

Abstract
Cx50 (connexin50), a member of the α-family of gap junction proteins expressed in the lens of the eye, has been shown to be essential for normal lens development. In the present study, we identified a CaMBD [CaM (calmodulin)-binding domain] (residues 141–166) in the intracellular loop of Cx50. Elevations in intracellular Ca2+ concentration effected a 95 % decline in gj (junctional conductance) of Cx50 in N2a cells that is likely to be mediated by CaM, because inclusion of the CaM inhibitor calmidazolium prevented this Ca2+-dependent decrease in gj. The direct involvement of the Cx50 CaMBD in this Ca2+/CaM-dependent regulation was demonstrated further by the inclusion of a synthetic peptide encompassing the CaMBD in both whole-cell patch pipettes, which effectively prevented the intracellular Ca2+-dependent decline in gj. Biophysical studies using NMR and fluorescence spectroscopy reveal further that the peptide stoichiometrically binds to Ca2+/CaM with an affinity of ~ 5 nM. The binding of the peptide expanded the Ca2+-sensing range of CaM by increasing the Ca2+ affinity of the C-lobe of CaM, while decreasing the Ca2+ affinity of the N-lobe of CaM. Overall, these results demonstrate that the binding of Ca2+/CaM to the intracellular loop of Cx50 is critical for mediating the Ca2+-dependent inhibition of Cx50 gap junctions in the lens of the eye.
Keywords: calcium, calmodulin, connexin50, gap junction, junctional conductance, protein–protein interaction
INTRODUCTION
The circulation of current fluid flow through the closely packed cells of the lens of the eye is essential for lens homoeostasis, and is highly dependent on functional cell–cell coupling. This is accomplished by gap junctions, relatively non-selective channels that are permeable to low-molecular-mass (<1 kDa) molecules that directly link the cytoplasms of adjacent cells [1–3]. The gap junction channel formed between two adjacent cells comprises two connexon hemichannels, one from each cell, joined in mirror symmetry. Each hemichannel is composed of six transmembrane Cx (connexin) proteins embedded in the plasma membrane of the cell. The mammalian lens comprises three different Cx proteins, namely Cx43, Cx46 and Cx50, that, like all other Cxs, have four transmembrane segments, a short N-terminal cytoplasmic region, one intracellular loop and two extracellular loops with a set of highly conserved cysteine residues; the major difference between all Cxs is in the sequences of their cytoplasmic loops and C-terminal tails [3].
Inhibition of cell–cell communication in lens by elevated [Ca2+]i (intracellular Ca2+ concentration) was first demonstrated as an increased internal electrical resistance [4] that was prevented by pre-incubation with CaM (calmodulin) antagonists. Elevated [Ca2+]i also inhibits cell–cell communication in both bovine [5] and sheep [6] lens primary cell cultures. We have demonstrated that lens cell–cell communication was half-maximally inhibited at ~ 300 nM [Ca2+]i [6] and was prevented by pre-incubation of lens cultures with CaM antagonists [7]. The rapid onset of this inhibition (within seconds) suggests that this is mediated by a direct interaction of CaM with one or more of the lens Cxs, rather than being mediated via the action of a CaM-dependent protein kinase. Indeed, Peracchia et al. [8] have demonstrated that CaM gates Cx32-containing gap junctions directly, and Török et al. [9] have identified two distinct CaM-binding amino acid sequences in Cx32, with the N- and C-lobes of CaM showing separate functions, suggesting trans-domain or trans-subunit bridging by CaM as a possible mechanism of gap junction gating [10]; notably, this CaM-binding motif in Cx32 is absent from the three lens Cxs.
To better understand the Ca2+-dependent inhibition of lens gap junctions, we identified high-affinity CaM-binding sites in the lens Cxs Cx43 and Cx44 (the sheep homologue of human Cx46). Specifically, these domains encompass residues 138–157 in Cx43 [11] and residues 129–148 in Cx44 (corresponding to residues 138–157 in Cx46) [12]. These sequences have conserved hydrophobic residues at positions 1, 5, 10 and 14 that matched well with two major CaM-binding classes, 1–10 and 1–14, which are similar to the CaM-binding motifs found in calcineurin, nitric oxide synthase, adenylate cyclase and skeletal myosin light chain kinase [13,14]. These CaMBDs (CaM-binding domains) are located at the C-terminal end of the loop region between the predicted Cx TM (transmembrane region) 2 and TM3 [2–3,15–18] and are highly conserved in all three lens Cxs.
The present study was conducted to determine whether Cx50, like Cx43 and Cx44, was also regulated by [Ca2+]i in the micromolar range, and, if so, whether this Ca2+ regulation was CaM-mediated. A bioinformatic analysis [14] of Cx50 resulted in the identification of a candidate CaMBD with high predictive score that was located in the same C-terminal end of the loop region between the predicted Cx TM2 and TM3 of Cx50. This region is highly conserved in all three lens Cxs. A peptide model, which proved to be successful in our studies with Cx43 and Cx44 [11,12], was then adopted to demonstrate the physical contacts between the predicted Cx50 CaMBD and CaM. Studies using high-resolution NMR and fluorescence spectroscopy clearly established a high-affinity Ca2+-dependent interaction between a domain in the intracellular loop of Cx50 and CaM. Our results provide important new insights into the molecular mechanism by which altered regulation of intracellular Ca2+ concentration in the lens affects the regulation of lens Cx50 gap junctions.
EXPERIMENTAL
Prediction of transmembrane topology and a CaMBD
On the basis of a hidden Markov model, the topology and orientation of the transmembrane helices of the mouse Cx50 were predicted using four different programs, including TMHMM, MEMSAT, SOSUI and HMMTOP [16,19–21]. ClustalW2 was used to align multiple mammalian gap junction polypeptide sequences in the α-family, including Cx43 (GenBank® accession number NP_000156; Gja1 human), Cx44 (GenBank® accession number AAD56220; Gja3 sheep), Cx46 (GenBank® accession number NP_068773; Gja3 human) and Cx50 (GenBank® accession number AAF32309; Gja8 human). The candidate CaMBD in Cx50 was predicted using the CaM Target Database developed on the basis of over 100 known CaM target sequences [14].
Peptides and proteins
The Cx50 peptide, Cx50p141–166 (S141SKGTKKFRLEGTLLRTYVCHIIFKT166), was synthesized by EZ Biolab and purified by preparative reversed-phase HPLC with a purity of >87 %. A scrambled peptide, SCx50p (FKLYKCISFGGTEITTRSHVLTKKRL), with the same composition of amino acids which shows no predicted CaM-binding capacity, was synthesized with a purity of >90 % and served as a negative control. The molecular masses of these two peptides were determined by MALDI (matrix-assisted laser-desorption/ionization)–TOF (time-of-flight)-MS in reflectron mode. To mimic the protein environment and remove extra charges, peptides were acylated at their N-termini and aminated at their C-termini.
Both 14N and 15N isotopically labelled recombinant rat CaMs were expressed and purified as described previously [11]. D-CaM (dansylated CaM) was prepared as described previously [22]. The concentration of CaM was determined using the ε276 of 3030 M−1 · cm−1 [23]. The amount of dye bound was determined using an ε335 of 3980 M−1· cm−1 for D-CaM. The concentration of modified CaM was determined using the BCA (bicinchoninic acid) assay (Pierce) with BSA or unmodified CaM as the standard. The modification of CaM was confirmed further by MALDI–TOF-MS in the linear mode.
Electrophysiological measurement of gj (junctional conductance)
Parental N2a cells were grown to 80 % confluence in a 12-well format and were transiently transfected with 1 μg of the pTracer-Cx50 cDNA plasmid for 4 h, lightly trypsinized and placed in 35-mm-diameter culture dishes overnight. Dual whole-cell patch-clamp experiments were performed the next day on GFP (green fluorescent protein)-expressing cell pairs to measure gj, and the sensitivity to [Ca2+]i was determined by permeabilizing the cells with 1 μM ionomycin and perfusing with ± 1.8 mM external CaCl2 saline solutions. A +20 mV Vj (transjunctional voltage) pulse was applied every 15 s to monitor gj during bath perfusion with nominally 0 or 1.8 mM [Ca2+]o (external Ca2+ concentration) saline solutions. The gj was normalized to the initial value (Gj = gj/gj, initial) for each Cx50 cell pair, and the results were pooled for each dataset. To examine the effect of CaM inhibitor or the candidate CaM-binding peptide on gj, Cx50-N2a cells were pre-treated with 1 μM CDZ (calmidazolium) (Calbiochem) for 10–15 min or 1 μM of the synthetic Cx50 mimetic peptides (Cx50p141–166 or SCx50p) were added to both whole-cell patch pipettes.
NMR spectroscopy
NMR experiments were performed with a Varian INOVA 600 MHz NMR spectrometer at 37 °C. (1H,15N)-HSQC (heteronuclear single-quantum coherence) data were collected with 2048 complex data points and a spectral width of ~ 14 p.p.m. in the 1H dimension, and 128 increments and a spectral width of 33 p.p.m. in the 15N dimension. A sample of 0.26 mM uniformly 15N-labelled CaM in buffer consisting of 5 mM Mes, 10 mM Bis-Tris (pH 6.5), 5 mM DTT (dithiothreitol), 0.1 mM sodium azide, 10 mM CaCl2 and 10 % 2H2O was titrated with peptide stock solutions (~ 5 mM) of the peptides Cx50p141–166 or SCx50p in the same buffer. NMR data were processed using NMRPipe [24] and analysed using Sparky 3 (T.D. Goddard and D.G. Kneller, University of California, San Francisco).
The pulse-field gradient diffusion NMR spectra were collected with a modified PG-SLED pulse sequence [25] on a 600 MHz Varian INOVA spectrometer. In each FID (free induction decay) were contained 8k complex data points with a spectral width of ~ 13 p.p.m. The pulse-field gradient level (G) was arrayed from ~ 0.2 to ~ 31.0 Gauss/cm with a pulse gradient time (δ) of 5 ms and a diffusion time (Δ) of 112.5 ms. The data were processed using FELIX (Accelrys). The relationship between the NMR signal intensity (A) and the diffusion constant (D) follows eqn (1):
| (1) |
where A0 is the signal intensity when pulse gradient is not used and γ is the gyromagnetic ratio of the proton. In the data processing, the signal intensity as a function of pulse-field gradient level was fitted with eqn (2) using KaleidaGraph 3.5 (Synergy). The values discussed in the present paper were all determined by fitting with a linear correlation coefficient ≥0.999.
| (2) |
where C is a constant [ = γδ(Δ − δ/3)D], the diffusion constant and the hydrodynamic radii R were obtained by comparing the C values of different molecules measured under identical conditions using eqns (3) and (4):
| (3) |
| (4) |
where D0, R0 and C0 are the diffusion constant, hydrodynamic radius and the measured C value of lysozyme. The hydrodynamic radius and diffusion constant of lysozyme have been reported previously [26]. The gradients were calibrated at 25 °C on the residual 1H signal in a sample of 99.9 % 2H2O, using the published value of (1.902 ± 0.002) × 10−9 m2 · s−1 for the self-diffusion coefficient of 1H2HO at 25 °C [27].
NMR samples contained 0.23 mM CaM in 10 % 2H2O, 100 mM KCl and 10 mM imidazole at pH 6.5, with 10 mM Ca2+ for Ca2+-bound. The intensities of the protein signals were integrated from the methylene and methyl region of ~ 2 p.p.m. spectral width ( −0.2–1.8 p.p.m.). The integrated regions for each species were carefully selected to avoid or reduce interferences from buffer signals. The integrated intensities were normalized further to minimize the experimental errors from phase adjustment and baseline correction during the processing.
Chemical-shift perturbations (Δ) of the (1H,15N)-HSQC spectra with and without peptide were calculated using both 1H and 15N chemical shifts (δ) as shown in eqn (5):
| (5) |
Fluorescence measurements
Steady-state fluorescence spectra were recorded using a QM1 fluorescence spectrophotometer (PTI) in a 1-cm-pathlength cell with a xenon short-arc lamp at 25 °C. The fluorescence emission spectra were acquired between 400 and 600 nm with an excitation wavelength at 335 nm for D-CaM. The slit widths were set at 4 nm for excitation and 8 nm for emission. A solution (0.8 ml) containing 0.25–2 μM D-CaM in 10 mM Tris/HCl (pH 7.4) and 100 mM KCl, with 5 mM Ca2+ or 5 mM EGTA was titrated by gradually adding 5–10 μl aliquots of the peptide stock solution (0.25–0.8 mM) in the same buffer. The binding constant of the synthetic peptide to modified CaM was obtained with a 1:1 binding model by fitting normalized florescence data as described previously [11,12].
The fluorescence anisotropy of D-CaM before and after addition of Cx50p141–166 was measured with excitation at 335 nm and emission at 500 nm. An integration time of at least 20 s (20 points/s) was used to record the fluorescence signal, and all measurements were repeated at least three times by following established protocols [28,29].
Owing to domain-specific distribution of phenylalanine and tyrosine residues in CaM, the equilibrium Ca2+-binding constants for CaM were determined by monitoring fluorescence of intrinsic phenylalanine (λex = 250 nm; λem = 280 nm;) for N-domain or tyrosine (λex = 277 nm; λem = 320) for C-domain at 25 °C as described previously [30]. For N- or C-domain Ca2+-binding constants, 5–8 μM CaM was titrated with 5–10 μl aliquots of 50 or 15 mM Ca2+ stock solution in Ca2+ equilibrium buffer with 50 mM Hepes (pH 7.4), 100 mM KCl, 5 mM NTA (nitrilotriacetic acid) and 0.05 mM EGTA. The same method was applied for the 1:1 CaM/peptide mixture. Free Ca2+ at each titration point was determined with the Ca2+ dye Oregon Green 488 BAPTA-5N [5-nitro-1,2-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid] (0.2 μM; Kd = −21.7 ± 2.5 μM [12]) at an emission of 520 nm with excitation of 495 nm. The free concentration was obtained using eqn (6):
| (6) |
where F is the fluorescence intensity of the dye at each titration point, [Ca2+]free is the concentration of free ionized calcium in solution, and Fmin and Fmax represent the dye fluorescence intensities of Ca2+-free form and Ca2+-saturated dye respectively. The Ca2+-binding affinity was obtained by fitting to the non-linear Hill equation:
| (7) |
where f is the fractional change of intrinsic fluorescence intensity, Kd is the Ca2+ dissociation constant, and h is the Hill coefficient.
CD spectroscopy
CD spectra were recorded in the far-UV (190–260 nm) range on a Jasco-810 spectropolarimeter at ambient temperature (25 °C) using a 0.1-cm-pathlength quartz cuvette. The measurements of CaM and the CaM–Cx50p141–166 complex (10 μM) were made in 10 mM Tris/HCl (pH 7.5) and 100 mM KCl with 5 mM CaCl2 or 5 mM EGTA. All spectra presented were averaged for at least 15 scans, and the background signal from the buffer was removed from the sample signals. The far-UV CD spectra of the peptide in different percentages of TFE (trifluoroethanol) were obtained using a 20 μM concentration of Cx50p141–166 in the same buffer. The secondary structures of the peptides were calculated with the online secondary-structure prediction server DICHROWEB [31]. The α-helical content of peptides was predicted using the Agadir algorithm [32–34].
Mass spectrometry
The MALDI–TOF-MS analysis was carried out on an Applied Biosystems 4800 plus MALDI–TOF/TOF analyser mass spectrometer. The data were acquired in a linear positive mode with SA (sinapinic acid) as matrix for CaM (50 μM) and the CaM–Cx50p141–166 complex. The molecular mass of Cx50p141–166 was also confirmed by MALDI–TOF-MS with CHCA (α-cyano-4-hydroxycinnamic acid) as matrix. CaM (50 μM) and Cx50p141–166 (50 μM) were mixed in 50 mM Tris/HCl (pH 7.5) and 100 mM KCl, and a 1 μl mixture was added with 10 μl of saturated SA solution and then dried on the MALDI plate for the measurement.
RESULTS
A putative CaMBD in the single cytoplasmic loop of Cx50
The CaM-binding site predication server, Calmodulin Target Database, was used to identify potential CaMBDs within Cx50, by taking into account the hydropathy, α-helical propensity, residue mass, residue charge, hydrophobic residue content and helical class [14]. The highest predictive score was assigned to a stretch of sequences (residues 141–166; Figure 1) in the C-terminal portion of the intracellular loop of Cx50. Sequence alignments with previously characterized CaMBDs in rodent Cx43 [11] and sheep Cx44 (human Cx46) [12], and secondary-structure prediction, identified common features among the three α-family Cxs that included: (i) the spacing of hydrophobic residues with bulky side chains, often used as anchor in CaM target complexes [35–37], followed a 1–5–10 pattern; (ii) all of the domains exhibited strict conservation in positions with positively charged residues, which help to drive the formation of CaM target complexes via electrostatic interactions [38]; and (iii) the CaMBDs were predicted to have a predominantly α-helical structure. Helical wheel analysis further suggests the clustering of hydrophobic residues on one side and the frequent occurrence of charged residues on the opposite side. A peptide corresponding to the predicted CaMBD was subsequently synthesized for the functional and biophysical studies described below.
Figure 1. Cxs membrane topology and the putative CaM-binding sites.
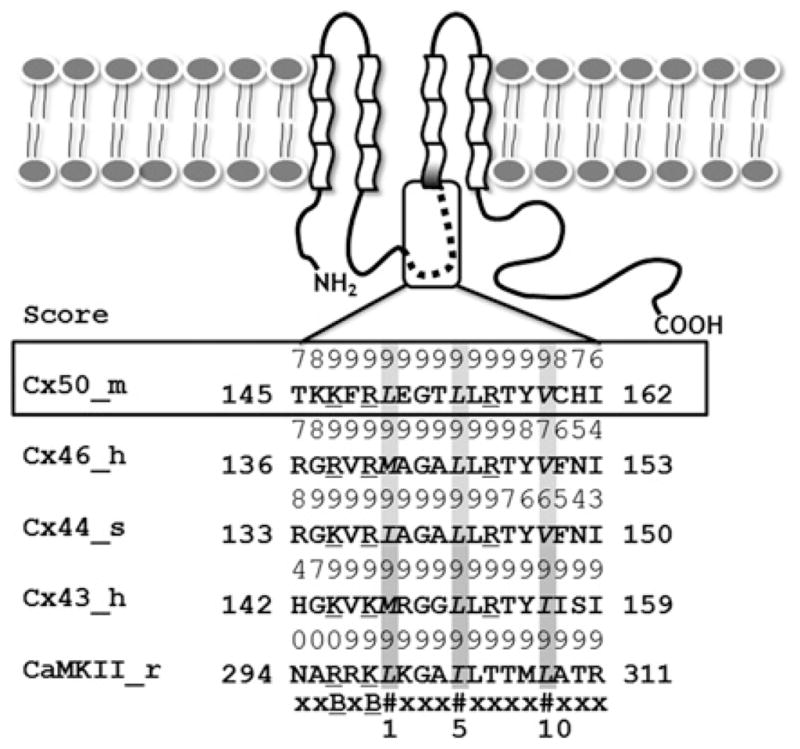
The α-class of Cxs are composed of four transmembrane segments, two extracellular loops, one cytoplasmic loop, a short N-terminus and a much longer C-terminal tail. The predicted CaM-binding sites are located in the second half of the intracellular loop between TM2 and TM3. The numeric score (1–9) represents the probability of an accurate prediction of high-affinity CaM-binding sites. The CaM-binding sequences identified in the α-class Cxs are similar to those of CaMKII. All of the aligned sequences fit the 1–5–10 CaM-binding mode subclass, where each number represents the presence of a hydrophobic residue. m, mouse; h, human; s, sheep; r, rat; #, hydrophobic residues (highlighted in grey); B, basic residues (underscored).
Ca2+-dependent uncoupling of Cx50 gap junctions requires CaM
To determine whether Cx50 gap junctions like those comprising Cx43 [11,39] and Cx46 [12] are regulated by [Ca2+]i, we first performed dual whole-cell patch-clamp experiments in Cx50-N2a cells. Perfusion with saline solution containing 1 μM ionomycin+1.8 mM [Ca2+]o induced a time-dependent reduction in Gj (Figure 2A and Table 1); we have shown previously that such treatment effects an elevation of [Ca2+]i in HeLa cells [40]. Omission of 1.8 mM CaCl2 from the bath saline prevented the Cx50 gap junction uncoupling response in the presence of 1 μM ionomycin.
Figure 2. Ca2+- and CaM-dependence of Cx50 gap junction uncoupling.
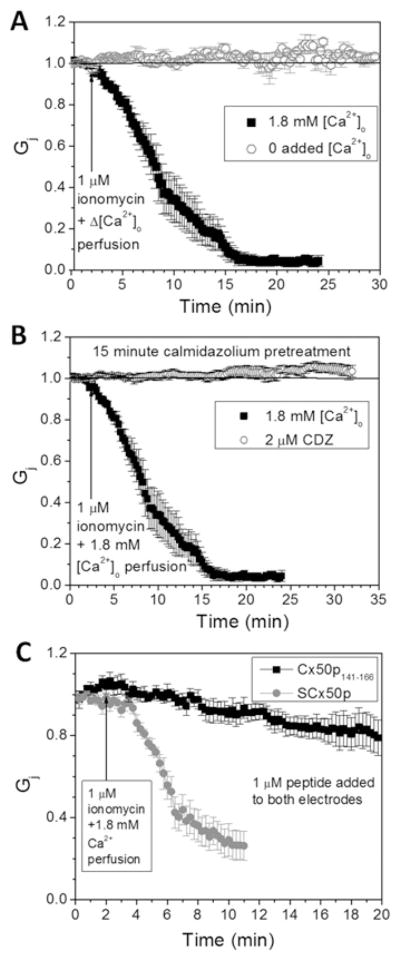
(A) Cx50-N2a cell pair superfusion with 1 μM ionomycin bath saline with or without 1.8 mM CaCl2 demonstrates a time-dependent uncoupling of Cx50 gap junctions that requires 1.8 mM [Ca2+]o. (B) Pre-treatment with 2 mM CDZ for 10–15 min before patch-clamp analysis completely prevented the 1.8 mM [Ca2+]o-dependent uncoupling response. (C) Inclusion of 1 μM Cx50p141–166 in both whole-cell patch pipettes prevented 80 % of the Ca2+/CaM-dependent decline in Cx50 Gj. In contrast, experiments performed with SCx50p prevented only 20 % of the 1.8 mM [Ca2+]o–CaM-dependent reduction in Cx50 Gj. The average ± S.E.M. initial gj values for all experimental groups are displayed in Table 1.
Table 1.
Electrophysiological measurement
| Experiment | Initial gj (nS) | N |
|---|---|---|
| 1.8 mM CaCl2 | 9.90 ± 2.90 | 5 |
| 0 mM CaCl2 | 14.74 ± 6.51 | 4 |
| 2 mM CDZ | 16.79 ± 2.95 | 4 |
| Cx50p141–166 | 7.05 ± 2.53 | 7 |
| SCx50p | 3.36 ± 1.80 | 6 |
gj = Ij/V j (Ohm’s law, junctional current divided by transjunctional voltage). N = number of cell pairs recorded from each dataset.
To test whether the Ca2+-dependent decrease in Cx50 Gj produced by addition of 1 μM ionomycin+1.8 mM [Ca2+]o required CaM, Cx50-N2a cell cultures were acutely (≤15 min) treated with CDZ (2 μM), a CaM inhibitor with a Kd of ~ 1 μM [41]. Pre-treatment with CDZ prevented the Ca2+-dependent decrease in Gj (Figure 2B).
A more specific test for a role of Cx50 CaMBD in mediating the Ca2+-dependent inhibition of this Cx was to add the 26-mer Cx50 mimetic peptide corresponding to residues 141–166 of the cytoplasmic loop to the Cx50-transfected N2A cells. A scrambled peptide containing a randomized sequence of the same amino acids was also utilized as a control. The addition of Cx50p141–166, but not the scrambled peptide, prevented the decline in Gj (Figure 2C). Overall, these electrophysiological data support the hypothesis that an elevation of [Ca2+]i can uncouple Cx50 gap junctions in a CaM-dependent manner mediated by the Cx50 141–166 domain.
To examine the molecular basis for the Ca2+-dependent decrease in Cx50 Gj, gap junction channel recordings were analysed in two poorly coupled cell pairs. Both experiments exhibited a maximum of three open channels (N) and their cumulative open probability (N · Po) declined from 1.21 open channels per unit time to 0 upon exposure to 1 μM ionomycin and 1.8 mM [Ca2+]o saline (Figure 3A). Unitary Cx50 gap junction channel currents remained constant (314 ± 18 pS, mean ± S.D., n = 14) during the decline in Po, indicative of a conformational gating type behaviour (Figure 3B).
Figure 3. Mechanistic basis for Ca2+/CaM-dependent Cx50 gap junction uncoupling.

(A) The product of the number of open channels (N) and open probability (P o) from two poorly coupled Cx50-N2a cell pairs is plotted as a function of time and 1 μM ionomycin +1.8 mM [Ca2+]o superfusion. N · Po declined from an average control value of 1.21 open channels to 0 within 2 min of ionomycin/CaCl2 saline superfusion. (B) Cx50 gap junction channel current recordings from one experiment are shown for the time points indicated by the circles in (A). The number and duration of the open channels declined progressively to 0 without any apparent reduction in the single-channel conductance (314 pS), indicative of closure of a Ca2+–CaM-dependent gate without a block of the ion-permeation pathway.
Ca2+-dependent specific interaction between Cx50 CaMBD and CaM revealed by NMR
After confirming the requirement for CaM in the Ca2+-dependent regulation of the Cx50 gap junctions, we further asked the question whether or not there is a direct interaction between the predicted CaMBD (or Cx50p141–166) and CaM. The formation of the CaM–Cx50p141–166 complex was first confirmed by MALDI–TOF-MS (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/435/bj4350711add.htm). In the presence of Ca2+, a distinct peak representing the CaM–Cx50p141–166 complex (theoretical molecular mass 19.93 kDa; experimental molecular mass 20.02 kDa) was detected.
To identify residue-specific changes in the conformation of CaM during complex formation with Cx50p141–166, we carried out (1H,15N)-HSQC NMR experiments. Unlabelled Cx50p141–166 stock solution was gradually titrated into 15N-labelled CaM. Under Ca2+-loaded conditions, the HSQC spectrum of Ca2+-saturated CaM (holo-CaM) underwent significant changes on addition of 1 mol. eq. of Cx50p141–166 (Figure 4A). In contrast, SCx50p, which is predicted to have no binding affinity for CaM, was similarly added to 15N-labelled holo-CaM. This scrambled peptide failed to induce chemical-shift changes in CaM (Figure 4B), therefore ruling out the possibility of non-specific interactions between peptide and CaM.
Figure 4. Monitoring the interaction between CaM and Cx50p141–166 by (1H,15N)-HSQC spectroscopy.

(A) An overlay of HSQC spectra of holo-CaM (black) with the spectrum of the holo-CaM–Cx50p141–166 (grey). (B) An overlay of the HSQC spectra of holo-CaM (black) with the spectrum of the holo-CaM–SCx50p (grey). (C) Chemical-shift perturbation in CaM induced by addition of 2-fold molar excess of Cx50p141–166. The weight-average chemical-shift change (Δδ) was calculated using eqn (5). Residues with Δδ> 0.1 p.p.m. were mapped to the three-dimensional structure of holo-CaM (PDB code 3CLN). (D) The chemical-shift change of Gly33 during titration of holo-CaM with Cx50p141–166. The disappearance of the peak (free form) was accompanied by the appearance of the corresponding peak (bound form) at a new position. (E) Structural basis of the difference in directionality of chemical shift change. The structure of the CaM–CaMKII complex (PDB code 1CDM) is shown with the peptide (inside) and the CaM residues (outside). Leu116 and Met145 of CaM are within 5 Å of Lys298p and Lys300p of the peptide respectively. These peptide positions correspond to Lys146p and Arg148p for Cx43, and Arg149p and Glu151p for Cx50, as can be seen in the superposition of their sequences. In Cx50, Glu151p may be pulled away from Met145 by Lys147p. In contrast, in Cx43, Arg148p may be pushed towards Met145 by Lys144p. These differences in peptide sequences can cause changes of chemical shifts in different directions.
By analysing the NMR chemical shifts of CaM–peptide complexes deposited in the Biological Magnetic Resonance Bank (BMRB codes 1634, 4270, 5480, 5770, 5893, 5896, 15470 and 16465), we identified some significant changes in chemical shifts [δ(CaM) − δ(complex) > 1 S.D.] that are common to all complexes such as the 1H shifts of Ala57 and Met71, and 15N shift of Ala128. In the classical CaM–CaMK (Ca2+/CaM-dependent protein kinase) IIα peptide structure with 1–10 binding mode, Met71 and Ala128 are in the hydrophobic pockets of CaM and are within 5 Å (1 Å = 0.1 nm) of the peptide. Ala57 occupies the second position of the second Ca2+-binding loop, suggesting that peptide binding leads to conformational change in this loop. In the CaM–Cx50p141–166 complex, Ala57 and Met71 have δH values of 0.29 and 0.05 p.p.m. respectively, whereas Ala128 has a δN of 0.21 p.p.m., consistent with Cx50p141–166 bound to Ca2+–CaM.
Upon the formation of a Ca2+/CaM–Cx50p141–166 complex, the most significant perturbations, with weighted average chemical shift changes greater than 0.1 p.p.m., were observed in the N-lobe (e.g. Ser17, Thr26, Thr29, Glu31, Leu39 and Ala57), the C-lobe (e.g. Glu82, Asp93, Leu105, Arg106, Ala147 and Lys148) and the linker region (e.g. Asp64 and Phe68), indicating that Cx50p141–166 causes a global conformation changes in CaM. A detailed residue–residue plot of chemical-shift perturbation is shown in Figure 4(C). The overall change of the N-lobe residues (2.74 p.p.m.) was greater than in the C-lobe residues (1.78 p.p.m.). During the titration, we observed the simultaneous appearance of two peaks representing the same residue. For example, a progressive disappearance of the amide signal of Gly33 representing unbound CaM was accompanied by the concomitant emergence of a new set of peaks from the CaM–Cx50p141–166 complex (Figure 4D). Such a slow exchange process, which occurs when the exchange rate of the bound and unbound states is smaller than the amide frequency difference, often heralds a high affinity protein–protein or protein–peptide association (i.e. interactions with submicromolar affinities) [12,42].
Probing the CaM–peptide complex state by diffusion NMR
Pulse-field gradient NMR was performed to determine the diffusion constant of the CaM–Cx50p141–166 complex, enabling us to assess the overall size of this complex in solution. The diffusion constant was determined to be (12.4 ± 0.4) × 107 cm/s (Figure 5). By assuming a spherical shape of the complex, the calculated size of the Ca2+/CaM–Cx50p141–166 complex was determined to be 17.3 ± 0.6 Å, a value comparable with the hydrodynamic radii of CaM in complex with well-known targets such as smMLCK (smooth muscle myosin light chain kinase) (17.9–21.8 Å), PDE (phosphodiesterase) (18.8–22.3Å) and CaMKI (21.2 Å) [43]. The hydrodynamic radius of CaM (22.6 ± 0.6 Å) was decreased by 23 % on formation of the CaM–Cx50p141–166 complex. This size conforms to a collapsed structure of the complex that involves the unwinding of the central helix within CaM to embrace the target peptide.
Figure 5. Hydrodynamic radii of CaM–Cx50p141–166 complex determination by pulse-field gradient NMR.
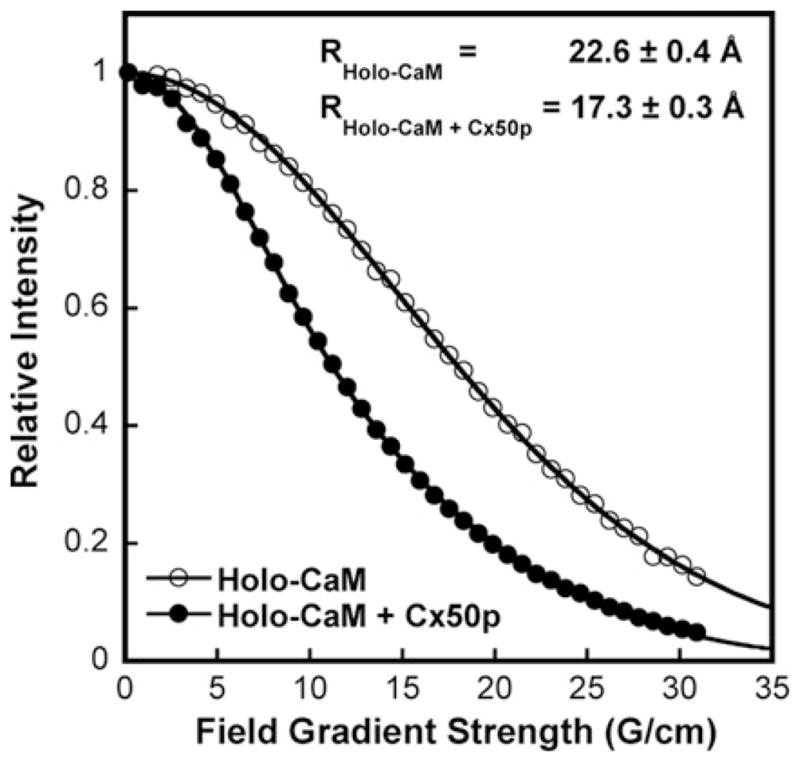
The NMR signal decay of holo-CaM (○) and holo-CaM–Cx50p141–166 complex (●) as a function of field strength. The calculated hydrodynamic radii of the CaM and complexes are indicated on the top.
Revealing CaM–peptide interactions with CD spectroscopy
CD spectra also confirmed the structural and conformational changes during the formation of the CaM–Cx50p141–166 complex. As indicated in Figure 6(A), the addition of Cx50p141–166 to holo-CaM at a 1:1 molar ratio resulted in a ~ 6 %increase in the negative ellipticity at 222 nm. The hydrophobic environment in the peptide-binding pocket of holo-CaM could be the reason for the increase in the negative ellipticity attributed to the peptide itself. In contrast, a 5 % decrease at 222 nm and a 20 % decrease at 208 nm in the CD signal intensity were observed after the addition of Cx50p141–166 to apo-CaM. Further exploring the interaction of apo-CaM with Cx50p141–166 using NMR was not possible because of limited solubility of the peptide in 5 mM EGTA (see Supplementary Figure S2 at http://www.BiochemJ.org/bj/435/bj4350711add.htm).
Figure 6. CD studies of the interaction between CaM and Cx50p141 – 166.

(A) Far-UV CD spectra of CaM in the presence of 5 mM EGTA (○, apo-CaM) or CaCl2 (□, holo-CaM), and a CaM–Cx50p141–166 (1:1) mixture with 5 mM EGTA (●, apo-CaM–Cx50p141–166) or CaCl2 (■, holo-CaM–Cx50p141–166). (B) Far-UV spectra of the synthetic peptide Cx50p141–166 with addition of TFE (0–80 %). The inset shows calculated α-helical content as a function of TFE concentration for the peptides Cx50p141–166 (■), Cx44p129–150 (▲) and Cx43p136–158 (●). (C) Far-UV spectra of the synthetic peptide Cx50p141–166 with (●) or without (○) addition of CaM.
We examined further the secondary structure of Cx50p141–166 under different solvent conditions. TFE is known to induce α-helical formation of peptides, and the helical content in the presence of TFE usually reflects its helical propensity [44]. Cx50p141–166 becomes predominantly helical (>90 %) at TFE ≥30 % (v/v) (Figure 6B, inset), suggesting that the Cx50 CaM-binding sequence readily adopts an α-helical configuration. Cx50, Cx44 and Cx43 exhibit 94, 55 and 33 % helical conformation respectively in the presence of 30 %TFE (Table 2).
Table 2.
Binding affinities to CaM and α-helicity of the Cx peptides
| Peptide |
Kd (nM)
|
α-Helicity (%)
|
||
|---|---|---|---|---|
| 5 mM Ca2+ | 5 mM EGTA | Prediction | Far-UV CD measurement | |
| Cx50p141–166 | 4.9 ± 0.6 | >8000 | 1.11 | 94 |
| Cx44p129–150 | 49 ± 3.0 | >5000 | 1.20 | 55 |
| Cx43p136–158 | 860 ± 20 | nd | 0.52 | 33 |
All of the experiments were conducted three times (n = 3). nd, not detectable. Kd measurements were made of D-CaM fluorescence in 50 mM Tris/HCl (pH 7.5) and 100 mM KCl. The prediction of the helical content of peptides used the Agadir algorithm. Far-UV CD measurements were conducted using 30 % TFE in 10 mM Tris/HCl (pH 7.5) and 100 mM KCl.
The peptide in the absence of CaM has only 7 % α-helix and 72 % random coil. The addition of peptide into CaM resulted in the addition of helicity as shown in Figure 6(A). Figure 6(C) shows that our difference CD spectrum of the CaM in the presence and absence of the peptide exhibits two negative minima at 208 and 222 nm. To understand the origin of the additional helicity due to the formation of the CaM–Cx50p141–166 complex, we analysed the helical contents in the X-ray structure of Ca2+-loaded calmodulin alone (PDB code 3CLN) and the complex structure of CaM–CaMKII peptide (PDB code 1CDM) since our chemical-shift studies suggest that CaM binds to Cx50p141–166 in a similar binding mode as to the peptide of CaMKII (Figure 4E). CaM does not exhibit any additional change in its helical content upon formation of the complex with a helicity of ~ 62 %. Thus the complex-induced helicity is attributed to the conversion of peptide to be ~ 20 % helical.
Determination of the CaM–peptide binding affinity by fluorescence spectroscopy
D-CaM has frequently been used to determine the binding affinity of the CaM–peptide interaction due to its sensitivity to the changes in the surrounding chemical environment and its ease of preparation [22]. The dansyl moiety has an emission maximum at ~ 500–510 nm, a range in which the signals from intrinsic aromatic residues are negligible. We first examined the dansyl fluorescence anisotropy changes to confirm the formation of complex in the low-micromolar range. Fluorescence anisotropy is dependent on the rotational correlation time of fluorophore in the sample, and is often used to reflect the hydrodynamic properties of biomolecules [45]. The increase in anisotropy often arises from slower tumbling of the fluorophore and thus reports the formation of a larger complex. Indeed, upon addition of 1 mol. eq. of Cx50p141–166, the anisotropy of the dansyl moiety within D-CaM increased from 0.083 to 0.125, suggesting the association of Cx50p141–166 with Ca2+–CaM (see Supplementary Figure S3 at http://www.BiochemJ.org/bj/435/bj4350711add.htm).
We then carried out the titration of peptide with D-CaM by monitoring the fluorescence emission between 400 and 600 nm. In the presence of Ca2+, D-CaM showed a fluorescence maximum at 500 nm. With the addition of Cx50p141–166, the dansyl fluorescence emission maximum blue-shifted to 474 nm with a concomitant enhancement of its fluorescence intensity (Figure 7A), implying that the dansyl group entered a more hydrophobic environment upon complex formation with peptide. In contrast, in the presence of EGTA (Figure 7B), although the dansyl fluorescence intensity also increased, the fluorescence maxima changed less than in the presence of Ca2+. The data confirm the CD signal change on the addition of peptide to apo-CaM. By fitting the titration curve with a 1:1 binding mode, we obtained a Kd of 4.9 ± 0.6 nM for the interaction between Ca2+–CaM and Cx50p141–166 (Table 2).
Figure 7. Interaction of Cx50p141–166 with D-CaM monitored by steady-state fluorescence.
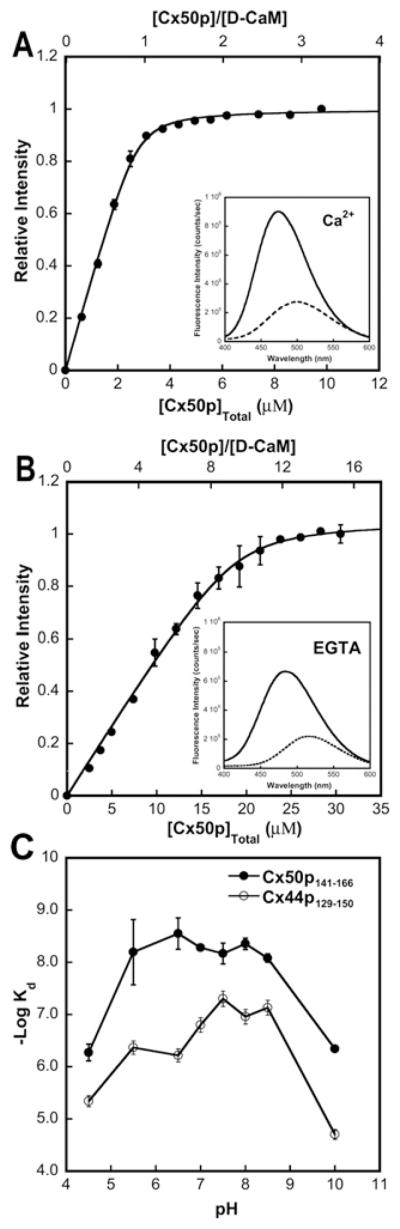
(A) The titration curve of D-CaM (2 μM) with Cx50p141–166 in the presence of 5 mM Ca2+ in a buffer consisting of 50 mM Tris/HCl (pH 7.5) and 100 mM KCl. The inset shows the fluorescence spectrum of D-CaM in the absence (broken line) or the presence (continuous line) of an equivalent molar concentration of Cx50p141–166. (B) Titration curve of D-CaM (2 μM) with Cx50p141–166 in the presence of 5 mM EGTA in a buffer consisting of 50 mM Tris/HCl (pH 7.5) and 100 mM KCl. The inset shows the fluorescence spectrum of D-CaM in the absence (broken line) or the presence (continuous line) of an equivalent molar concentration of Cx50p141–166. (C) pH-dependence of Cx50p141–166 and Cx44p129–150 binding to CaM. The binding affinities were derived from the peptide titration curves of D-CaM at various pH values. All experiments were conducted in triplicate.
To investigate further the possible role of pH on CaM–peptide association, the effect of varying pH on the binding affinity of CaM for Cx50p141–166 was also examined. As shown in Figure 7(C), the binding affinity (represented as − log Kd) exhibited a pH-dependent change between pH 5.0 and 10.0. The highest binding affinity was obtained at pH 6.5. The observed pH-dependence of the interaction between CaM and Cx50p141–166 indicates that electrostatic interactions are likely to be the main force driving CaM–peptide complex formation.
Binding of Cx50p141–166 expands the [Ca2+]i-sensing range of CaM
We next performed the Ca2+ equilibrium titration of CaM to obtain macroscopic Ca2+-binding constants for both the N- and C-lobes of CaM by monitoring domain-specific fluorescence changes as described previously [29,30]. The decrease in phenylalanine fluorescence (λex = 250 nm; λem = 280 nm) and the increase in tyrosine fluorescence (λex = 277 nm; λem = 320 nm) report Ca2+ binding to the N-lobe and the C-lobe of CaM respectively. This allows us to examine the effect of peptide binding on the domain-specific Ca2+-sensing capability of CaM. As shown in Figure 8 and summarized in Table 3, the addition of Cx50p141–166 led to a rightward shift of the titration curve, representing the Ca2+ binding to the N-lobe, and concomitantly caused a leftward shift of the titration curve, reflecting the Ca2+ binding to the C-lobe of CaM. Specifically, the Ca2+ affinity of the N-lobe of Cx50p141–166-bound CaM decreased 2-fold, whereas the Ca2+ affinity of the C-lobe increased by ~ 20 %. As a result of Cx50p141–166 binding, CaM responded to subtle changes in [Ca2+]i over a broader range of calcium concentrations.
Figure 8. Domain-specific equilibrium Ca2+ titration of CaM (○) and CaM in complex with Cx50p141–166 (●).

(A) The intrinsic phenylalanine fluorescence (λex = 250 nm; λem = 280 nm) was monitored to report the equilibrium Ca2+-binding constants of the N-lobe of CaM. (B) The intrinsic tyrosine fluorescence (λex = 277 nm; λem = 320 nm) was monitored to report the equilibrium Ca2+-binding constants of the C-lobe of CaM. The free ionized Ca2+ concentration was measured using the Ca2+ indicator dye Oregon Green 488 BAPTA-5N. All experiments were conducted in triplicate in 50 mM Hepes (pH 7.5), 100 mM KCl, 5 mM NTA and 0.5 mM EGTA.
Table 3.
Effects of Cx peptides binding on the metal-binding properties of CaM
| Peptide | N-domain (sites I and II)
|
C-domain (sites III and IV)
|
||
|---|---|---|---|---|
| Kd (μM) | h | Kd (μM) | h | |
| None | 12.0 ± 0.2 | 1.4 ± 0.1 | 3.78 ± 0.03 | 2.14 ± 0.03 |
| Cx50p141–166 | 22.7 ± 0.6 | 0.8 ± 0.1 | 3.18 ± 0.03 | 2.74 ± 0.07 |
| Cx44p129–150 | 11.6 ± 0.1 | 1.5 ± 0.1 | 0.93 ± 0.02 | 2.20 ± 0.10 |
| Cx43p136–158 | 14.5 ± 0.1 | 1.6 ± 0.2 | 1.16 ± 0.02 | 2.10 ± 0.10 |
All of the experiments were conducted three times (n = 3). Kd and the Hill coefficient (h) were obtained by fitting the titration curve to eqn (7). Phenylalanine fluorescence (λex = 250 nm; λem = 280 nm) reports the Ca2+ binding to the N-domain of CaM. Tyrosine fluorescence (λex = 277 nm; λem = 320 nm) reports the Ca2+ binding to the C-domain of CaM.
DISCUSSION
Regulation of Cxs by intracellular Ca2+ and CaM
The results of the present study demonstrate that intracellular calcium is important in the regulation of Cx50 gap junctions [40]. Furthermore, using the CaM inhibitor CDZ as well as a peptide fragment encompassing the predicted CaM-binding region, we were able to demonstrate that this Ca2+-dependent inhibition of Cx50 gap junctions is mediated by the ubiquitous intracellular Ca2+ receptor CaM. Thus CaM appears to mediate the Ca2+-dependent regulation of all three major α-Cxs in the lens, i.e. Cx43, Cx46 and Cx50. Our results also demonstrate that this inhibition of Cx50 gap junction channels by Ca2+ is due to a decrease in channel open time probability as opposed to a reduction in single-channel conductance (Figure 3A). These records provide the first direct evidence for the gated closure of individual Cx gap junction channels by Ca2+–CaM. The average observed gj value of 314 pS is indicative of Cx50, since it is at least double the observed gj for Cx40, Cx43 or Cx45 gap junctions in our Cx50-N2a cell cultures and consistent with the reported 210–290 pS values from reconstituted lens fibre cell membranes or exogenously expressed Cx50 gap junction channels ([46–50], and Q. Xu, Y. Chen, J.J. Yang and R.D. Veenstra, unpublished work).
The possible role of CaM in the chemical gating of the β-family Cxs was first proposed by Peracchia and Wang [51]. CaM co-localizes with Cx32 [9] and directly gates Cx32-containing gap junctions [52]. Török et al. [9] reported that fluorescently labelled CaM derivatives bind to synthetic peptides spanning most of the cytoplasmic sequences from two regions of Cx32: an N-terminal sequence comprising residues 1–21 (Kd = 27 nM) or residues 1–19 (Kd = 1.1 μM); and a C-terminal sequence comprising residues 216–230 (Kd = 2.1 μM) or residues 208–226 (Kd = 3.5 μM). Both exhibit Ca2+-dependent CaM-binding properties [9,10]. They also identified two distinct CaM-binding amino acid sequences in Cx32 within the N- and C-lobes of CaM showing separate functions, suggesting trans-domain or trans-subunit bridging by CaM as a possible mechanism of gap junction gating [52]. In addition, Ahmad et al. [53] using in vitro translation approaches have shown that oligomerization of Cx32 is CaM-dependent, since CaM interacts with Cxs at an early stage of gap junction assembly [8,9]. The C-terminal tail of the CaM-binding site of Cx32 is likely to be involved in the assembly of Cxs and is Ca2+-dependent [8]. Furthermore, Blodow et al. [54] reported that CaM antagonists suppress gap junction coupling of Cx26 in isolated Hensen cells of the guinea pig cochlea. Taken together, [Ca2+]i and CaM play vital roles in regulating gap junctions mediated by both the α- and β-family of Cxs.
Comparison of CaM-binding affinity with the α-family Cxs
Using high-resolution NMR, CD and fluorescence studies, we have shown using a peptide model that CaM interacts with the predicted cytosolic CaMBD of Cx50. Our current studies, as well as previous studies, have clearly shown that CaM exhibits a Ca2+-dependent interaction of all three peptide fragments encompassing the intracellular loop regions of all three lens Cxs. Fluorescence spectroscopy revealed conformational changes of both the peptide and CaM following formation of the CaM–peptide complex. NMR studies demonstrated that the peptide binds to CaM with a 1:1 stoichiometry. It also indicated that the peptide induces structural changes in both the N- and C-terminal domains, as well as in the linker region of CaM and the binding of Cx peptides to CaM reflects a classical embracing mode of interaction. Overall, for these three Cxs, binding of the peptide to CaM decreases the apparent Kd of Ca2+ for CaM, and the Hill coefficient increased. In addition, Cx43 exhibits weaker affinity for CaM than does either Cx50 or Cx44.
Consistent with the CaM–Cx peptide binding affinity studies using fluorescence spectroscopy, our NMR chemical-shift analysis also supports a lower affinity for the CaM–Cx43p136–158 complex. The addition of Cx43p136–158 and Cx50p141–166 results in similar changes in the CaM spectra as shown in Supplementary Figure S4 (at http://www.BiochemJ.org/bj/435/bj4350711add.htm). For most signals, the directionalities of the changes are the same, reflecting the same 1–5–10 binding mode of these peptides to CaM. However, we have observed the slow exchange process for Gly33 in Cx50 shown in Figure 4(D). A similar scenario was observed for Thr29, Ala57 and Thr117 for the CaM–Cx44p129–150 complex [12]. Such a slow exchange process occurs when the exchange rate of the bound and unbound states is smaller than the amide frequency difference, and often heralds a high-affinity protein–protein or protein–peptide association (i.e. interactions with submicromolar affinities) [42] that is consistent with the CaM-binding affinity to Cx50p141–166 and Cx44p129–150. In contrast, the NMR signals of CaM upon addition of Cx43p136–158 exhibits a gradual change in chemical shifts characteristic of the fast exchange regime, suggesting a weaker binding affinity.
Implications for the factors contributing to the CaM-binding affinity to Cxs
There are several positively charged residues, such as Lys147, Arg149 and Arg156, in Cx50 that are conserved in all α-family members. To identify the key factors responsible for the binding affinity of CaM to Cxs, we have determined the CaM-binding affinity of Cx50p141–166 to CaM as a function of pH. The peptide-binding affinity is decreased at pH values lower than 5.5 and greater than 8.5. This trend is very similar to our previous results reported for Cx44 [12]. Both Cx44p129–150 and Cx50p141–166 behave similarly when pH is varied, but CaM binding to Cx50p141–166 is ~ 1 order of magnitude stronger than to the Cx44p129–150. Such a similar pH-dependence suggests that electrostatic interactions following the protonation of aspartate and glutamate and deprotonation of lysine are important for this complex formation.
The difference in CaM-binding affinity of Cx50p141–166 and Cx44p129–150 may originate from the intrinsic sequences encoded in the CaM-binding regions of different Cxs. As shown in Figure 1, there are several differences in the predicted regions especially with variations between conserved residue positions 1 and 5. Cx43 contains two flexible glycine residues instead of the helix-forming residues in Cx44 and Cx50. Such sequence variation reflects their ability to form α-helices that are important for their interaction with CaM [14]. As shown in Figure 6(B) and Table 2, Cx43p136–158 has the lowest α-helix-formation capacity. The addition of TFE (>60 %) induces the highest α-helical content in the Cx50p141–166 and Cx44p129–150. In Figure 6(B), the α-helicity of Cx50p141–166 is higher than that of Cx43p136–158 and Cx44p129–150 reported previously [11,12]. The calculated α-helical content of peptides in TFE exhibits the rank order Cx50p141–166>Cx44p129–150>Cx43p136–158. Interestingly, negatively charged Glu151 located close to Arg149 in Cx50 may stabilize the α-helical conformation of this peptide. In contrast, Cx43 has a positively charged arginine residue at the same location that may disable α-helical conformation. The α-helical content of the CaM-binding peptides observed is consistent with the trend for the CaM-binding affinity results from our fluorescence experiments (Table 2). Both Cx44 and Cx50p141–166 also bind CaM in the absence of Ca2+, as revealed by fluorescence data. The CaM-binding affinities to Cx44 and Cx50 in EGTA are 100- and 1000-fold weaker than those in the presence of Ca2+ respectively. The apo-form of CaM does not interact with Cx43p136–158, which has the least α-helical content. Therefore our results reveal that the α-helicity of the CaM-binding peptide is an important factor in predicting the CaM-binding affinity of these Cxs.
Our detailed NMR analysis has also revealed the differential interactions of CaM with Cx50p141–166 and Cx43p136–158. We observed that, in general, the N-lobe of CaM–Cx50p141–166 has larger chemical-shift changes, whereas the C-lobe has smaller changes than those of CaM–Cx43p136–158, although such interactions belong to a similar 1–5–10 binding mode. In addition, chemical shifts of residues such as Phe12, Lys13, Lys94, Leu116, Ile130 and Met145 of CaM–Cx50p141–166 changed in different directions compared with those of CaM–Cx43p136–158 (see Supplementary Figure S4). Among these residues, Lys94 and Ile130 are on the third and fourth Ca2+-binding loops respectively, and these loops could be flexible and adopt different conformations, leading to different directions of change in chemical shifts. In the structure of CaM–CaMKIIα (PDB code 1CDM), Leu116 interacts with Lys298, which corresponds to Lys146 in Cx43 and Arg149 in Cx50. Similarly, Met145 interacts with Lys300, which corresponds to Arg148 in Cx43 and Glu151 in Cx50 (Figure 4E). Such changes in directions of the chemical shifts of CaM are likely to be due to the intrinsic properties such as the α-helical propensity of the sequences in the CaM-binding regions of Cxs.
Structural and disease implications
The intracellular region regulated by CaM has been reported to be involved in several disease-related mutations. ODDD (oculodentodigital dysplasia) is caused by mutations in Cx43 such as G138R, G138S, G143S, G143D, K144E, V145G, M147T, R148G, R1481, T154A and T154N [55], overlapping with the CaM-binding region identified previously [11]. There have been no reports of point mutations in the CaMBD of Cx50 associated with cataract formation. However, Beahm and Hall [56] have reported that the Cx50 H161N mutant does not form detectable hemichannels, but forms gap junctions indistinguishable from wild-type. Interestingly, Toloue et al. [57] identified three nonfunctional mutations in Cx50, T157C, H161C and E169C, that failed to function homotypically or in heterotypic pairings with wild-type Cx50. The threonine residue at position 157 is highly conserved and this residue has been shown previously to be functionally essential for all Cxs tested, including Cx26, Cx43 and Cx50 [58]. Mutation of a conserved threonine residue in the third transmembrane helix of α- and β-Cxs creates a dominant-negative closed gap junction channel [58]. It is possible that such mutations may compromise the regulation of this Cx by CaM. The testing of such mutational effect on Cx50 function is currently underway in this laboratory.
The molecular scenario of CaM-mediated regulation of the α-class of gap junctions seems to be quite different from the β-class, probably due to the fact that the α-class Cxs lack the N-terminal glycine hinge and possess longer intracellular loops and C-termini than the β-class Cxs. Indeed, the CaM-targeting sequences in the α-class Cxs, such as rodent Cx43, sheep Cx44 (or human Cx46) and mouse Cx50, as revealed in the present study, are in the cytoplasmic loop connecting TM2 and TM3, in close proximity to the cytoplasmic loop–TM3 interface. The apparent juxtaposition of the Cx32 N-terminal CaMBD to TM1 and of the Cx43 cytoplasmic loop CaMBD to TM3 may facilitate the closing of the Cx channel pore since both TM1 and TM3 are suggested to be involved in forming the transmembrane pore while the N-terminus loops back to form a pore cytoplasmic vestibule.
The recently determined three-dimensional structure of the Cx26 gap junction channel at 3.5 Å resolution provides insights into the gating mechanism of the β-class of gap junctions at atomic resolution [59]. In this tsuzumi (Japanese drum)-shaped structure, the permeation pathway is mainly defined by the short NTH (N-terminal helix) (residues 2–10) that lines the pore funnel of gap junction channel and TM1 that constitutes the pore-lining helix. This structure suggests that the molecular identity of the channel ‘plug’ observed using electron microscopy [60] is likely to be the NTH. However, owing to the invisibility of both the cytoplasmic loop and the C-terminus in the crystal structure, both of which play important roles in the chemical gating of gap junctions [61], it is still too premature to explain the chemical gating mechanism with the current structure.
Given that the N-terminal CaMBD in Cx32 overlaps with the NTH that is believed to form the channel ‘plug’ in β-class gap junctions, it is conceivable that the Ca2+-dependent binding of CaM to NTH might function as ‘cork’ to block the channel by the complex or by inducing conformational changes in the NTH. However, since the NTH is lined interior to the channel to form a funnel, its accessibility to CaM in the assembled channel remains to be confirmed.
Despite the absence of X-ray structural information about the intracellular loop of Cxs [59], we have provided detailed structural information about the association of CaM with all three α-family Cx members. Our results provide the first direct evidence that CaM binds to a specific region of the lens gap junction Cxs Cx43, Cx44 and Cx50 in a Ca2+-dependent manner [11,12]. Our data suggest a common conformational gating mechanism by which the Ca2+-dependent inhibition of the α-class of gap junction proteins is mediated by the direct association of an intracellular loop region of these proteins with Ca2+–CaM [6,7,39,40].
Supplementary Material
Figure S1 Monitoring CaM–Cx50p141–166 complex formation by MS
The MALDI–TOF-MS spectra of the free form of CaM (~17 kDa) and the complex form of CaM–Cx50p141–166 (~20 kDa). The inset shows the molecular mass of Cx50p141–166.
Figure S2 Monitoring the interaction between apo-CaM and Cx50p141–166 by (1H,15N)-HSQC spectroscopy
An overlay of HSQC spectra of apo-CaM (red) with the spectrum of the apo-CaM–Cx50p141–166 (green).
Figure S3 Dansyl fluorescence anisotropy
D-CaM (1 μM) was titrated with Cx50p141–166 in 50 mM Tris/HCl (pH 7.5), 100 mM KCl and 5 mM CaCl2. The fluorescence anisotropy were measured at λex = 335 nm and λem = 495 nm with an integration time of 20 s. The inset shows the anisotropy change for D-CaM (free), and the complex of D-CaM–Cx50p141–166 (bound).
Figure S4 Comparison of the α-family of Cxs
An overlay of the HSQC spectra of holo-CaM (yellow), holo-CaM–Cx50p141–166 (green) and holo-CaM–Cx43p136–158 (violet). Arrows indicate signals that shifted to the same direction because of identical binding modes, whereas rectangles highlight some exceptions.
Acknowledgments
FUNDING
This work is supported in part by the National Institutes of Health [grant numbers EY-05684 (to C.F.L. and J.J.Y.), HL-042220 (to R.D.V.) and GM-081749 (to J.J.Y.)].
Abbreviations used
- BAPTA-5N
5-nitro-1,2-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid
- [Ca2+]i
intracellular Ca2+ concentration
- [Ca2+]o
external Ca2+ concentration
- CaM
calmodulin
- apo-CaM
Ca2+-chelated CaM
- D-CaM
dansylated CaM
- holo-CaM
Ca2+-saturated CaM
- CaMBD
CaM-binding domain
- CaMK
Ca2+/CaM-dependent protein kinase
- CDZ
calmidazolium
- CHCA
α-cyano-4-hydroxycinnamic acid
- Cx
connexin
- DTT
dithiothreitol
- gj
junctional conductance
- HSQC
heteronuclear single-quantum coherence
- MALDI
matrix-assisted laser-desorption/ionization
- NTA
nitrilotriacetic acid
- NTH
N-terminal helix
- SA
sinapinic acid
- TFE
trifluoroethanol
- TM
transmembrane region
- TOF
time-of-flight
- Vj
transjunctional voltage
Footnotes
AUTHOR CONTRIBUTION
Jenny Yang, Charles Louis and Richard Veenstra conceived the project and designed experiments. Yanyi Chen, Yubin Zhou, Xianming Lin, Hing-Cheung Wong, Qin Xu and Jie Jiang collected and analysed data. Yanyi Chen, Jenny Yang, Charles Louis and Richard Veenstra wrote the paper with input from Yubin Zhou, Hing-Cheung Wong, Jie Jiang, Siming Wang and Monica Lurtz.
References
- 1.Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 2.Evans WH, Martin PE. Gap junctions: structure and function. Mol Membr Biol. 2002;19:121–136. doi: 10.1080/09687680210139839. [DOI] [PubMed] [Google Scholar]
- 3.Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- 4.Gandolfi SA, Duncan G, Tomlinson J, Maraini G. Mammalian lens inter-fiber resistance is modulated by calcium and calmodulin. Curr Eye Res. 1990;9:533–541. doi: 10.3109/02713689008999593. [DOI] [PubMed] [Google Scholar]
- 5.Crow JM, Atkinson MM, Johnson RG. Micromolar levels of intracellular calcium reduce gap junctional permeability in lens cultures. Invest Ophthalmol Visual Sci. 1994;35:3332–3341. [PubMed] [Google Scholar]
- 6.Churchill GC, Lurtz MM, Louis CF. Ca2+ regulation of gap junctional coupling in lens epithelial cells. Am J Physiol Cell Physiol. 2001;281:C972–C981. doi: 10.1152/ajpcell.2001.281.3.C972. [DOI] [PubMed] [Google Scholar]
- 7.Lurtz MM, Louis CF. Calmodulin and protein kinase C regulate gap junctional coupling in lens epithelial cells. Am J Physiol Cell Physiol. 2003;285:C1475–C1482. doi: 10.1152/ajpcell.00361.2002. [DOI] [PubMed] [Google Scholar]
- 8.Peracchia C, Sotkis A, Wang XG, Peracchia LL, Persechini A. Calmodulin directly gates gap junction channels. J Biol Chem. 2000;275:26220–26224. doi: 10.1074/jbc.M004007200. [DOI] [PubMed] [Google Scholar]
- 9.Török K, Stauffer K, Evans WH. Connexin 32 of gap junctions contains two cytoplasmic calmodulin-binding domains. Biochem J. 1997;326:479–483. doi: 10.1042/bj3260479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodd R, Peracchia C, Stolady D, Török K. Calmodulin association with connexin32-derived peptides suggests trans-domain interaction in chemical gating of gap junction channels. J Biol Chem. 2008;283:26911–26920. doi: 10.1074/jbc.M801434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, Yang W, Lurtz MM, Ye Y, Huang Y, Lee HW, Chen Y, Louis CF, Yang JJ. Identification of the calmodulin binding domain of connexin 43. J Biol Chem. 2007;282:35005–35017. doi: 10.1074/jbc.M707728200. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y, Yang W, Lurtz MM, Chen Y, Jiang J, Huang Y, Louis CF, Yang JJ. Calmodulin mediates the Ca2+-dependent regulation of Cx44 gap junctions. Biophys J. 2009;96:2832–2848. doi: 10.1016/j.bpj.2008.12.3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Day DH. CaMBOT: profiling and characterizing calmodulin-binding proteins. Cell Signalling. 2003;15:347–354. doi: 10.1016/s0898-6568(02)00116-x. [DOI] [PubMed] [Google Scholar]
- 14.Yap KL, Kim J, Truong K, Sherman M, Yuan T, Ikura M. Calmodulin target database. J Struct Funct Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- 15.Jones DT, Taylor WR, Thornton JM. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry. 1994;33:3038–3049. doi: 10.1021/bi00176a037. [DOI] [PubMed] [Google Scholar]
- 16.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 17.Tusnady GE, Simon I. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol. 1998;283:489–506. doi: 10.1006/jmbi.1998.2107. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann K, Stoffel W. TMbase: a database of membrane spanning protein segments. Biol Chem Hoppe-Seyler. 1993;374:166. [Google Scholar]
- 19.Mitaku S, Hirokawa T, Tsuji T. Amphiphilicity index of polar amino acids as an aid in the characterization of amino acid preference at membrane–water interfaces. Bioinformatics. 2002;18:608–616. doi: 10.1093/bioinformatics/18.4.608. [DOI] [PubMed] [Google Scholar]
- 20.Jones DT. Do transmembrane protein superfolds exist? FEBS Lett. 1998;423:281–285. doi: 10.1016/s0014-5793(98)00095-7. [DOI] [PubMed] [Google Scholar]
- 21.Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 22.Johnson JD, Wittenauer LA. A fluorescent calmodulin that reports the binding of hydrophobic inhibitory ligands. Biochem J. 1983;211:473–479. doi: 10.1042/bj2110473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallace RW, Tallant EA, Cheung WY. Assay of calmodulin by Ca2+-dependent phosphodiesterase. Methods Enzymol. 1983;102:39–47. doi: 10.1016/s0076-6879(83)02006-6. [DOI] [PubMed] [Google Scholar]
- 24.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 26.Lee HW, Yang W, Ye Y, Liu ZR, Glushka J, Yang JJ. Isolated EF-loop III of calmodulin in a scaffold protein remains unpaired in solution using pulsed-field-gradient NMR spectroscopy. Biochim Biophys Acta. 2002;1598:80–87. doi: 10.1016/s0167-4838(02)00338-2. [DOI] [PubMed] [Google Scholar]
- 27.Mills R. Self-diffusion in normal and heavy water in the range 1–45°. J Phys Chem. 1973;77:685–688. [Google Scholar]
- 28.LaPorte DC, Keller CH, Olwin BB, Storm DR. Preparation of a fluorescent-labeled derivative of calmodulin which retains its affinity for calmodulin binding proteins. Biochemistry. 1981;20:3965–3972. doi: 10.1021/bi00517a004. [DOI] [PubMed] [Google Scholar]
- 29.Theoharis NT, Sorensen BR, Theisen-Toupal J, Shea MA. The neuronal voltage-dependent sodium channel type II IQ motif lowers the calcium affinity of the C-domain of calmodulin. Biochemistry. 2008;47:112–123. doi: 10.1021/bi7013129. [DOI] [PubMed] [Google Scholar]
- 30.VanScyoc WS, Sorensen BR, Rusinova E, Laws WR, Ross JB, Shea MA. Calcium binding to calmodulin mutants monitored by domain-specific intrinsic phenylalanine and tyrosine fluorescence. Biophys J. 2002;83:2767–2780. doi: 10.1016/S0006-3495(02)75286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. II Helix macrodipole effects and rational modification of the helical content of natural peptides. J Mol Biol. 1995;245:275–296. doi: 10.1006/jmbi.1994.0023. [DOI] [PubMed] [Google Scholar]
- 33.Munoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. III Temperature and pH dependence. J Mol Biol. 1995;245:297–308. doi: 10.1006/jmbi.1994.0024. [DOI] [PubMed] [Google Scholar]
- 34.Munoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Biol. 1994;1:399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- 35.Nicol S, Rahman D, Baines AJ. Ca2+-dependent interaction with calmodulin is conserved in the synapsin family: identification of a high-affinity site. Biochemistry. 1997;36:11487–11495. doi: 10.1021/bi970709r. [DOI] [PubMed] [Google Scholar]
- 36.Yamauchi E, Nakatsu T, Matsubara M, Kato H, Taniguchi H. Crystal structure of a MARCKS peptide containing the calmodulin-binding domain in complex with Ca2+–calmodulin. Nat Struct Biol. 2003;10:226–231. doi: 10.1038/nsb900. [DOI] [PubMed] [Google Scholar]
- 37.Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of X-ray structures. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 38.Andre I, Kesvatera T, Jonsson B, Akerfeldt KS, Linse S. The role of electrostatic interactions in calmodulin–peptide complex formation. Biophys J. 2004;87:1929–1938. doi: 10.1529/biophysj.104.040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lurtz MM, Louis CF. Intracellular calcium regulation of connexin43. Am J Physiol Cell Physiol. 2007;293:C1806–C1813. doi: 10.1152/ajpcell.00630.2006. [DOI] [PubMed] [Google Scholar]
- 40.Yang DI, Louis CF. Molecular cloning of ovine connexin44 and temporal expression of gap junction proteins in a lens cell culture. Invest Ophthalmol Visual Sci. 2000;41:2658–2664. [PubMed] [Google Scholar]
- 41.Illiano S, Nagao T, Vanhoutte PM. Calmidazolium, a calmodulin inhibitor, inhibits endothelium-dependent relaxations resistant to nitro-L-arginine in the canine coronary artery. Br J Pharmacol. 1992;107:387–392. doi: 10.1111/j.1476-5381.1992.tb12756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fielding L. NMR methods for the determination of protein–ligand dissociation constants. Curr Top Med Chem. 2003;3:39–53. doi: 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- 43.Weljie AM, Yamniuk AP, Yoshino H, Izumi Y, Vogel HJ. Protein conformational changes studied by diffusion NMR spectroscopy: application to helix–loop–helix calcium binding proteins. Protein Sci. 2003;12:228–236. doi: 10.1110/ps.0226203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang JJ, Buck M, Pitkeathly M, Kotik M, Haynie DT, Dobson CM, Radford SE. Conformational properties of four peptides spanning the sequence of hen lysozyme. J Mol Biol. 1995;252:483–491. doi: 10.1006/jmbi.1995.0513. [DOI] [PubMed] [Google Scholar]
- 45.Ludescher RD, Johnson ID, Volwerk JJ, de Haas GH, Jost PC, Hudson BS. Rotational dynamics of the single tryptophan of porcine pancreatic phospholipase A2, its zymogen, and an enzyme/micelle complex: a steady-state and time-resolved anisotropy study. Biochemistry. 1988;27:6618–6628. doi: 10.1021/bi00417a061. [DOI] [PubMed] [Google Scholar]
- 46.Veenstra RD, Wang HZ, Beyer EC, Brink PR. Selective dye and ionic permeability of gap junction channels formed by connexin45. Circ Res. 1994;75:483–490. doi: 10.1161/01.res.75.3.483. [DOI] [PubMed] [Google Scholar]
- 47.Beblo DA, Wang HZ, Beyer EC, Westphale EM, Veenstra RD. Unique conductance, gating, and selective permeability properties of gap junction channels formed by connexin40. Circ Res. 1995;77:813–822. doi: 10.1161/01.res.77.4.813. [DOI] [PubMed] [Google Scholar]
- 48.Donaldson P, Kistler J. Reconstitution of channels from preparations enriched in lens gap junction protein MP70. J Membr Biol. 1992;129:155–165. doi: 10.1007/BF00219511. [DOI] [PubMed] [Google Scholar]
- 49.Srinivas M, Costa M, Gao Y, Fort A, Fishman GI, Spray DC. Voltage dependence of macroscopic and unitary currents of gap junction channels formed by mouse connexin50 expressed in rat neuroblastoma cells. J Physiol. 1999;517:673–689. doi: 10.1111/j.1469-7793.1999.0673s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu X, Berthoud VM, Beyer EC, Ebihara L. Functional role of the carboxyl terminal domain of human connexin 50 in gap junctional channels. J Membr Biol. 2002;186:101–112. doi: 10.1007/s00232-001-0139-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peracchia C, Wang XC. Connexin domains relevant to the chemical gating of gap junction channels. Braz J Med Biol Res. 1997;30:577–590. doi: 10.1590/s0100-879x1997000500003. [DOI] [PubMed] [Google Scholar]
- 52.Dodd R, Peracchia C, Stolady D, Török K. Calmodulin association with connexin32-derived peptides suggests trans-domain interaction in chemical gating of gap junction channels. J Biol Chem. 2008;283:26911–26920. doi: 10.1074/jbc.M801434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmad S, Martin PE, Evans WH. Assembly of gap junction channels: mechanism, effects of calmodulin antagonists and identification of connexin oligomerization determinants. Eur J Biochem. 2001;268:4544–4552. doi: 10.1046/j.1432-1327.2001.02380.x. [DOI] [PubMed] [Google Scholar]
- 54.Blodow A, Ngezahayo A, Ernst A, Kolb HA. Calmodulin antagonists suppress gap junction coupling in isolated Hensen cells of the guinea pig cochlea. Pflügers Arch. 2003;446:36–41. doi: 10.1007/s00424-002-1004-9. [DOI] [PubMed] [Google Scholar]
- 55.Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, Koivisto PA, Van Maldergem L, Boyadjiev SA, Bodurtha JN, Jabs EW. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724–733. doi: 10.1002/humu.20958. [DOI] [PubMed] [Google Scholar]
- 56.Beahm DL, Hall JE. Hemichannel and junctional properties of connexin 50. Biophys J. 2002;82:2016–2031. doi: 10.1016/S0006-3495(02)75550-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toloue MM, Woolwine Y, Karcz JA, Kasperek EM, Nicholson BJ, Skerett IM. Site-directed mutagenesis reveals putative regions of protein interaction within the transmembrane domains of connexins. Cell Commun Adhes. 2008;15:95–105. doi: 10.1080/15419060802013463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beahm DL, Oshima A, Gaietta GM, Hand GM, Smock AE, Zucker SN, Toloue MM, Chandrasekhar A, Nicholson BJ, Sosinsky GE. Mutation of a conserved threonine in the third transmembrane helix of α- and β-connexins creates a dominant-negative closed gap junction channel. J Biol Chem. 2006;281:7994–8009. doi: 10.1074/jbc.M506533200. [DOI] [PubMed] [Google Scholar]
- 59.Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 60.Oshima A, Tani K, Hiroaki Y, Fujiyoshi Y, Sosinsky GE. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc Natl Acad Sci USA. 2007;104:10034–10039. doi: 10.1073/pnas.0703704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peracchia C, Wang XG, Peracchia LL. Chemical gating of gap junction channels. Methods. 2000;20:188–195. doi: 10.1006/meth.1999.0936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Monitoring CaM–Cx50p141–166 complex formation by MS
The MALDI–TOF-MS spectra of the free form of CaM (~17 kDa) and the complex form of CaM–Cx50p141–166 (~20 kDa). The inset shows the molecular mass of Cx50p141–166.
Figure S2 Monitoring the interaction between apo-CaM and Cx50p141–166 by (1H,15N)-HSQC spectroscopy
An overlay of HSQC spectra of apo-CaM (red) with the spectrum of the apo-CaM–Cx50p141–166 (green).
Figure S3 Dansyl fluorescence anisotropy
D-CaM (1 μM) was titrated with Cx50p141–166 in 50 mM Tris/HCl (pH 7.5), 100 mM KCl and 5 mM CaCl2. The fluorescence anisotropy were measured at λex = 335 nm and λem = 495 nm with an integration time of 20 s. The inset shows the anisotropy change for D-CaM (free), and the complex of D-CaM–Cx50p141–166 (bound).
Figure S4 Comparison of the α-family of Cxs
An overlay of the HSQC spectra of holo-CaM (yellow), holo-CaM–Cx50p141–166 (green) and holo-CaM–Cx43p136–158 (violet). Arrows indicate signals that shifted to the same direction because of identical binding modes, whereas rectangles highlight some exceptions.