-2 expression is functionally associated with gastric hypermotility[47]. Because atropine prevented both gastric hypermotility and COX-2 expression in response to indomethacin[21,47] and because gastric microvascular permeability increased in association with gastric hypermotility[8,63], the upregulation of COX-2 expression may result from mild mucosal injury and/or vascular injury caused by gastric hypermotility. However, the cells responsible for COX-2 expression induced by COX-1 inhibition also remain to be identified. In addition, other possible actions, such as inhibition of phosphorylative oxidation, injury of mitochondrial membrane and cell apoptotic change, have been demonstrated as the cellular mechanisms of NSAID-induced gastropathy[66-70], although these effects are shared by NSAIDs, including aspirin that does not cause gastric damage through parenteral administration. Further study is certainly needed to clarify these points, and these approaches should contribute to the development of gastric-sparing NSAIDs that are devoid of ulcerogenic properties.
BIOGRAPHY
Professor Koji Takeuchi received his PhD degree from the University of Tokyo, Tokyo, Japan. He had an extensive 4-year postdoctoral training at Department of Physiology and Cell Biology, University of Texas, Houston and Department of Surgery, Harvard Medical School, Boston, United States. He is presently Professor and Chairman of the Department of Pharmacology and Experimental Therapeutics, Dean of the Graduate School and Vice President of the Kyoto Pharmaceutical University, Kyoto, Japan. His research interest covers numerous areas of GI pharmacology and physiology, and one of his most notable contributions is the understanding of mucosal defense, focusing on the regulation of acid/bicarbonate secretion, the influences of non-steroidal anti-inflammatory drugs and prostaglandins; their mode of action, cyclooxygenase isoforms, receptors that drive physiological responses, and their role in mucosal injury, protection and healing. He has had 448 papers published on peer-reviewed journals, including 45 book chapters, and gave numerous presentations at national and international meetings. Professor Koji Takeuchi has enjoyed quite a few prestigious academic awards and honors.
Footnotes
Peer reviewer: Andrzej S Tarnawski, MD, PhD, DSc (Med), Professor of Medicine, Chief Gastroenterology, VA Long Beach Health Care System, University of California, Irvine, CA 5901 E. Seventh Str., Long Beach, CA 90822, United States
S- Editor Cheng JX L- Editor Ma JY E- Editor Xiong L
References
- 1.Lanza FL. Endoscopic studies of gastric and duodenal injury after the use of ibuprofen, aspirin, and other nonsteroidal anti-inflammatory agents. Am J Med. 1984;77:19–24. doi: 10.1016/s0002-9343(84)80014-5. [DOI] [PubMed] [Google Scholar]
- 2.Wang JY, Yamasaki S, Takeuchi K, Okabe S. Delayed healing of acetic acid-induced gastric ulcers in rats by indomethacin. Gastroenterology. 1989;96:393–402. doi: 10.1016/0016-5085(89)91563-1. [DOI] [PubMed] [Google Scholar]
- 3.Konturek PK, Brzozowski T, Konturek SJ, Dembiński A. Role of epidermal growth factor, prostaglandin, and sulfhydryls in stress-induced gastric lesions. Gastroenterology. 1990;99:1607–1615. doi: 10.1016/0016-5085(90)90464-c. [DOI] [PubMed] [Google Scholar]
- 4.Ukawa H, Yamakuni H, Kato S, Takeuchi K. Effects of cyclooxygenase-2 selective and nitric oxide-releasing nonsteroidal antiinflammatory drugs on mucosal ulcerogenic and healing responses of the stomach. Dig Dis Sci. 1998;43:2003–2011. doi: 10.1023/a:1018846912032. [DOI] [PubMed] [Google Scholar]
- 5.Whittle BJ. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis, and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology. 1981;80:94–98. [PubMed] [Google Scholar]
- 6.Takeuchi K, Ueki S, Okabe S. Importance of gastric motility in the pathogenesis of indomethacin-induced gastric lesions in rats. Dig Dis Sci. 1986;31:1114–1122. doi: 10.1007/BF01300266. [DOI] [PubMed] [Google Scholar]
- 7.Mersereau WA, Hinchey EJ. Prevention of phenylbutazone ulcer in the rat by glucose: role of a glycoprivic receptor system. Am J Physiol. 1982;242:G429–G432. doi: 10.1152/ajpgi.1982.242.4.G429. [DOI] [PubMed] [Google Scholar]
- 8.Okada M, Niida H, Takeuchi K, Okabe S. Role of prostaglandin deficiency in pathogenetic mechanism of gastric lesions induced by indomethacin in rats. Dig Dis Sci. 1989;34:694–702. doi: 10.1007/BF01540340. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi K, Nishiwaki H, Okada M, Niida H, Okabe S. Bilateral adrenalectomy worsens gastric mucosal lesions induced by indomethacin in the rat. Role of enhanced gastric motility. Gastroenterology. 1989;97:284–293. doi: 10.1016/0016-5085(89)90063-2. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi K, Ueshima K, Hironaka Y, Fujioka Y, Matsumoto J, Okabe S. Oxygen free radicals and lipid peroxidation in the pathogenesis of gastric mucosal lesions induced by indomethacin in rats. Relation to gastric hypermotility. Digestion. 1991;49:175–184. doi: 10.1159/000200718. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi K, Takehara K, Ohuchi T. Diethyldithiocarbamate, a superoxide dismutase inhibitor, reduces indomethacin-induced gastric lesions in rats. Digestion. 1996;57:201–209. doi: 10.1159/000201341. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi K, Kato S, Nishiwaki H, Hirata T. Analysis of pathogenic elements involved in gastric lesions induced by non-steroidal anti-inflammatory drugs in rats. J Gastroenterol Hepatol. 1997;12:360–367. [Google Scholar]
- 13.Asako H, Kubes P, Wallace J, Gaginella T, Wolf RE, Granger DN. Indomethacin-induced leukocyte adhesion in mesenteric venules: role of lipoxygenase products. Am J Physiol. 1992;262:G903–G908. doi: 10.1152/ajpgi.1992.262.5.G903. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill GP, Ford-Hutchinson AW. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett. 1993;330:156–160. doi: 10.1016/0014-5793(93)80263-t. [DOI] [PubMed] [Google Scholar]
- 15.Kargman S, Charleson S, Cartwright M, Frank J, Riendeau D, Mancini J, Evans J, O’Neill G. Characterization of Prostaglandin G/H Synthase 1 and 2 in rat, dog, monkey, and human gastrointestinal tracts. Gastroenterology. 1996;111:445–454. doi: 10.1053/gast.1996.v111.pm8690211. [DOI] [PubMed] [Google Scholar]
- 16.Futaki N, Yoshikawa K, Hamasaka Y, Arai I, Higuchi S, Iizuka H, Otomo S. NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen Pharmacol. 1993;24:105–110. doi: 10.1016/0306-3623(93)90018-s. [DOI] [PubMed] [Google Scholar]
- 17.Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka A, Araki H, Komoike Y, Hase S, Takeuchi K. Inhibition of both COX-1 and COX-2 is required for development of gastric damage in response to nonsteroidal antiinflammatory drugs. J Physiol Paris. 2001;95:21–27. doi: 10.1016/s0928-4257(01)00005-5. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka A, Araki H, Hase S, Komoike Y, Takeuchi K. Up-regulation of COX-2 by inhibition of COX-1 in the rat: a key to NSAID-induced gastric injury. Aliment Pharmacol Ther. 2002;16 Suppl 2:90–101. doi: 10.1046/j.1365-2036.16.s2.22.x. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka A, Hase S, Miyazawa T, Takeuchi K. Up-regulation of cyclooxygenase-2 by inhibition of cyclooxygenase-1: a key to nonsteroidal anti-inflammatory drug-induced intestinal damage. J Pharmacol Exp Ther. 2002;300:754–761. doi: 10.1124/jpet.300.3.754. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi K, Tanaka A, Kato S, Amagase K, Satoh H. Roles of COX inhibition in pathogenesis of NSAID-induced small intestinal damage. Clin Chim Acta. 2010;411:459–466. doi: 10.1016/j.cca.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 22.Filaretova L, Tanaka A, Komoike Y, Takeuchi K. Selective cyclooxygenase-2 inhibitor induces gastric mucosal damage in adrenalectomized rats. Inflammopharmacology. 2002;10:413–422. [Google Scholar]
- 23.Kato S, Ogawa Y, Kanatsu K, Okayama M, Watanabe T, Arakawa T, Takeuchi K. Ulcerogenic influence of selective cyclooxygenase-2 inhibitors in the rat stomach with adjuvant-induced arthritis. J Pharmacol Exp Ther. 2002;303:503–509. doi: 10.1124/jpet.102.040659. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi S, Fujita T, Yamamoto A. Role of cyclooxygenase-2 in Helicobacter pylori- induced gastritis in Mongolian gerbils. Am J Physiol Gastrointest Liver Physiol. 2000;279:G791–G798. doi: 10.1152/ajpgi.2000.279.4.G791. [DOI] [PubMed] [Google Scholar]
- 25.Komoike Y, Takeeda M, Tanaka A, Kato S, Takeuchi K. Prevention by parenteral aspirin of indomethacin-induced gastric lesions in rats: mediation by salicylic acid. Dig Dis Sci. 2002;47:1538–1545. doi: 10.1023/a:1015867119014. [DOI] [PubMed] [Google Scholar]
- 26.Ueki S, Takeuchi K, Okabe S. Gastric motility is an important factor in the pathogenesis of indomethacin-induced gastric mucosal lesions in rats. Dig Dis Sci. 1988;33:209–216. doi: 10.1007/BF01535735. [DOI] [PubMed] [Google Scholar]
- 27.Mashita Y, Taniguchi M, Yokota A, Tanaka A, Takeuchi K. Oral but not parenteral aspirin upregulates COX-2 expression in rat stomachs. a relationship between COX-2 expression and PG deficiency. Digestion. 2006;73:124–132. doi: 10.1159/000094098. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki K, Araki H, Komoike Y, Takeuchi K. Permissive role of neutrophils in pathogenesis of indomethacin-induced gastric lesions in rats. Med Sci Monit. 2000;6:908–914. [PubMed] [Google Scholar]
- 29.Suemasu S, Tanaka K, Namba T, Ishihara T, Katsu T, Fujimoto M, Adachi H, Sobue G, Takeuchi K, Nakai A, et al. A role for HSP70 in protecting against indomethacin-induced gastric lesions. J Biol Chem. 2009;284:19705–19715. doi: 10.1074/jbc.M109.006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garrick T, Buack S, Bass P. Gastric motility is a major factor in cold restraint-induced lesion formation in rats. Am J Physiol. 1986;250:G191–G199. doi: 10.1152/ajpgi.1986.250.2.G191. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi K, Okada M, Niida H, Okabe S. Possible mechanisms involved in gastric hypermotility caused by indomethacin in the rat. Role of glycoprivic response. Dig Dis Sci. 1990;35:984–992. doi: 10.1007/BF01537247. [DOI] [PubMed] [Google Scholar]
- 32.Yamaguchi T. Relationship between gastric mucosal hemodynamics and gastric motility. Gastroenterol Jpn. 1990;25:299–305. doi: 10.1007/BF02779442. [DOI] [PubMed] [Google Scholar]
- 33.Anthony A, Sim R, Dhillon AP, Pounder RE, Wakefield AJ. Gastric mucosal contraction and vascular injury induced by indomethacin precede neutrophil infiltration in the rat. Gut. 1996;39:363–368. doi: 10.1136/gut.39.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace JL, Granger DN. Pathogenesis of NSAID gastropathy: are neutrophils the culprits? Trends Pharmacol Sci. 1992;13:129–131. doi: 10.1016/0165-6147(92)90046-9. [DOI] [PubMed] [Google Scholar]
- 35.Morise Z, Granger DN, Fuseler JW, Anderson DC, Grisham MB. Indomethacin induced gastropathy in CD18, intercellular adhesion molecule 1, or P-selectin deficient mice. Gut. 1999;45:523–528. doi: 10.1136/gut.45.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trevethick MA, Bahl AK, Clayton NM, Strong P, Sanjar S, Harman IW. Neutrophil infiltration does not contribute to the ulcerogenic effects of indomethacin in the rat gastric antrum. Agents Actions. 1994;43:39–43. doi: 10.1007/BF02005762. [DOI] [PubMed] [Google Scholar]
- 37.Melarange R, Gentry C, Toseland CD, Smith PH, Fuller J. Neutropenia does not prevent etodolac- or indomethacin-induced gastrointestinal damage in the rat. Dig Dis Sci. 1995;40:2694–2703. doi: 10.1007/BF02220462. [DOI] [PubMed] [Google Scholar]
- 38.Santucci L, Fiorucci S, Di Matteo FM, Morelli A. Role of tumor necrosis factor alpha release and leukocyte margination in indomethacin-induced gastric injury in rats. Gastroenterology. 1995;108:393–401. doi: 10.1016/0016-5085(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki K, Araki H, Mizoguchi H, Furukawa O, Takeuchi K. Prostaglandin E inhibits indomethacin-induced gastric lesions through EP-1 receptors. Digestion. 2001;63:92–101. doi: 10.1159/000051876. [DOI] [PubMed] [Google Scholar]
- 40.Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology. 2008;135:41–60. doi: 10.1053/j.gastro.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 41.Araki H, Ukawa H, Sugawa Y, Yagi K, Suzuki K, Takeuchi K. The roles of prostaglandin E receptor subtypes in the cytoprotective action of prostaglandin E2 in rat stomach. Aliment Pharmacol Ther. 2000;14 Suppl 1:116–124. doi: 10.1046/j.1365-2036.2000.014s1116.x. [DOI] [PubMed] [Google Scholar]
- 42.Milenov K, Golenhofen K. Contractile responses of longitudinal and circular smooth muscle of the canine stomach to prostaglandins E and F2alpha. Prostaglandins Leukot Med. 1982;8:287–300. doi: 10.1016/0262-1746(82)90051-8. [DOI] [PubMed] [Google Scholar]
- 43.Ding M, Kinoshita Y, Kishi K, Nakata H, Hassan S, Kawanami C, Sugimoto Y, Katsuyama M, Negishi M, Narumiya S, et al. Distribution of prostaglandin E receptors in the rat gastrointestinal tract. Prostaglandins. 1997;53:199–216. doi: 10.1016/s0090-6980(97)00015-4. [DOI] [PubMed] [Google Scholar]
- 44.Morimoto K, Sugimoto Y, Katsuyama M, Oida H, Tsuboi K, Kishi K, Kinoshita Y, Negishi M, Chiba T, Narumiya S, et al. Cellular localization of mRNAs for prostaglandin E receptor subtypes in mouse gastrointestinal tract. Am J Physiol. 1997;272:G681–G687. doi: 10.1152/ajpgi.1997.272.3.G681. [DOI] [PubMed] [Google Scholar]
- 45.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 46.Armstrong RA. Investigation of the inhibitory effects of PGE2 and selective EP agonists on chemotaxis of human neutrophils. Br J Pharmacol. 1995;116:2903–2908. doi: 10.1111/j.1476-5381.1995.tb15943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi K, Tanaka A, Hayashi Y, Kubo Y. Functional mechanism underlying COX-2 expression following administration of indomethacin in rat stomachs: importance of gastric hypermotility. Dig Dis Sci. 2004;49:180–187. doi: 10.1023/b:ddas.0000017436.05273.fd. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka A, Hase S, Miyazawa T, Ohno R, Takeuchi K. Role of cyclooxygenase (COX)-1 and COX-2 inhibition in nonsteroidal anti-inflammatory drug-induced intestinal damage in rats: relation to various pathogenic events. J Pharmacol Exp Ther. 2002;303:1248–1254. doi: 10.1124/jpet.102.041715. [DOI] [PubMed] [Google Scholar]
- 50.Santucci L, Fiorucci S, Giansanti M, Brunori PM, Di Matteo FM, Morelli A. Pentoxifylline prevents indomethacin induced acute gastric mucosal damage in rats: role of tumour necrosis factor alpha. Gut. 1994;35:909–915. doi: 10.1136/gut.35.7.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DiPasquale G, Welaj P. Letter: Ulcerogenic potential of indomethacin in arthritic and non-arthritic rats. J Pharm Pharmacol. 1973;25:831–832. doi: 10.1111/j.2042-7158.1973.tb09953.x. [DOI] [PubMed] [Google Scholar]
- 52.Schleyerbach R, Wedde H. Alterations in the gastro-intestinal functions during the development of adjuvant disease in rats. Agents Actions. 1984;15:392–397. doi: 10.1007/BF01972377. [DOI] [PubMed] [Google Scholar]
- 53.Kato S, Tanaka A, Kunikata T, Nishijima M, Takeuchi K. Changes in gastric mucosal ulcerogenic responses in rats with adjuvant arthritis: role of nitric oxide. Aliment Pharmacol Ther. 1999;13:833–840. doi: 10.1046/j.1365-2036.1999.00548.x. [DOI] [PubMed] [Google Scholar]
- 54.Kato S, Takeuchi K. Alteration of gastric ulcerogenic and healing responses in rats with adjuvant-induced arthritis. Jpn J Pharmacol. 2002;89:1–6. doi: 10.1254/jjp.89.1. [DOI] [PubMed] [Google Scholar]
- 55.Mathur PP, Smyth RD. The relationship between serum gastrin, gastric ulceration and basal acid output in the polyarthritic rat. J Pharmacol Exp Ther. 1980;212:333–336. [PubMed] [Google Scholar]
- 56.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka A, Hatazawa R, Takahira Y, Izumi N, Filaretova L, Takeuchi K. Preconditioning stress prevents cold restraint stress-induced gastric lesions in rats: roles of COX-1, COX-2, and PLA2. Dig Dis Sci. 2007;52:478–487. doi: 10.1007/s10620-006-9394-8. [DOI] [PubMed] [Google Scholar]
- 58.Tanigawa T, Watanabe T, Hamaguchi M, Sasaki E, Tominaga K, Fujiwara Y, Oshitani N, Matsumoto T, Higuchi K, Arakawa T. Anti-inflammatory effect of two isoforms of COX in H. pylori-induced gastritis in mice: possible involvement of PGE2. Am J Physiol Gastrointest Liver Physiol. 2004;286:G148–G156. doi: 10.1152/ajpgi.00137.2003. [DOI] [PubMed] [Google Scholar]
- 59.Cronstein BN, Montesinos MC, Weissmann G. Salicylates and sulfasalazine, but not glucocorticoids, inhibit leukocyte accumulation by an adenosine-dependent mechanism that is independent of inhibition of prostaglandin synthesis and p105 of NFkappaB. Proc Natl Acad Sci USA. 1999;96:6377–6381. doi: 10.1073/pnas.96.11.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robert A. Gastric cytoprotection by sodium salicylate. Prostaglandins. 1981;21 Suppl:139–146. doi: 10.1016/0090-6980(81)90130-1. [DOI] [PubMed] [Google Scholar]
- 61.Fiorucci S, de Lima OM, Mencarelli A, Palazzetti B, Distrutti E, McKnight W, Dicay M, Ma L, Romano M, Morelli A, et al. Cyclooxygenase-2-derived lipoxin A4 increases gastric resistance to aspirin-induced damage. Gastroenterology. 2002;123:1598–1606. doi: 10.1053/gast.2002.36558. [DOI] [PubMed] [Google Scholar]
- 62.Souza MH, de Lima OM, Zamuner SR, Fiorucci S, Wallace JL. Gastritis increases resistance to aspirin-induced mucosal injury via COX-2-mediated lipoxin synthesis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G54–G61. doi: 10.1152/ajpgi.00525.2002. [DOI] [PubMed] [Google Scholar]
- 63.Takeuchi K, Tanaka A, Kato S, Aihara E, Amagase K. Effect of (S)-4-(1-(5-chloro-2-(4-fluorophenyoxy)benzamido)ethyl) benzoic acid (CJ-42794), a selective antagonist of prostaglandin E receptor subtype 4, on ulcerogenic and healing responses in rat gastrointestinal mucosa. J Pharmacol Exp Ther. 2007;322:903–912. doi: 10.1124/jpet.107.122978. [DOI] [PubMed] [Google Scholar]
- 64.Takeuchi K, Okada M, Ebara S, Osano H. Increased microvascular permeability and lesion formation during gastric hypermotility caused by indomethacin and 2-deoxy-D-glucose in the rat. J Clin Gastroenterol. 1990;12 Suppl 1:S76–S84. doi: 10.1097/00004836-199001001-00014. [DOI] [PubMed] [Google Scholar]
- 65.Takeuchi K, Miyazawa T, Matsumoto M, Hayashi Y. Both selective COX-1 and COX-2 inhibitors aggravate gastric damage induced in rats by 2-deoxy-D-glucose. relation to gastric hypermotility and COX-2 expression. Digestion. 2003;68:71–79. doi: 10.1159/000074518. [DOI] [PubMed] [Google Scholar]
- 66.Tarnawski A, Stachura J, Gergely H, Hollander D. Gastric microvascular endothelium: a major target for aspirin-induced injury and arachidonic acid protection. An ultrastructural analysis in the rat. Eur J Clin Invest. 1990;20:432–440. doi: 10.1111/j.1365-2362.1990.tb01881.x. [DOI] [PubMed] [Google Scholar]
- 67.Cherkasskaia MD, Iasaĭtis AA. [Effect of acetylsalicylic and 2,3-dihydroxybenzoic acids on liver mitochondrial respiration in rats] Vopr Med Khim. 1976;22:443–448. [PubMed] [Google Scholar]
- 68.Tanaka K, Tomisato W, Hoshino T, Ishihara T, Namba T, Aburaya M, Katsu T, Suzuki K, Tsutsumi S, Mizushima T. Involvement of intracellular Ca2+ levels in nonsteroidal anti-inflammatory drug-induced apoptosis. J Biol Chem. 2005;280:31059–31067. doi: 10.1074/jbc.M502956200. [DOI] [PubMed] [Google Scholar]
- 69.Pal C, Bindu S, Dey S, Alam A, Goyal M, Iqbal MS, Maity P, Adhikari SS, Bandyopadhyay U. Gallic acid prevents nonsteroidal anti-inflammatory drug-induced gastropathy in rat by blocking oxidative stress and apoptosis. Free Radic Biol Med. 2010;49:258–267. doi: 10.1016/j.freeradbiomed.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 70.Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, Dutta S, Sarkar S, Kumar R, Maity P, et al. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem. 2011;286:39387–39402. doi: 10.1074/jbc.M111.279893. [DOI] [PMC free article] [PubMed] [Google Scholar]