

Abstract
Background
In IRCU it is uncertain whether variation of urinary protein, especially non-albumin protein (NAlb-P), is due to the presence of stones or reflects alteration of oxidative metabolism.
Aims
To validate in a tripartite cross-sectional study of 187 ambulatory male patients, undergoing a standardized laboratory programme, whether stones impact on N-Alb-P or the state of oxidative metabolism interferes with IRCU pathophysiology.
Methods
In part 1 the strata low and high of fasting urinary excretion rate per 2 h of N-Alb-P, malonedialdehyde, hypoxanthine, xanthine, pH and other urine components were compared, and association with renal stones in situ evaluated; in part 2 the co-variation of oxidatively modulated environment, fasting urinary pH, calcium (Ca) salt crystallization risk and the number of patients with stones in situ was examined; in part 3, the nucleation of Ca oxalate and Ca phosphate was tested in undiluted postprandial urine of patients and related to the state of oxidative metabolism.
Results
In part 1, N-Alb-P excretion > 4.3 mg was associated with increase of blood pressure, excretion of total protein, hypoxanthine (a marker of tissue hypoxia), malonedialdehyde (a marker of lipid peroxidation), sodium, magnesium, citrate, uric acid, volume, pH, and increase of renal fractional excretion of both NAlb-P and uric acid; when stones were present, urinary pH was elevated but other parameters were unaffected. Significant predictors of N-Alb-P excretion were malonedialdehyde, fractional N-Alb-P and hypoxanthine. In part 2, urine pH > 6.14 was associated with unchanged blood pressure and plasma vasopressin, increase of blood pH, urinary volume, malonedialde hyde, fractional excretion of N-Alb-P, uric acid, Ca phosphate, but not Ca oxalate, supersaturation; this spectrum was accompanied by decrease of concentration of urinary total and free magnesium, total and complexed citrate, plasma uric acid (in humans the major circulating antioxidant) and insulin; the number of stone-bearing patients was increased. Significant predictors of urine pH were body mass index, plasma insulin and uric acid (negative), and urinary xanthine (positive). In part 3 low plasma uric acid, not high urinary malonedialdehyde or high ratio malonedialdehyde/uric acid was significantly associated with diminished Ca but not oxalate tolerance, with the first nucleating crystal type being mostly Ca phosphate (hydroxyapatite), in the rest Ca oxalate dihydrate; uricemia correlated marginally positively (p = 0.055) with Ca tolerance of urine, stronger with blood pressure and insulin, and negatively with urinary xanthine, fractional N-Alb-P, volume, sodium.
Conclusions
In IRCU 1) not renal stones in situ, but disturbed oxidative metabolism apparently modulates nephron functionality, ending up in higher renal NAlb-P release, urinary volume, sodium and pH of fasting urine; 2) etiologically unknown decline of uricemia may represent antioxidant deficiency and cause a risk of hydroxyapatite crystallization and stone formation in a weakly acidic or alkaline inhibitor-deficient and NAlb-P-rich milieu; 3) several observations, linking oxidative and systemic metabolism, are compatible with Ca stone initiation beyond tubules.
Keywords: Calcium urolithiasis, Stones in situ, Urinary protein, Oxidative and systemic metabolism, Urinary pH, Calcium salt crystallization
Introduction
In the complex pathophysiology of IRCU (for full definition see ref. [1]) the cause of variation of urinary protein, especially the non-albumin protein (N-Alb-P) fraction, and its possible contribution to calcium (Ca) salt crystallization and Ca stone development are incompletely understood. The literature in this area, including pioneering work [2,3], lacks information on pre-existence and nature of tissue damage that could underlie the variation of N-Alb-P and other urine components, with the development of Ca salt crystallization risk and stones being secondary events. The picture appears even more complex, as in numerous patients the renal capacity to acidify urine was found defective and accompanied by higher proteinuria [1], and as renal stones in situ can act as a foreign body leading to abrasion of tubule-lining epithelium, shedding of protein and other substances, able to modulate urinary pH [4]. On the other hand, there is now growing evidence that alteration of oxidative metabolism is widespread among various forms of nephrolithiasis, including IRCU [5-9]. More specifically, ascorbic acid and alpha-tocopherol, both documented antioxidants, were lowered in red blood cells of IRCU [6,9], and there was decrease of plasma total antioxidative substances (TAS) that was mainly due to low plasma uric acid and albumin (Alb) concentration, in human blood the two major reactive oxygen species (ROS) scavenging antioxidants [see ref. [5]]; in addition, urinary and cellular levels of malonedialdehyde (MDA), a marker of ROS excess, were found increased [6,7]. ROS excess in general manifests as per-oxidation of lipoproteins [6,10], in IRCU as damage of renal cell membranes [6] and nuclear deoxyribonucleic acid (DNA) [11,12], and increase of proteinuria [13]. However, although the latter would help understand why in addition to incorporation of Ca oxalate (CaOx) and Ca phosphate (CaPi) the one of proteins (so-called matrix) is characteristic for Ca stones [14], numerous steps in the sequence of events leading to Ca stones under conditions of altered oxidative metabolism are far from clear. After all, it is unknown if analysing readily available biomaterials such as urine and blood for substances so far less recognized in IRCU adds to insight into pathways linking oxidative and systemic metabolism to urinary protein, pH, Ca crystal and stone forming processes and stones. To arrive at a more realistic consideration of IRCU pathophysiology, there is a need to clarify whether there are effects that can be ascribed to stones in situ, and others that can independently impact on Ca salt crystallization. Historically, for the latter to occur in urine, the pre-requisites are seen in sufficient degree of supersaturation and crystal nucleation as modulated by inhibitors and promoters (only on this basis can crystals grow and agglomerate, processes over decades considered as the first stages in stone formation [2,3]).
From the screening of a large body of data from IRCU patients of our laboratory the impression was that in fasting urine, i.e., not compromised by prior ingestion and metabolism of nutrients and obtained under strictly controlled conditions (see below), there was enormous scatter of urinary protein, especially the N-Alb-P fraction (of which several members modulate crystallization of stone substances [15]), but also variation of urinary volume and pH, and body size (body mass index; BMI). Therefore, the present work was carried out to find a basis from which hypotheses can be formulated that are testable by future more in depth investigating controlled studies. For the first time two defined periods of a daily cycle were selected for studying the patients in the laboratory, allowing to rule out unspecific influences. Answers to the following questions were sought: 1) Is excretion of urinary N-Alb-P affected by renal stones in situ, or is there covariation of this urine protein fraction with markers of oxidative metabolism? 2) Are there links between oxidative and systemic metabolism, urinary pH, N-Alb-P concentration, volume, Ca salt supersaturation and risk of crystallization, and the number of patients bearing stones? 3) Do excess of oxidants, deficit of antioxidants or imbalance of the two alter the propensity of urine to nucleate CaOx or CaPi, if not, are there links to systemic metabolism?
Materials and methods
Study participants
From our ambulatory stone clinic 187 consecutively examined adult male stone patients with defined IRCU [for stone analysis and other details see ref. [1]], of whom several anthropometric features, the actual presence of stones and clinical chemistry data of relevance for lithogenicity of urine were available, were included. All had normal renal function (plasma creatinine < 124 μm/l) but at least one Ca stone recurrence in the past, with the last dating back more than 1 month, IRCU was diagnosed on the basis of disease history and stone analysis, showing that Ca, Ox and Pi were the only constituents (see ref. [1]). Patients were defined as "stone-free" (SF, n = 93) when at the time of laboratory investigation concretions were not detectable by meticulously carried out clinical techniques (X-ray, including tomography, ultrasound, etc.) in the renal pelvis, calyces, papillae or further upstream parenchyma, or as "stone-bearing" (one stone or more, but no stone nests; SB, n = 94). Exclusion criteria were females (for reasons see ref. [5]), non-European ethnology, residence outside North Bavaria, accompanying diseases (primary hyperparathyroidism, documented essential hypertension, diabetes mellitus, renal tubular acidosis), oxaluria > 0.5 mmol in daily urine (precluding the possibility that ROS excess and subsequently elevated MDA originate from Ox excess in urine [7,16]), hematuria (dipstickpositive urine), signs of urinary tract obstruction and/or infection with urease-producing germs, and cases with potential post-renal sources of urinary protein (cystitis, prostatitis, etc.). It is emphasized that delineation of members of urinary N-Alb-P fraction in terms of structure, amount and function was not among the goals of present work. All patients had not taken specific anti-stone medication during the previous 6 weeks. A defined control group was not studied, but from a small group of adult males without a history of stones limits of normalcy for several variables are given. Upon written information all subjects gave their consent to the envisaged laboratory investigations, and the study was carried out in accordance with the principles of the Declaration of Helsinki.
Laboratory program
Details of the standardized examination protocol (including analysis of daily, fasting, postprandial urine, and fasting blood, etc.) as practiced in our outpatient stone clinic have been described [17]. For the present work the composition of fasting blood and urine and postprandial urine (after intake of a purine- and oxalate-free but Ca-rich and urine acidifying meal of fixed composition [1,18]) was studied. After an overnight fasting period of 12-14 h, diuresis was stimulated in the laboratory by drinking 2 × 300 ml distilled water (generally resulting in urine flow of 1 - 2 ml/min), blood pressure was measured with the patient in a recumbent position, and forearm venous blood withdrawn into heparinized prechilled tubes. After puncture of an ear-lobe for blood gas analysis, the bladder was voided to give urine from a timed period of 2 h; thereafter the meal was ingested, and urine collected from a timed 3 h period. Aliquots of plasma and paper-filtered (Whatman no. 3) urine were prepared and either analysed on the same day or stored at -80°C.
Study design, data compilation
The study was tripartite, retrospective and observational, cross-sectional and correlative. The overlap of participants in the present and previous studies, using different strategies and outcomes [1,5,18], was 80 - 95%. As a search for the mechanism(s) underlying ROS production and ROS nature was not in the focus of present work, overproduction of ROS (by cells during hypoxia-induced ATP degradation [19]) was taken as reflected by increase of urinary hypoxanthine, and ROS excess-mediated damage of lipids and lipid-containing cell membranes, other molecules and genes [11,12,20] as reflected by increase of urinary MDA; low plasma uric acid, the dominant antioxidant component of human plasma [5,21], was taken as indicating antioxidant deficiency. In part 1 variables as observed in fasting urine of the pool group (SF + SB patients, n = 187) were stratified according to the median urinary N-Alb-P excretion (strata Low and High), the protein fraction known to contain crystallization-inhibiting and -promoting species [15,22,23]; these data were complemented by general features, including a score roughly reflecting the activity of stone-forming processes (ASFP) in the past 2-year period [24], urinary excretion rate of volume, minerals, MDA and the uric acid precursor oxypurines hypoxanthine and xanthine, renal fractional excretion (FE; indicating the state of the kidney to retain or release substances) of N-Alb-P (FE-N-Alb-P), uric acid (FE-Uric acid) and oxalate (FE-oxalate), fasting plasma levels of uric acid and total antioxidants (TAS), vasopressin and insulin. Also in part 1, and using the same database as for stratification of N-Alb-P excretion (see above), SB were contrasted with SF patients. Part 2 comprised, besides general features and the state of oxidative metabolism, FE data and those on the concentration of substances that govern the propensity of urine to crystallize: the physico-chemical supersaturation with CaOx and CaPi, the latter in the form of both the molar Ca/Pi ratio (the formation of amorphous CaPi solid begins at 0.015 M [1]) and the Ca-rich hydroxyapatite (HAP; molar Ca/Pi ratio of crystals 1.60), magnesium (Mg) and citrate (Cit) in terms of total concentration and after splitting into moieties able to form soluble complexes or remaining as free ions (F-Mg, F-Cit; discussedassmall-molecularCaOxand CaPi crystallization inhibitors [25,26]). Thereafter, the patients were stratified according to median pH of fasting urine (strata Low and High). In part 3 the nucleation of Ca salts, i.e., appearance of solid CaOx or CaPi visible by light microscopy, was probed in postprandial urine by means of the maximally tolerable total concentration of Ca (T-Ca) and Ox (T-Ox). This urine was chosen because in it deficient acidification in response to intake of an acid meal is frequent among IRCU patients, accompanied by unchanged Ca/Pi but dramatic rise of HAP supersaturation [1]; therefore, and in accordance with studies on CaPi solubility in urine-like solutions with varying pH [27], the formation of HAP and other CaPi solids at urine pH 6.0 upon addition of Ca in excess of Ca in native urine should be indicated by T-Ca. In contrast, at the CaOx supersaturation level prevailing at the native urine pH and Ox added in excess of Ox in native urine, CaOx solid formation should inform about T-Ox. These data were assigned to three sets of patients, with each set subdivided into the strata Low and High: Fasting urinary MDA excretion, fasting plasma uric acid concentration, the ratio urinary MDA/plasma uric acid (as crude measure of imbalance of oxidative metabolism); several additional parameters served as complementary data. For the sake of transparency and space a considerable body of data is presented in APPENDIX.
Analyses
For analyzing blood, plasma and urine routine methods or well-established techniques [5,17] were utilized, including those 14 analytes required for estimation of urinary supersaturation (see below) with stone substances. Exceptions were: urinary pH (glass electrode); colorimetric determination of plasma TAS (using commercial reagents, supplied by Randox, Krefeld, Germany) which follows the principle outlined by Miller et al. [28]; high-performance liquid chromatography measurement of MDA and oxypurines in native urine [24], Ox in plasma ultrafiltrate [29], thawed and acidified (pH ≤1.5) urine [29]; urinary total protein by colorimetry [30], urinary Alb by immuno-nephelometry (using antibody OSAL 10, BN II analyzer, Dade Behring, Marburg, Germany); plasma vasopressin and insulin by radioimmunoassay (kit from Immuno-Biological Laboratory, Hamburg, Germany, and inhouse assay, respectively). Crystal nucleation was assessed by the earlier described small-scale (0.5 ml undiluted urine) light microscopy and image analysis-guided technique (for details see ref. [31]), using addition of microliter amounts of analytical grade sodium oxalate stock solution at original pH (for T-Ox), or addition of microliter amounts of analytical grade Ca chloride stock solution at prefixed pH 6.0 (for T-Ca). The morphology and nature of formed solids were documented by scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) elemental analysis (for details, techniques and equipment see ref. [18]).
Calculations, statistics
N-Alb-P in plasma and urine was taken as difference between total protein and Alb (note that in present work urinary Alb was not further considered). FE and creatinine clearance were calculated conventionally. From EQUIL-2 [32] the free energy (DG) that drives urine supersaturation with CaOx, HAP and uric acid toward nucleation and crystal growth in aqueous solution was calculated, and from the same procedure the concentration of soluble Mg and Cit complexes, F-Mg and F-Cit, were obtained. The molar Ca/Pi ratio relative to concentration of Na, F-Mg, F-Cit, and urinary volume was given to illustrate possible interdependencies. Despite the wide scatter of individual values the decadic logarithm of numerical values mostly gave symmetric data, allowing application of a two-tailed Student's t-test, otherwise the Wilcoxon signed-rank test was used. Categorical data were examined by the Chi2 test. When p ≤0.05, differences between strata were considered significant. For practical reasons results are given as mean values (with SE or range). Simple and multiple regression analyses (with adjustment for confounding variables) were established. The software STATISTICA (Statsoft, Tulsa, OK, USA) was used.
Results
Urinary N-Alb-P, stones in situ (part 1)
According to Table 1-A, in the High vs. Low patients urinary N-Alb-P excretion was approx. 6 times and total protein approx. 4 times higher, whereas the plasma levels of total protein, Alb and N-Alb-P were unchanged (data not shown); there was in addition increase of urinary volume, Na, K, Mg, Cit, creatinine clearance, and FE-N-Alb-P; in contrast, excretion of Ox, Ca and Pi was unchanged. Also in the High patients, there was slightly but significantly higher systolic and diastolic blood pressure, increase of urinary pH, excretion of the uric acid precursor oxypurines hypoxanthine and xanthine, uric acid, FE-Uric acid and MDA, but decrease of plasma concentration of uric acid and TAS, this indicating deficiency of antioxidants [5,28] not studied here; BMI, ASFP and the number of SB (stones present) and SF (stones absent) patients were statistically indistinguishable among strata, as was plasma K and vasopressin (mean values 4.4 pM/l and 4.6 pM/l, assumed molecular mass 1084 Da). Table 1-B, giving data from the same variables as shown in Table 1-A separately for SF (column Absent) and SB (column Present) patients, shows that in the latter group urine pH was elevated, but that other variables differed only insignificantly. Significant (p≤0.05) simple correlations were listed in APPENDIX I (Table 5, code 1 - 10), resulting in summed r2 0.74 (code 1 - 7, without FE-N-Alb-P) and r2 0.63 (code8-10). Using log N-Alb-P excretion as outcome measure, multivariate regression analysis identified log MDA and log FE-N-Alb-P (R2 0.91, p < 0.00001 after adjustment for covariates) and log hypoxanthine (adjusted R2 0.42, p < 0.000001) as the dominant predictors; together these variables can explain more than the full range of variation of N-Alb-P excretion rate.
Table 1.
Characteristics of IRCU patients with low and high N-Alb-P excretion in fasting urine (A), absence or presence of renal stones in situ (B), and of all patients (C)
| A. N-Alb-P; median Low |
4.3 mg High |
High vs. Low | B. Stones Absent |
Present | C. All Present vs. Absent |
Mean | Range | Normal1 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| N+ | ≤Median | >Median | p | p | ||||||
| Urine | ||||||||||
| Total protein; mg | 187 | 3.3 (0.2); 94 | 13 (2); 93++ | < 0.001x | 6.2 (0.5); 94++ | 9.7 (2.1); 93++ | 0.24x | 7.93 | 0.8-161 | < 50 |
| N-Alb-P; mg | 187 | 1.7 (0.4); 94++ | 11 (1.9); 93 | < 0.001x | 5.1 (0.4); 94 | 7.3 (2.1); 93 | 0.19x | 6.19 | 0-161 | < 7.0 |
| Cr*-Clearance; ml/min | 187 | 108 (2); 94 | 115 (3); 93 | 0.03 | 112 (3); 94 | 111 (2); 93 | 0.44 | 111 | 41-187 | > 50 |
| 103 × FE-N-Alb-P; % | 187 | 0.90 (0.0); 94 | 3.4 (0.5); 93 | < 0.001 | 1.8 (0.0); 94 | 2.7 (0.0); 93 | 0.13x | 2.2 | 0.08-45 | |
| Volume; ml | 187 | 161 (11); 94 | 290 (17); 93 | < 0.001 | 229 (15); 94 | 221 (16); 93 | 0.25x | 225 | 60-720 | 50-600 |
| Na; mM | 187 | 10 (0.4); 94 | 15 (0.8); 93 | < 0.001 | 13 (0.6); 94 | 12 (0.7); 93 | 0.43 | 12.5 | 2-47 | < 20 |
| K; mM | 187 | 6.7 (0.2); 94 | 9.0 (0.3); 93 | < 0.001 | 7.8 (0.3); 94 | 7.9 (0.3); 93 | 0.41 | 7.8 | 1.8-17 | 2-8 |
| Ca; μM | 187 | 298 (19); 93 | 334 (16); 94 | 0.07x | 307 (17); 94 | 325 (18); 93 | 0.23x | 316 | 29-1261 | < 625 |
| Mg; μM | 187 | 231 (10); 94 | 277 (10); 93 | 0.002 | 257 (10); 93 | 250 (12); 94 | 0.34 | 253 | 48-776 | > 280 |
| Pi; mM | 187 | 1.15 (0.06); 94 | 1.23 (0.07); 93 | 0.22x | 1.19 (0.06); 94 | 1.19 (0.07); 93 | 0.47x | 1.19 | 0.10-3.9 | < 3 |
| Ox; μM | 179 | 22 (1.4); 88 | 23 (2.1); 91 | 0.34 | 23 (1); 89 | 24 (2); 90 | 0.32 | 22 | 1.3-193 | < 37 |
| Cit; μM | 185 | 253 (13); 93 | 311 (14); 92 | 0.001 | 290 (14); 93 | 272 (13); 92 | 0.16 | 279 | 44-732 | > 132 |
| pH | 187 | 5.92 (0.09); 94 | 6.35 (0.07); 93 | < 0.001 | 5.95 (0.08); 94 | 6.33 (0.07); 93 | < 0.001 | 6.14 | 4.4-7.6 | 5.2-7.9 |
| General features | ||||||||||
| Age; y | 187 | 42 (1.2); 94 | 41 (1.2); 93 | 0.25 | 41 (1.2); 94 | 42 (1.2); 93 | 0.29 | 41.6 | 21-67 | > 22 |
| ASFP; score | 187 | 35 (4); 94 | 40 (4); 93 | 0.18 x | 34 (4); 94 | 41 (4); 93 | 0.08x | 36 | 1-271 | 1 |
| Renal stones; Absent/Present | 187 | 47/47; 94 | 46/47; 93 | 94/0 | 0/93 | |||||
| Systolic blood pressure; mmHg | 152 | 126 (2); 78 | 132 (3); 74 | 0.004 | 129 (2); 75 | 129 (3); 77 | 0.44 | 129 | 90-220 | ≤130 |
| Diastolic blood pressure; mmHg | 152 | 82 (1); 78 | 86 (2); 74 | 0.03 | 83 (1); 75 | 84 (2); 77 | 0.32 | 84 | 60-150 | ≤80 |
| BMI; kg/(m)2 | 187 | 26.4 (0.3); 94 | 26.2 (0.4); 93 | 0.39 | 26.2 (0.3); 94 | 26.5 (0.4); 94 | 0.25 | 26.3 | 19-40 | ≤25 |
| P**-Insulin; μU/ml | 184 | 17 (1); 92 | 16 (1); 92 | 0.26 | 16 (1); 91 | 17 (1); 93 | 0.36 | 16 | 1-83 | < 20 |
| P-K; mM/l | 106 | 4.2 (0.03); 62 | 4.2 (0.05);44 | 0.46 | 4.2 (0.04); 49 | 4.2 (0.04); 57 | 0.43 | 4.2 | 3.6-5.1 | 3.8-5.0 |
| U**-Hypoxanthine; μM | 75 | 9.9 (0.8); 29 | 13 (0.7); 46 | < 0.001x | 11.5 (0.8); 39 | 12.7 (0.8); 36 | 0.08x | 12.1 | 2.8-25 | < 30 |
| U-Xanthine; μM | 75 | 4.0 (0.4); 29 | 7.8 (0.5); 46 | < 0.001x | 6.4 (0.6); 39 | 6.3 (0.5); 36 | 0.45x | 6.3 | 1.2-17 | > 3 |
| U-Uric acid; μM | 187 | 357 (16); 94 | 433 (14); 93 | < 0.001 | 403 (12); 94 | 386 (18); 93 | 0.21x | 395 | 44-1351 | < 400 |
| U-FE-Uric acid; % | 186 | 7.4 (0.3); 94 | 8.8 (0.4); 92 | < 0.001 | 8.3 (0.3); 94 | 7.8 (0.4); 92 | 0.16 | 8.1 | 0.7-24 | < 11 |
| P-Uric acid; μM/l | 187 | 374 (7); 94 | 342 (7); 93 | < 0.001 | 353 (7); 94 | 363 (7); 93 | 0.17 | 358 | 160-631 | < 446 |
| P-TAS; mM/l | 57 | 1.37 (0.02); 21 | 1.31 (0.01); 36 | 0.007 | 1.34 (0.02); 30 | 1.32 (0.02); 27 | 0.20 | 1.33 | 1.18-1.49 | 1.29-1.61 |
| U-MDA; nM | 176 | 116 (5); 89 | 148 (7); 87 | < 0.001 | 135 (6); 88 | 129 (6); 88 | 0.24 | 132 | 25-345 | < 110 |
| U-MDA/P-Uric acid; nM/mM × l -1 | 176 | 316 (14); 89 | 486 (22); 87 | < 0.001x | 373 (20); 88 | 397 (20); 88 | 0.19x | 385 | 74-1051 | < 375 |
All excretions ratesare per 2 h. If not otherwise indicated, data are mean values (SE). For further informations see text.
+: total number of patients; ++: number of patients in subgroups; *: creatinine; **: U and P indicate urine and plasma, respectively; x: based on log10; nd: not determined; 1: limits or range observed in 10 - 20 healthy adult male subjects in the authors' laboratory, and from literature.
Table 5.
Appendix I. Simple correlation of variables in fasting urine (U) and plasma (P) of Results, parts 1-3; n: paired observations; r: coefficient; p: level of significance
| Code | Varia bles | n | r | p | Code | Variables | n | r | p |
|---|---|---|---|---|---|---|---|---|---|
| Part 1 | Continuation: | ||||||||
| Dependent: Log N-Alb-P | |||||||||
| Influential: | 16 | U-FE-Uric acid | 186 | 0.18 | 0.015x | ||||
| 1 | U-MDA | 170 | 0.35 | < 0.001x | 17 | U-HAP | 172 | 0.42 | < 0.001 |
| 2 | U-Volume | 176 | 0.53 | < 0.001x | 18 | U-Hypoxanthine | 75 | 0.29 | 0.01x |
| 3 | U-Na | 181 | 0.39 | < 0.001x | 19 | U-Xanthine | 75 | 0.50 | < 0.001x |
| 4 | U-Mg | 181 | 0.24 | 0.0 01x | 20 | P-Uric acid | 187 | -0.23 | 0.001 |
| 5 | U-Cit | 179 | 0.26 | 0.0 01x | 21 | P-TAS | 57 | -0.28 | 0.03 |
| 6 | U-FE-N-Alb -P | 181 | 0.95 | < 0.001x | |||||
| 7 | U-pH | 181 | 0.25 | 0.0 01 | Part 3 | ||||
| 8 | U-Hypoxanthine | 75 | 0.40 | 0.0 01x | Dependent: Log T-Ox | ||||
| 9 | U-Xanthine | 75 | 0.66 | < 0.001x | Influential: | ||||
| 10 | P-Uric acid | 187 | -0.18 | 0.0 2x | 22 | T-Ca | 49 | -0.26 | 0.07x |
| Part 2 | Dependent: Log T-Ca | ||||||||
| Dependent: U-pH | Influential: | ||||||||
| Influential: | 23 | P-Uric acid | 49 | 0.28 | 0.055x | ||||
| 11 | BMI | 187 | -0.27 | < 0.001 | |||||
| 12 | P-Insuli n | 184 | -0.29 | < 0.001x | Dependent: Log (T-Ca/102 x T-Ox) | ||||
| 13 | U-Cit | 185 | -0.14 | 0.0 5x | Influential: | ||||
| 14 | U-Volume | 187 | 0.19 | 0.0 1x | 24 | P-Uric acid | 49 | 0.28 | 0.051x |
| 15 | U-FE-N-Alb -P | 181 | 0.26 | < 0.001x | |||||
For dimension of variables see Tables 1 (part 1), 2 (part 2), 3 (part 3); x: based on log data.
Urinary ph (Part 2)
In the High vs. Low stratum(Table 2)the number of patients with BMI > 25.0 was uneven (High: n = 54; Low: n = 67), and there was decrease of unclassified BMI. Also according to Table 2, the stratum High patients exhibited decrease of plasma insulin and uric acid; plasma TAS of 34 out of 57 patients studied was < 1.35 mM/l (which is the lower limit of normalcy [5]); unchanged were systolic and diastolic blood pressure, urinary N-Alb-P concentration and the plasma levels of total protein, Alb, N-Alb-P (data not shown); there was increase of urinary hypoxanthine, xanthine and MDA excretion, FE-N-Alb-P and volume, but decrease of urinary concentration of Na, total Mg and F-Mg, total Cit and complexed Cit, uric acid, Ox, supersaturation with uric acid; most importantly, there was unchanged urinary Ca/Pi with dramatic increase of HAP in association with borderline significant decrease of CaOx supersaturation and significant increase of the number of SB patients. Urinary pH failed to correlate with MDA (n = 176, r2 0.01). Significant (p < 0.05) correlations were listed in APPENDIX I (Table 5), resulting in summed r2 0.49 (code 11 - 17), and summed r2 0.46 (code 18 - 21). Using urinary pH as outcome, multivariate regression analysis identified BMI and plasma log insulin as negative predictors (adjusted R2 0.14, p < 0.00006), log plasma uric acid as negative, log xanthine excretion and log FE-N-Alb-P (adjusted R2 0.28, p < 0.00001) as positive predictors. Together these variables can explain approx. 42% of variation of urinary pH.
Table 2.
Characteristics of IRCU patients with either low or high pH in fasting urine, and of all patients
| pH; median 6.14 | ||||||
|---|---|---|---|---|---|---|
| Low | High | High vs. Low | All | |||
| N | ≤Median | > Median | p | Mean | Range | |
| General features | ||||||
| ASFP; score | 187 | 40 (5); 93 | 35 (3); 94 | 0.49x | a | b |
| Renal stones; Absent/Present | 187 | 57/36 | 37/57 | 0.03xx | ||
| BMI; kg/(m)2 | 187 | 27.0 (0.4); 93 | 25.6 (0.3); 94 | 0.001 | a | b |
| P*-Insulin; μU/ml | 184 | 20 (1); 90 | 13 (1); 94 | < 0.001 | a | b |
| Systolic blood pressure; mm Hg | 152 | 130 (3); 74 | 128 (2); 78 | 0.24 | a | b |
| Diastolic blood pressure; mm Hg | 152 | 84 (2); 74 | 83 (1); 78 | 0.29 | a | b |
| U*-Hypoxanthine; μM | 75 | 11 (0.9); 40 | 13 (0.6); 35 | 0.03x | a | b |
| U-Xanthine; μM | 75 | 5.2 (0.5); 40 | 7.7 (0.5); 35 | < 0.001x | a | b |
| P-Uric acid; μM/l | 187 | 374 (7), 93 | 342 (7); 94 | 0.001 | a | b |
| P-TAS; mM/l | 57 | 1.35 (0.02); 29 | 1.32 (0.02); 28 | 0.14 | a | b |
| P-Oxalate; μM | 61 | 1.72 (0.09); 32 | 1.71 (0.08); 29 | 0.46 | 1.9 | 1.1-3.8 |
| U-MDA; nM | 176 | 124 (5); 89 | 140 (7); 87 | 0.03 | a | b |
| B*-pH | 185 | 7.40 (0.00); 91 | 7.41 (0.00); 94 | 0.02 | 7.401 | 7.35-7.49 |
| B-Bicarbonate; mM/l | 185 | 23.2 (0.2); 91 | 23.7 (0.2); 94 | 0.07 | 23.52 | 18-31 |
| U-MDA/P-Uric acid; nM/mM × l-1 | 176 | 345 (17); 90 | 426 (22); 86 | 0.002 | a | b |
| Urine | ||||||
| pH | 187 | 5.46 (0.04); 93 | 6.81 (0.04); 94 | < 0.001 | a | b |
| Volume; ml | 187 | 194 (14); 93 | 256 (17); 94 | 0.003 | a | b |
| Cr**-Clearance; ml/min | 187 | 127 (4); 93 | 119 (3); 94 | 0.08 | a | b |
| N-Alb-P; mg/l | 181 | 32 (5); 89 | 34 (5); 92 | 0.24x | 33 | 2-435 |
| 103 × FE-N-Alb-P; % | 187 | 1.9 (0.3); 91 | 2.5 (0.5); 96 | 0.002x | a | b |
| FE-Uric acid; % | 186 | 7.7 (0.3); 92 | 8.5 (0.4); 94 | 0.07 | a | b |
| FE-Oxalate; % | 61 | 108 (8); 32 | 120 (9); 29 | 0.16 | 113 | 40-245 |
| Na; mM/l | 187 | 77 (4); 93 | 66 (4); 94 | 0.03 | 72 | 13-201 |
| Ca; mM/l | 187 | 2.1 (0.2); 93 | 1.9 (0.2); 94 | 0.18x | 2.0 | 0.2-11 |
| Pi; mM/l | 187 | 8.6 (0.7); 93 | 7.7 (0.8); 94 | 0.20x | 8.2 | 0.4-66 |
| Ox; μM/l | 179 | 144 (10); 87 | 110 (8); 92 | 0.003x | 130 | 3-470 |
| Uric acid; mM/l | 187 | 2.7 (0.1); 93 | 2.2 (0.1); 94 | 0.005 | 2.4 | 0.39-6.1 |
| Uric acid; DG | 187 | 5.26 (0.16); 85 | 4.45 (0.17); 87 | < 0.001 | 0.82 | 8.13 |
| Ca/Pi; mM/mM | 187 | 0.39 (0.04); 93 | 0.40 (0.06); 94 | 0.49 | 0.40 | 0.05-3.7 |
| Total Mg; mM/l | 187 | 1.75 (0.13); 93 | 1.38 (0.08); 94 | 0.005 | 1.6 | 0.21-4.58 |
| Complexed Mg; mM/l | 172 | 0.83 (0.06); 85 | 0.71 (0.06); 87 | 0.08 | 0.77 | 0.1-2.8 |
| F-Mg; mM/l | 172 | 0.95 (0.06); 85 | 0.70 (0.05); 87 | 0.001 | 0.82 | 0.1-2.8 |
| Total Cit; mM/l | 185 | 1.93 (0.13); 93 | 1.61 (0.13); 92 | 0.03x | 1.8 | 0.18-5.9 |
| Complexed Cit; mM/l | 172 | 1.63 (0.12); 85 | 1.33 (0.11); 87 | 0.03 | 1.5 | 0.2-5.2 |
| F-Cit; mM/l | 172 | 0.29 (0.03); 85 | 0.31 (0.03); 87 | 0.31 | 0.30 | 0.02-1.5 |
| CaOx; DG | 172 | 1.0 (0.11); 85 | 0.78 (0.13); 87 | 0.06 | 0.91 | -2.7-3.5 |
| HAP; DG | 172 | 1.9 (0.32); 85 | 3.7 (0.28); 87 | < 0.001 | 2.8 | -4.6-8.6 |
Excretion rates are per 2 h. N: number of participating patients; for the number of patients in strata, data for a and b and other information, see Table 1 and text.
*: P, U and B indicate plasma, urine and blood, respectively; **: creatinine; x: based on log10; xx:Chi2 9.0 (3 degrees of freedom); 1, 2: limits of normalcy in the authors' laboratory are ≥7.35, ≥18, respectively.
In analogy to in vitro work, showing that the higher the pH, the higher is HAP supersaturation [27], from data of Table 2 a possible sequence of events in urine has been derived: Figure 1, ➀ - ➂ shows that in the patients with high urine pH (stratum High) the increase of urinary volume and decreasing concentration of Na and F-Mg are central to the increase of (Ca/Pi)/Na ➀ and (Ca/Pi)/F-Mg ➁, whereas (Ca/Pi)/F-Cit was resistant to the change of volume ➂; volumecorrelated directly with FE-N-Alb-P (Figure 1, B), (Ca/Pi)/Na (Figure 1, C), indirectly with Na concentration (Figure 1, D) and CaOx DG (Figure 1, E); conversely, HAP DG correlated directly with (Ca/Pi)/F-Mg (Figure 1, F). In these plots segregation of SB from SF patients was not recognizable (Figure 1B-F), but the data allow to postulate that a rise of HAP crystallization risk is characteristic for urine with higher pH, occurring in association with high urine volume and low small-molecular crystallization inhibitors (see also below).
Figure 1.
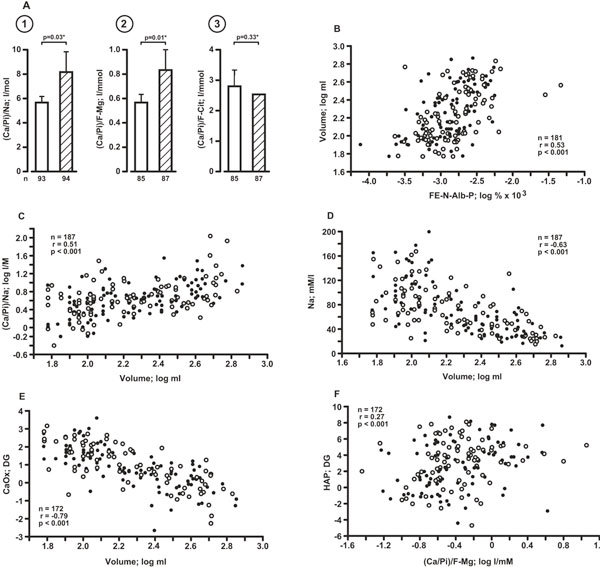
A: Ca/Pi ratio in fasting 2 h urine as influenced by the prevailing concentration of Na (➀), F-Mg (➁)and F-Cit(➂); light and hatched columns are patients with urine pH ≤6.14 and > 6.14, respectively; n: number of patients; *: based on log data. Mean values and (bars) SEM. B - F: Variables correlating with urinary volume (B-E) and HAP DG, respectively; note that negative values of DG (CaOx, HAP) indicate solubility of the Ca salt under study [32], and that segregation of SF (filled circles) and SB (open circles) patients is not recognizable.
Broadening this thinking, APPENDIX II (Figure 3) (a - i) gives further information from additional significant (p < 0.05 -p < 0.001) simple correlations (n = 172 - 181 paired observations): positively correlated were HAP DG and log N-Alb-P concentration (a), log volumeand (Ca/Pi)/TotalMg(b),(Ca/Pi)/Total Cit(c); negatively correlated were log volume and log N-Alb P concentration (d), insulin and HAP DG (e), blood bicarbonate (f), blood pH (g), BMI and blood bicarbonate (h); BMI and plasma uric acid correlated positively (i) (for correlation of BMI and insulin see Table 4); note that in h and i the majority of patients exhibited BMI > 25.0 (dashed line), and that modest clustering of SB patients was restricted to those with insulin in the low-normal range (for example in e: n = 39 SB and n = 27 SF; the dashed line in e, f, g marks the upper limit of normal insulin (20 μU/ml [33]), the stippled line mean insulin within the normal range).
Figure 3.
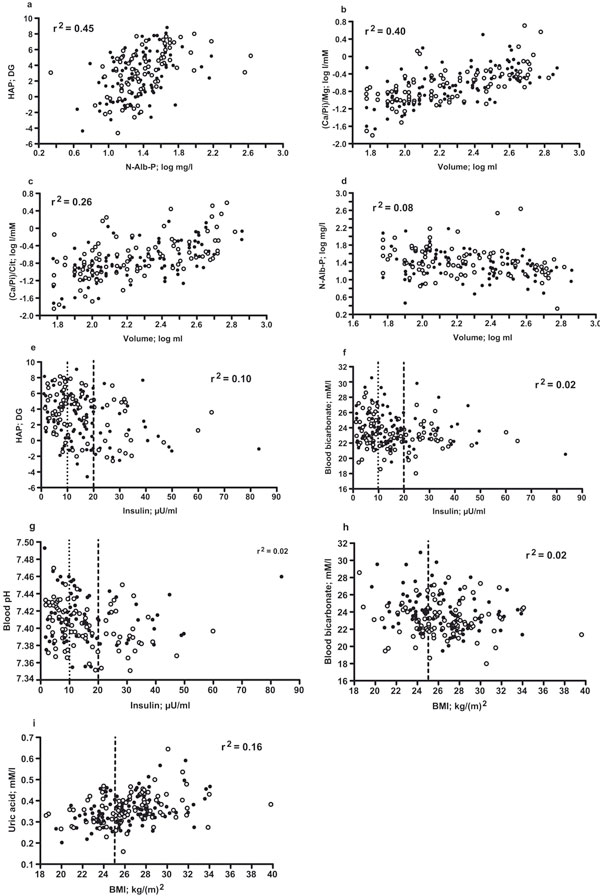
Appendix II. Extra relationships of parameters in urine (a-d), urine and plasma (e), blood, plasma and BMI (f-i) of part 2 (for abbreviations and symbols see Results section and Figure 1).
Table 4.
Simple correlations of parameters of oxidative metabolism, precursor oxypurines of uric acid, several complementary data from Table 3, and blood pressure (BP)
| Dependent1) | |||||||
|---|---|---|---|---|---|---|---|
| U-FE-N-Alb-P | U-Xanthine | U-Volume | U-Na | P-Insulin | Systolic BP | Diastolic BP | |
| Log (% × 103) | Log μM | Log ml | Log mM | Log μU/ml | mm Hg | mm Hg | |
| Influential1) | |||||||
| BMI | -0.20; 181 | - 15; 75 | -0.10; 187 | -0.10; 187 | 0.48; 184 | 0.47; 152 | 0.49; 152 |
| U-Hypoxanthine | 0.26; 75 | 0.69; 75 | 15; 75 | - 13; 75 | -0.01; 73 | 0.43; 54 | 0.27; 54 |
| U-MDA | 0.30; 170 | 0.35; 74 | 0.54; 176 | 0.34; 176 | 06; 177 | 0.23; 144 | 0.22; 144 |
| P-Uric acid | -0.26; 184 | -0.35; 75 | -0.18; 187 | -0.19; 187 | 0.28; 184 | 0.22; 152 | 0.21; 152 |
| U-MDA/P-Uric acid | 0.38; 170 | 0.46; 74 | 0.57; 176 | 0.39; 176 | 07; 176 | 0.13; 144 | 0.12; 144 |
| P-TAS | -0.39; 57 | -0.24; 57 | -0.20; 57 | -0.21; 57 | 0.37; 57 | 0.08; 41 | 0.18; 41 |
U-urine; P-plasma. Coefficients in bold (p ≤0.05) are followed by number of pairs.
1) For other abbreviations see text, for dimensions Tables 3 and 1 (log data are used, except BMI and TAS).
Nucleation of ca salts (Part 3)
Table 3 represents low and high (below and above the median) fasting urinary MDA, plasma uric acid and the ratio of these parameters, their association with postprandial urine T-Ox, T-Ca, T-Ca/102xT-Ox, pH, supersaturation with CaOx and HAP, and with several complementary parameters from the fasting 2 h period (see Tables 1 and 2). Despite the presence of high MDA and high MDA/Uric acid, both signalling oxidant excess (7, 13), T-Ox was unchanged. Conversely, when plasma uric acid, signalling antioxidant deficiency [21], was low, T-Ca and T-Ca/102xT-Ox were decreased too; this latter constellation was associated with elevation of FE-N-Alb-P, volume, Na in fasting urine, and decrease of fasting plasma insulin and BMI.
Table 3.
A - Low and High fasting urinary (U) MDA excretion, plasma (P) concentration of uric acid, ratio U-MDA/P-Uric acid; B-Ca saltcrystal nucleation and other data in postprandialurine; C - complementary data from fasting period
| A | B | C | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FASTING | POSTPRANDIAL | COMPLEMENTARY | ||||||||||
| Lowx | T-Oxa | T-Cab | T-Ca/ | pHc | CaOx | HAP | FE-N-Alb-P | U-Volume | U-Na | Insulin | BMI | |
| Highxx | 102 × T-Ox | |||||||||||
| mM/l | mM/l | ΔG | ΔG | % × 103 | ml | mM | μU/ml | kg/(m)2 | ||||
| U-MDA; nM1) | 89(2);88 | 0.52(0.03);25 24(3);20 | 0.51(0.08);20 | 5.46(0.09);29 1.3(0.1);84 | 3.1(0.3);84 | 1.5(0.1);85 | 167(13);88 | 11(0.5);88 | 16(1);86 | 26.0(0.3);88 | ||
| 175(5);88 | 0.53(0.03);35 20(3);26 | 0.44(0.07);26 | 5.57(0.08);44 0.5(0.1);80 | 2.7(0.3);80 | 2.9(0.6);85 | 283(17);88 | 14(0.8);88 | 18(1);87 | 26.7(0.4);88 | |||
| p | < 0.001 | 0.41d | 0.18d | 0.23d | 0.17 | < 0.001 | 0.17 | 0.002d | < 0.001 | < 0.001 | 0.15 | 0.08 |
| P-Uric acid; | 301(10);94 | 0.54(0.03);30 16(2);25 | 0.34(0.06);25 | 5.72(0.10);36 0.8(0.1);88 | 3.5(0.3);88 | 2.9(0.6);91 | 252(17);94 | 13(0.6);94 | 14(1);93 | 25.4(0.3);94 | ||
| μM -1-1 2) | 425(10);93 | 0.52(0.02);33 27(3);24 | 0.59(0.08);24 | 5.35(0.07);43 1.0(0.1);84 | 2.1(0.3);84 | 1.5(0.1);90 | 197(14);93 | 12(0.7);93 | 18(1);91 | 27.2(0.3);93 | ||
| p | < 0.001 | 0.29d | 0.007d | 0.007d | 0.001 | 0.18 | 0.001 | < 0.008d | 0.007 | 0.03 | 0.003d | < 0.001 |
| U-MDA/P-Uric acid; | 244(6);88 | 0.52(0.03);27 25(3);20 | 0.55(0.09);20 | 5.41(0.08);33 1.3(0.1);78 | 2.8(0.4);78 | 1.4(0.1);85 | 157(12);88 | 11(0.5);88 | 17(1);86 | 26.6(0.3);88 | ||
| nM/mM -l-1 3) | 525(18);88 | 0.54(0.03);33 19(3);26 | 0.41(0.06);26 | 5.62(0.08);40 0.6(0.1);86 | 3.0(0.3);86 | 3.0(0.7);88 | 293(17);88 | 14(0.8);88 | 16(1);87 | 26.1(0.4);88 | ||
| P | < 0.001 | 0.26d | 0.09d | 0.13d | 0.04 | < 0.001 | 0.38 | < 0.001d | < 0.001 | < 0.001 | 0.18 | 0.13 |
Mean values (SE), followed by number of observations. For further information see Table 2 and text.
x, xx: indicate ≤ and > median, respectively; 1), 2), 3): medians are 123, 349 and 337, respectively; a: at original pH; b: at prefixed urine pH 6.0; c: original urine pH; d: based on log10 data.
Figure 2 shows the morphology (A-1 - C-1) and mineral nature (A-2 - C-2) of the formed solids. In the majority of the 49 samples studied at prefixed urine pH 6.0 the first appearing crystal was spheroidal CaPi (molar ratio approx. 1.6, synonymous HAP, see A-1; n = 31), followed by rhomboidal CaOx dihydrate crystals (weddellite) together with small amounts of CaPi (molar ratio approx. 1.0, synonymous amorphous and poorly crystallized CaPi, see B-1; n = 18); note that in only one of the latter samples plate-like brushite (Ca/P 1.0) was formed (not depicted). At original urine pH, weddellite was exclusively detected (C-1) but no phosphorus peak (C-2). Potassium and chloride peaks were detectable in A-2 - C-2, but both peaks were sizable in B-2. APPENDIX, I (Table 5) (codes 22-24) shows that Log T-Ox and Log T-Ca tended to correlate negatively, whereas Log T-Ca and Log plasma uric acid, Log (T-Ca/102 × T-Ox) and Log plasma uric acid approached the level of significance. Neither correlated T-Ox, T-Ca, T-Ca/102 × T-Ox significantly with the original urinary pH and Ca salt supersaturation in postprandial urine, nor with any of the complementary parameters of the fasting period. On the other hand, upon rearrangement of data of Tables 2 and 3, numerous correlations were identified, linking BMI, hypoxanthine, TAS and the three parameters of oxidative metabolism to a series of dependent variables (Table 4): each of the former variables impacted significantly on FE-N-Alb-P (positively or negatively), but xanthine and uricemia correlated negatively, xanthine and hypoxanthine positively.
Figure 2.
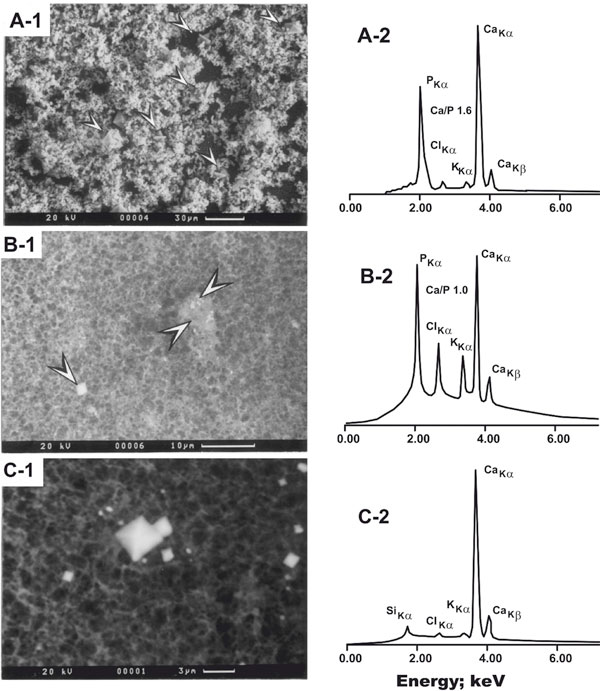
Electron micrographs of selected precipitates as developed in postprandial urine (A-1 - C-1), the pertinent energy peaks of elements, and the calcium/phosphorus peak ratio (inserted as Ca/P; A-2 and B-2). Note that in A-1 - C-1 the scale is different, and that in B2 (coexistence of mature CaOx crystals and poorly crystallized CaPi) the peaks of potassium (KKα) and chloride (ClKα) are impressive (for further information see text). A-1: Massive accumulation of mostly spheroidal CaPi crystals and interspersed rhomboidal CaOx dihydrate crystals (arrowheads). Note that Ca/P is approx. 1.6, indicative of hydroxyapatite; the situation is typical for Ca excess at urine pH 6.0 of patients in whom the first appearing crystal was CaPi. B-1 and B-2: Co-precipitation of CaOx (left arrowhead) and poorly crystallized CaPi (right arrowheads); note that Ca/P is approx. 1.0; the situation is typical for Ca excess at urine pH 6.0 of patients in whom the first appearing crystal was CaOx. C-1 and C-2: Rhomboidal CaOx dihydrate (weddellite), typically formed upon induction of Ox excess at original urine pH (for more information see text and Table 3). Note the absence of a P peak. Reproduced from [18] with kind permission of Walter De Gruyter, Berlin, Germany.
Discussion
General remarks
The disclosure that Ox in the physiological range is nontoxic to renal tissue [34], contrasting with earlier reports on a role as oxidant [16], forces to uncover the true etiology of IRCU. Although the present uncontrolled study cannot elicit the latter, it raises questions as to how Ox-independently disturbed oxidative metabolism can be integrated with IRCU pathophysiology. We can show that above pH 6.14 of fasting urine SB outnumber SF patients significantly, contrasting with earlier work [1] showing that the correlation between fasting urine pH and the number of SB patients was only of borderline significance (p = 0.059). We ascribe this discrepancy to the extremely careful clinical search for presence of stones; in addition, it indicates that, provided the urine pH is only weakly acid or alkaline, this will optimize renal crystal and stone forming processes. Thus, it would be unrealistic to assume that stones per se elicit the higher pH via, for example, diminished bicarbonate reabsorption by tubules [4]; even more important, reduction in size and total disappearance of stones did not improve the functional abnormality of tubules [35]. Therefore, the rise of urine pH, although etiologically insufficiently explicable by data presented in part 2, may be more complex and include diminished renal proton (H) generation, hyperfiltration of bicarbonate by glomeruli (see Tables 1, 2) and onset of defense mechanisms, together facilitating stone formation (see [36] and below). Because in the literature there is abundance of articles dealing with renal diseases as modified by oxidants and antioxidants, but virtually no reports with similar objectives as in present work, comments are made rather than discussing more in depth.
Variation of urinary N-Alb-P - guide to a role of ros in IRCU?
In IRCU, known as a disorder of affluence and approx. 50% recurrence rate within 5 years from the onset, the consumption of food with high protein content, producing acid ash and lowering urine pH, is frequent; despite, repercussions on Ca stone forming processes are poorly understood [1]. In animals fed a high protein diet increase of ROS production by mesangial cells has been interpreted as sequel of discrepant oxygen demand and supply, but only incommensurate increase of antioxidant defense mechanisms, ending up in glomerulo-tubulo-interstitial tissue damage [37]. Amplification of chronic kidney disease from obesity has been ascribed to association with oxidative stress [38], and overweight and obesity are frequent in IRCU (APPENDIX, II (Figure 3) (h, i)). Lipid uptake and lipoprotein degradation by human proximal tubular cells cause proteinuric disease [39], and renal lipid accumulation underlies impairment of Na+/H+ exchange [40]. Peroxidation of cell membrane lipids, MDA accumulation and cell death of renal epithelial cells are sequela of prior increase of formation of hydrogen peroxide (H2O2) [41,42]. Members of the ROS family also affect tubular enzymes with functions for the transport of Na and other ions, although the site(s) of action along the nephron [43,44] and involvement of vasopressin-independently impaired re-uptake of water via aquaporins [45], leading to enhanced diuresis, are uncertain. Kidney cortical cells exposed to hypoxia undergo ATP depletion [46], and H2O2 inhibits ATP-dependent proton extrusion mechanisms of tubular epithelial cells [47], in turn contributing to cellular acidosis and K loss via urine (presumably in exchange for H+ (Table 1)). The cells of ascending loop of Henle are most vulnerable due to limited receipt of blood and oxygen, but yet need to sustain high ATP production for electrolyte transport and synthesis of proteins [42] (note that among urinary N-Alb-P the quantitatively dominant fraction is Tamm-Horsfall protein, together with osteopontin powerful inhibitors of calcinosis and stones [48], but unclarified susceptibility to ROS excess and resulting state of inhibitor and/or promoter function in crystal and stone formation of IRCU [49]). Similarly, whether a rise of blood pressure is caused by oxidative stress (Tables 1, 4), hence becomes part of IRCU pathophysiology, is unsolved. In humans with chronic progressive kidney disease due to type 2 diabetes mellitus (mostly in association with insulin resistance of organs and hyperinsulinemia), blockade of angiotensin II receptors reduces oxidative damage of proteins and lipids, and markers of inflammation and fibrosis [50]; in that setting [50] blood pressure remained unchanged, and urinary MDA and protein turned out to be superior markers vis-à-vis measurement in plasma. From experiments in normotensive obese rats it was concluded that oxidative stress triggers the onset of kidney lesions in the absence of hyperglycemia, inflammation and hypertension [51]. Thus, if one accepts that increase of urinary hypoxanthine reflects transient, periodic or permanent tissue hypoxia, followed by deficient cellular ATP production, excess of ROS and MDA (as marker of deterioration of renal cell membrane function and exaggeration of release of N-Alb-P and probably other macromolecules into urine), then a good deal of our data (Tables 1A and 3; APPENDIX, I (Table 5), codes 1-10) would be in agreement with information from work in basic science and ROS markers in related clinical disorders [51,52].
CaPi nucleation and the making of Ca stones - key is an oxidatively modified environment?
Since the early reports that Ca stone patients, but not none-stone-forming humans, bear light microscopically visible calcified interstitial areas close to renal papilla [53], and that the vast majority of analyzed stones harbours a CaPi core [54], an unsolved conundrum exists as to whether CaPi or CaOx crystals appear first and at which renal anatomical site (tubular fluid, urine, interstitium?). With knowledge available to date, a more sound interpretation of the chain of events appears possible. In saturated urine-like solutions, containing Ca, Pi and macromolecules, heterogeneous nucleation of CaPi in the form of (Ca-rich) octacalcium phosphate is the dominant mechanism, provided the pH is weakly acidic or alkaline and kept constant together with Ca and Pi concentration (for details see [23,27]). Once in an oxidatively modified urinary environment the pH is high (Table 2) and there develops an additional need for dissipation of Ca ions from nephrons (note that with use of a closed crystallization system [31] the Ca concentrations tolerated by urine (Table 3) become unphysiologically high), either via excretion of Ca-poor CaPi or CaOx crystals in urine or, alternatively, via interstitial deposition as HAP (containing 10 moles Ca per mole), this latter appears as the preferred strategy and therefore may be fundamental for IRCU [53]. In this sense, Ca crystals in urine and Ca deposits in tissue may function as Ca sink. Figure 2, A-1-C-1, illustrates that at least in urine, with the pH varying within a broader range than in blood, heterogeneous nucleation [23,27] may run in both directions, depending on pH and abundance of reaction partners: Ca ions ⇋ Ca poor CaPi forms ⇋ Ca-richer CaPi forms ⇋ HAP ⇋ CaOx. If correct, this interpretation means that the situation in Figure 2B-1 is pivotal for Ca crystal nucleation, because it leaves the possibility of HAP formation along a rise of pH (Figure 2A-1), and of virtually complete CaPi dissolution and isolated presence of CaOx crystals once urine pH becomes increasingly acidic (Figure 2C-1); on the other hand, this view is limited by the sensitivity of the instrumentation in use for the detection of CaPi solid. Regarding the metabolic environment in interstitium, bicarbonate enrichment of blood (Table 2) may hint towards leakage of basolateral cell membranes for this anion, mainly of distal tubular cells, owing to a genetic defect [55] or acquired insufficient ATPase-dependent H+ generation. As mentioned above, clinically inapparent (intracellular) acidosis may be a so far neglected feature of IRCU, all the more as the elemental peaks of chloride and potassium (Figure 2, A-2-C-2) may - in the case of chloride - reflect malregulation of renal-tubular acid-base status, which is discussed as a possible factor in Ca stone etiology [57], and - in the case of potassium - is reminiscent of the transition of amorphous CaPi to HAP, a process that in calcifiying tissues is under the control of both cellular acid-base status [55] and matrix proteins [56]. In the light of present and other recent work the question "is ROS excess a primary event or secondary to interaction of renal epithelium with HAP [58]" deserves more conclusive addressing: 1) In IRCU HAP was found within the basolateral membrane of, and juxtapositioned outside, the thin part of loop of Henle [59,60], and alkalization of interstitium is pre-requisite for HAP formation [36]; 2) the inverse correlations of blood bicarbonate with insulin and BMI (APPENDIX, II (Figure 3), f and h), together with the observation that oxidatively modified metabolism in the form of a trend towards systemic alkaline tide (rise of blood bicarbonate and pH) coincides with a higher pressure to form stones (Table 2), are strong hints that ROS excess, extracellular bicarbonate accumulation and extratubular HAP development may be interrelated. Furthermore, once HAP invades epithelial cells and protrudes into tubular lumen [61] cellular particles serve as scaffolds and allow overgrowth of HAP either by CaPi, CaOx or both, depending on urine volume and the associated state of saturation of tubular fluid with these Ca salts [61,62]. The latter would imply that urinary physicochemical equilibria in fact determine the predominance of minerals in Ca stones (CaOx or CaPi) [54,62], but only indirectly. To reconcile this scenario with insufficient ROS neutralization, needs to consider the role of uric acid in this respect.
Plasma uric acid -friend or foe of IRCU?
The degree of uricemia, when within normal limits (Table 1, [33]), in the past failed to attract the interest of most researchers of IRCU pathophysiology. Even more, the roles of uric acid synthesis catalyzing xanthine oxidase as source of ROS and of uric acid as ROS scavenger in renal diseases of humans are controversially judged [[63-67]. In general, enzyme-substrate relationship dictates that uric acid biosynthesis is the higher, the greater the accumulation of precursor oxypurines. In present work, the negative correlation of urinary xanthine excretion and uricemia, but positive correlation of excretion of hypoxanthine and xanthine (Table 4) may be indirect proof of a primary defective xanthine oxidase activity. If yes, the nature of this finding is unknown. Therefore, once ROS are insufficiently buffered due to lowering of uric acid and possibly other scavengers [5,6] as reflected by plasma TAS [28], oxidative damage of vitally important tissues, pancreatic B-cells included [68,69], may enhance perturbations of acid-base [70] and mineral [71-73] homeostasis, ending up in renal stones.
Conclusions
Summarizing the data from the present uncontrolled tripartite study of constituents of urine and blood, and urinary crystallization risk, there is the impression that stone formation arises from disturbances of oxidative metabolism, secondary malregulation of functionality of nephrons and interaction with interstitial tissue, rather than from primarily disturbed physicochemical supersaturation of tubular fluid and urine. Therefore, re-evaluation of IRCU pathophysiology is recommended. In future controlled studies emphasis may be placed on 1) the nature of oxidative and impact on systemic metabolism; 2) the nature and function of proteins, especially N-Alb-P in urine and renal tissue (proteomics?); 3) the determinants of (in this order) urinary volume, Na, pH, and the pH inside renal cells of Ca stone patients.
Appendix
Note added in proof
Since the submission of present work 4 articles were published [see ref. [74-77], highlighting several aspects of IRCU pathophysiology and able to substantiate the idea that this disease is of cellular origin (electrolytes, proteins, genes), and probably shares defects seen in other related clinical disorders. For clinical calcium stone research a change of paradigms appears worthwhile, encompassing a shift from physico-chemistry of urine to renal soft tissue and cell research at the level of biochemistry and molecular biology, as well as interrelationships with systemic metabolism.
Note
* Dedicated to the late Professor Winfried Vahlensieck, former, Head of Department of Urology, University of Bonn, Germany, commemorating his pioneering work in clinical research of urolithiasis and the initiation of the Bonn - Vienna Urolithiasis Symposia series.
Acknowledgements
Over years the project was financially supported by the Erlangen University Hospital research Fund, the Wilhelm Sander Foundation, Munich, and the Deutsche Forschungsgemeinschaft, Bonn.
We owe thanks to the co-workers of the Department of Mineralogy, University of Erlangen-Nürnberg, for stone analysis, introduction into scanning electron microscopy and interpretation of crystal morphology. We are grateful to our coworkers for expert technical cooperation (M. Manoharan, K. Schwille) and expert secretarial work (ML. Rasenack), and to numerous physicians for referring patients to our laboratory.
References
- Schwille PO, Wipplinger J. Idiopathic recurrent calcium urolithiasis (IRCU): An acid meal challenge uncovers inappropriate pH of postprandial fasting and daily urine. A cross-sectional study of male patients providing insight into post-and pre-load urinary stone substances, crystallization risk, presence of renal stones, renal transport and systemic metabolic factors. Eur J Med Res. 2008;23:332–342. [PubMed] [Google Scholar]
- Finlayson B, Reid F. The expectation of free and fixed particles in urinary stone disease. Invest Urol. 1978;15:442–448. [PubMed] [Google Scholar]
- Kok DJ, Khan SR. Calcium oxalate nephrolithiasis, a free or fixed particle disease. Kidney Int. 1994;46:847–854. doi: 10.1038/ki.1994.341. [DOI] [PubMed] [Google Scholar]
- Jaeger P, Portmann L, Ginalski JM, Jaquet HF, Temler F, Burckhardt P. Tubulopathy in nephrolithiasis: consequence rather than cause? Kidney Int. 1986;29:563–571. doi: 10.1038/ki.1986.35. [DOI] [PubMed] [Google Scholar]
- Schwille PO, Manoharan M, Schmiedl A. Is idiopathic recurrent calcium urolithiasis in males a cellular disease? Laboratory findings in plasma, urine and erythrocytes, emphasizing the absence and presence of stones, oxidative and mineral metabolism: an observational study. Clin Chem Lab Med. 2005;43:590–600. doi: 10.1515/CCLM.2005.103. [DOI] [PubMed] [Google Scholar]
- Anuradha CV, Selvam R. Increased lipid peroxidation in the erythrocytes of kidney stone formers. Ind J Biochem Biophys. 1989;26:39–42. [PubMed] [Google Scholar]
- Huang HS, Ma MC, Chen CF, Cheng J. Lipid peroxidation and its correlations with urinary levels of oxalate, citric acid and osteopontin in patients with renal calcium ox-alate stones. Urology. 2003;62:1123–1128. doi: 10.1016/S0090-4295(03)00764-7. [DOI] [PubMed] [Google Scholar]
- Tungsanga K, Sriboonlue P, Futrakul P, Yachantha C, Tosukhowong P. Renal tubular cell damage and oxidative stress in renal stone patients and the effect of potassium citrate treatment. Urol Res. 2005;33:65–69. doi: 10.1007/s00240-004-0444-4. [DOI] [PubMed] [Google Scholar]
- Manoharan M, Schwille PO, Schmiedl A. In: Urolithiasis. Rodgers AL, Hibbert BE, Hess B, Khan SR, Preminger GM, editor. University of Cape Town; 2000. Are plasma and red blood cell (RBC) levels of antioxidative vitamins and uric acid disordered in idiopathic calcium urolithiasis (ICU)? A preliminary report; pp. 547–549. [Google Scholar]
- Esterbauer H. In: Oxidative Stress, Lipoproteins and Cardiovascular Dysfunction. Rice-Evans C, Bruckdorfer KR, editor. Portland Press London; 1995. The chemistry of oxidation of lipoproteins; pp. 55–79. [Google Scholar]
- Kehrer JP. Free radicals as mediators of tissue injury and disease. Crit Rev Toxicol. 1993;23:21–48. doi: 10.3109/10408449309104073. [DOI] [PubMed] [Google Scholar]
- Boonla C, Wunsuwan R, Tungsanga K, Tosukhowong P. Urinary 8-hydroxydeoxyguanosine is elevated in patients with nephrolithiasis. Urol Res. 2007;35:185–191. doi: 10.1007/s00240-007-0098-0. [DOI] [PubMed] [Google Scholar]
- Solin ML, Ahola H, Halthia A, Ursini F, Montine T, Roveri A, Kerjaschki D, Holthöfer H. Lipid peroxidation in human proteinuric disease. Kidney Int. 2001;59:481–487. doi: 10.1046/j.1523-1755.2001.059002481.x. [DOI] [PubMed] [Google Scholar]
- Boyce WH. In: Renal Stone Research Symposium. Hodgkinson A, Nordin BEC, editor. Churchill, London; 1969. Macromolecular components of kidney calculi in urine; pp. 181–190. [Google Scholar]
- Wesson JA, Ganne V, Beshensky AM, Kleinman JG. Regulation by macromolecules of calcium oxalate crystal aggregation in stone formers. Urol Res. 2005;33:206–212. doi: 10.1007/s00240-004-0455-1. [DOI] [PubMed] [Google Scholar]
- Scheid C, Koul H, Hill WA, Luber-Narod J, Kennington L, Honeyman T, Jonassen J, Menon M. Oxalate toxicity in LLC-PK1 cells: role of free radicals. Kidney Int. 1996;49:413–419. doi: 10.1038/ki.1996.60. [DOI] [PubMed] [Google Scholar]
- Schwille PO. In: Renal Tract Stone: Metabolic Basis and Clinical Practice. Wickham JEA, Buck A, editor. Churchill Livingstone, Edinburgh, London, Melbourne, New York; 1990. Rümenapf G: Idiopathic calcium urolithiasis - clinical problems and suggested approaches in an ambulatory stone clinic; pp. 217–238. [Google Scholar]
- Schwille PO, Schmiedl A, Manoharan M. Is calcium ox-alate nucleation in postprandial urine of males with idiopathic recurrent urolithiasis related to calcium phosphate nucleation and the intensity of stone formation? Studies allowing insight into a possible role of urinary free citrate and protein. Clin Chem Lab Med. 2004;42:283–293. doi: 10.1515/CCLM.2004.052. [DOI] [PubMed] [Google Scholar]
- Saugstad OD. Hypoxanthine as an indicator of hypoxia: its role in health and disease through free radical production. Pediatr Res. 1988;23:143–150. doi: 10.1203/00006450-198802000-00001. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Invited review: Oxygen during intermittent hypoxia: cellular and molecular mechanisms. J Appl Physiol. 2001;90:1986–1994. doi: 10.1063/1.1388173. [DOI] [PubMed] [Google Scholar]
- Rosell M, Regenström J, Kallner A, Hellenius ML. Serum urate determines antioxidant capacity in middle-aged men-a controlled randomized trial, and exercise intervention study. J Int Med. 1999;246:219–226. doi: 10.1046/j.1365-2796.1999.00522.x. [DOI] [PubMed] [Google Scholar]
- Grover PK, Resnick MI. Evidence for the presence of abnormal proteins in the urine of recurrent stone formers. J Urol. 1995;153:1716–1721. doi: 10.1016/S0022-5347(01)67511-6. [DOI] [PubMed] [Google Scholar]
- Campbell AA, Ebrahimpour A, Perez L, Smesko SA, Nancollas GH. The dual role of polyelectrolytes and proteins as mineralization promoters and inhibitors of calcium oxalate monohydrate. Calcif Tissue Int. 1989;45:122–128. doi: 10.1007/BF02561411. [DOI] [PubMed] [Google Scholar]
- Manoharan M, Schwille PO. Oxypurines, protein, glucose and the functional state of blood vasculature are markers of renal calcium stone-forming processes? Observations in men with idiopathic recurrent calcium urolithiasis. Clin Chem Lab Med. 2002;40:266–277. doi: 10.1515/CCLM.2002.043. [DOI] [PubMed] [Google Scholar]
- TenHuisen KS, Brown PW. Effects of magnesium on the formation of calcium-deficient hydroxyapatite from CaHPO4 · 2H2O and Ca4(PO4)2O. J Biomed Mat Res. 1997;36:306–314. doi: 10.1002/(SICI)1097-4636(19970905)36:3<306::AID-JBM5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Schwille PO, Schmiedl A, Hermann U, Fan J, Gottlieb D, Manoharan M, Wipplinger J. Magnesium, citrate, magnesium citrate and magnesium-alkali citrate as modulators of calcium oxalate crystallization in urine: observations in patients with recurrent idiopathic calcium urolithiasis. Urol Res. 1999;27:117–126. doi: 10.1007/s002400050097. [DOI] [PubMed] [Google Scholar]
- Nancollas GH, Zawacki SJ. Calcium phosphate mineralization. Conn Tiss Res. 1989;21:239–246. doi: 10.3109/03008208909050013. [DOI] [PubMed] [Google Scholar]
- Miller NJ, Rice-Evans C, Davies MJ, Gopinathan V, Milner A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin Sci. 1993;84:407–412. doi: 10.1042/cs0840407. [DOI] [PubMed] [Google Scholar]
- Schwille PO, Manoharan M, Rümenapf G, Wölfel G, Berens H. Oxalate measurement in the picomol range by ion chromatography: values in fasting plasma and urine of controls and patients with idiopathic calcium urolithiasis. J Clin Chem Clin Biochem. 1989;27:87–96. doi: 10.1515/cclm.1989.27.2.87. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Roseburgh NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Fan J, Schwille PO, Manoharan M, Gottlieb D, Schmiedl A. Calcium oxalate nucleation growth and aggregation in undiluted urine of healthy controls and patients with idiopathic recurrent calcium urolithiasis evaluated by a onetime small-volume test procedure. Effects of alkali citrate. Clin Lab. 1995;41:139–144. [Google Scholar]
- Werness PG, Brown CM, Smith LH. EQUIL-2: a basic computer program for calculation of urinary saturation. J Urol. 1985;134:1242–1244. doi: 10.1016/s0022-5347(17)47703-2. [DOI] [PubMed] [Google Scholar]
- Laposata M, editor. New England Journal of Medical Books. 1992. SI unit conversion guide; pp. 1–109. [Google Scholar]
- Schepers MS, van Ballegooijen ES, Bangma CH, Verkoelen CF. Oxalate is toxic to renal tubular cells only at supra-physiologic concentrations. Kidney Int. 2005;68:1660–1669. doi: 10.1111/j.1523-1755.2005.00576.x. [DOI] [PubMed] [Google Scholar]
- Pabico RC, McKenna BA, Freeman RB. Renal tubular dysfunctions in patients with idiopathic calcium nephrolithiasis. Min Electrol Metab. 1987;13:462–468. [PubMed] [Google Scholar]
- Halperin ML, Cheema Dhadli S, Kamel KS. Physiology of acid-base balance: links with kidney stone prevention. Sem Nephrol. 2006;26:441–446. doi: 10.1016/j.semnephrol.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Klahr S. Oxygen radicals and renal diseases. Min Electrol Metab. 1997;23:140–143. [PubMed] [Google Scholar]
- Ramos LF, Shintani A, Ikizler TA, Himmelfarb J. Oxidative stress and inflammation are associated with adiposity in moderate to severe CKD. J Am Soc Nephrol. 2008;19:593–598. doi: 10.1681/ASN.2007030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong AC, Moorhead JF. Tubular lipidosis: epiphenomenon of pathogenic lesion in human renal disease? Kidney Int. 1994;45:753–762. doi: 10.1038/ki.1994.100. [DOI] [PubMed] [Google Scholar]
- Bobulescu IA, Dubree M, Zhang J, Mc Leroy P, Moe OW. Effects of renal lipid accumulation on proximal tubule Na+/H+ exchange and ammonium secretion. Am J Physiol Renal Physiol. 2008;294:F1315–F1322. doi: 10.1152/ajprenal.00550.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan AM, Fitzpatrick S, Wang C, Wheeler DC, Lieberthal W. Lipid peroxidation contributes to hydrogen peroxide induced cytotoxicity in renal epithelial cells. Kidney Int. 1996;49:88–93. doi: 10.1038/ki.1996.12. [DOI] [PubMed] [Google Scholar]
- Salahudeen AK. Role of lipid peroxidation in H2O2-induced renal epithelial (LLC-PK1) cell injury. Am J Physiol. 1995;268(1 Pt 2):F30–38. doi: 10.1152/ajprenal.1995.268.1.F30. [DOI] [PubMed] [Google Scholar]
- Shah SV, Baliga R, Rajapurkar M, Fonseca VA. Oxidants in chronic kidney disease. J Am Soc Nephrol. 2007;18:16–28. doi: 10.1681/ASN.2006050500. [DOI] [PubMed] [Google Scholar]
- Garvin JL, Ortiz PA. The role of reactive oxygen species in the regulation of tubular function. Acta Physiol Scand. 2003;179:225–232. doi: 10.1046/j.0001-6772.2003.01203.x. [DOI] [PubMed] [Google Scholar]
- Heymann JB, Engel A. Aquaporins: Phylogeny, structure, and physiology of water channels. News Physiol Sci. 1999;14:187–193. doi: 10.1152/physiologyonline.1999.14.5.187. [DOI] [PubMed] [Google Scholar]
- Kim YK, Lee SK, Ha MS, Woo JS, Jung JS. Differential role of reactive oxygen in chemical hypoxia-induced cell injury in opossum kidney cells and rabbit cortical slices. Exp Nephrol. 2002;10:275–284. doi: 10.1159/000063702. [DOI] [PubMed] [Google Scholar]
- Kaufman DS, Goligorsky MS, Nord ER, Graber ML. Pertubation of cell pH regulation by H2O2 in renal epithelial cells. Arch Biochem Biophys. 1993;302:245–254. doi: 10.1006/abbi.1993.1206. [DOI] [PubMed] [Google Scholar]
- Mo L, Liaw L, Evan AP, Sommer AJ, Lieske JC, Wu XR. Renal calcinosis and stone formation in mice lacking osteopontin, Tamm-Horsfall protein, or both. Am J Physiol Renal Physiol. 2007;293:F1935–F1942. doi: 10.1152/ajprenal.00383.2007. [DOI] [PubMed] [Google Scholar]
- Cao LC, Boevé ER, Kok DJ, de Bruijn WC, de Water R, Romijn JC, Verkoelen CF, Schröder FM. In: Urolithiasis. Pak CYC, Resnik MI, Preminger GM, editor. Millet the Printer, Dallas; 1996. Oxidative modification of Tamm-Horsfall protein by hydrogen peroxide and ascorbate-Fe(III) pp. 337–338. [Google Scholar]
- Agarwal R. Proinflammatory affects of oxidative stress in chronic kidney disease: role of additional angiotensin II blockade. Am J Physiol Renal Physiol. 2003;284:F863–F869. doi: 10.1152/ajprenal.00385.2002. [DOI] [PubMed] [Google Scholar]
- Poirier B, Lannaud-Bournoville M, Conti M, Bazin R, Michel O, Bariéty J, Chevalier J, Myara I. Oxidative stress occurs in absence of hyperglycemia and inflammation in the onset of kidney lesions in normotensive obese rats. Nephrol Dial Transplant. 2000;15:2–3. doi: 10.1093/ndt/15.4.467. [DOI] [PubMed] [Google Scholar]
- Banupriya C, Ratnaker C, Doureradjou P, Mondal N, Vishnu B, Koner BC. Can urinary excretion rate of malonedialdehyde, uric acid and protein predict the severity and impending death in perinatal asphyxia? Clin Biochem. 2008;41:968–973. doi: 10.1016/j.clinbiochem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Randall A. The origin and growth of renal calculi. Ann Surg. 1937;105:1009–1027. doi: 10.1097/00000658-193706000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring LC. Observations on the analysis of ten thousand urinary calculus. J Urol. 1962;88:545–555. doi: 10.1016/S0022-5347(17)64842-0. [DOI] [PubMed] [Google Scholar]
- Bruce LJ, Unwin RJ, Wrong O, Tanner MJA. The association between familial distal renal tubular acidosis and mutations in the red cell anion exchanger (Band 3, AE 1)gene. Biochem Cell Biol. 1998;76:723–728. doi: 10.1139/bcb-76-5-723. [DOI] [PubMed] [Google Scholar]
- Wiesmann HP, Plate U, Zierold K, Höhling HJ. Potassium is involved in apatite biomineralization. J Dent Res. 1998;77:1654–1657. doi: 10.1177/00220345980770081401. [DOI] [PubMed] [Google Scholar]
- Silva IV, Morales MM, Lopes AG. Cl C5 chloride channel and kidney stones: what is the link? Braz J Med Biol Res. 2001;34:315–321. doi: 10.1590/S0100-879X2001000300004. [DOI] [PubMed] [Google Scholar]
- Escobar C, Byer KJ, Khaskeli H, Khan SR. Apatite induced renal epithelial injury: insight into the pathogenesis of kidney stones. J Urol. 2008;180:379–387. doi: 10.1016/j.juro.2008.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan EP, Lingeman JE, Coe FL, Parks JH, Bledsoe SB, Shao Y. et al. Randall's plaque of patients with nephrolithiasis begins in basement membranes of thin-loops of Henle. J Clin Invest. 2003;111:607–616. doi: 10.1172/JCI17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan AP, Coe FL, Lingeman JE, Worcester E. Insights on the pathology of kidney stone formation. Urol Res. 2005;33:383–389. doi: 10.1007/s00240-005-0488-0. [DOI] [PubMed] [Google Scholar]
- Low RK, Stoller ML, Schreiber CK. Metabolic and urinary risk factors associated with Randall's papillary plaques. J Endourol. 2000;14:507–510. doi: 10.1089/end.2000.14.507. [DOI] [PubMed] [Google Scholar]
- Evan AP, Coe FL, Lingeman JE, Shao Y, Sommr AJ, Bledsoe SB. et al. Mechanism of formation of human calcium oxalate renal stones on Randall's plaque. Anat Rec (Hoboken) 2007;290:1315–1323. doi: 10.1002/ar.20580. [DOI] [PubMed] [Google Scholar]
- Tan S, Radi R, Gaudier F, Evans RA, Rivera A, Kirk KA, Parks DA. Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatr Res. 1993;34:303–307. doi: 10.1203/00006450-199309000-00013. [DOI] [PubMed] [Google Scholar]
- Glantzounis GK, Tsimoyiannis EC, Kappas AM, Galavis DA. Uric acid and oxidative stress. Curr Pharm Des. 2005;11:4145–4151. doi: 10.2174/138161205774913255. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Kalaiselvi P, Sektivel R, Pragasam V, Muthu V, Varalakshmi P. Uric acid: an abettor or protector in calcium oxalate urolithiasis? Biochemical study in stone formers. Clin Chim Acta. 2005;353:45–51. doi: 10.1016/j.cccn.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Sem Nephrol. 2005;25:43–49. doi: 10.1016/j.semnephrol.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. 2008;19:1204–1211. doi: 10.1681/ASN.2007101075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou N, Torii S, Saito N, Hosaka M, Takeuchi T. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and C-Jun N-terminal kinase, and mitogen-activated protein kinase phosphates. Endocrinology. 2008;149:1654–1665. doi: 10.1210/en.2007-0988. [DOI] [PubMed] [Google Scholar]
- Duarte AI, Proenca T, Oliveira CR, Santos MS, Rego AC. Insulin restores metabolic function in cultured cortical neutrons subjected to oxidative stress. Diabetes. 2006;55:2863–2870. doi: 10.2337/db06-0030. [DOI] [PubMed] [Google Scholar]
- Klisic J, Hu MC, Nief V, Reyes L, Fuster D, Moe OW, Ambühl PM. Insulin activates Na+/H+ exchange 3: biphasic response and glucocorticoid dependence. Am J Physiol Renal Physiol. 2002;283:F532–F539. doi: 10.1152/ajprenal.00365.2001. [DOI] [PubMed] [Google Scholar]
- Ter Maaten JC, Voorburg A, Heine RJ, Ter Wee PM, Donker AJ, Gans RO. Renal handling of urate and sodium during acute physiological hyperinsulinemia in healthy subjects. Clin Sci. 1997;92:51–58. doi: 10.1042/cs0920051. [DOI] [PubMed] [Google Scholar]
- Ferranini E, Galvan AQ, Gastaldelli A, Camastra S, Sironi AM, Toschi E et al. Insulin: new roles for an ancient hormone. Eur J Clin Invest. 1999;29:842–852. doi: 10.1046/j.1365-2362.1999.00536.x. [DOI] [PubMed] [Google Scholar]
- We apologize that for the sake of space further articles in this context cannot be cited, and that the list of references is but a selection out of many excellent publications in the field
- Parks JH, Coe FL, Evan AP, Worcester EM. Urine pH in renal calcium stone formers who do and do not increase stone phosphate content with time. Nephrol Dial Transplant. 2009;24:130–6. doi: 10.1093/ndt/gfn420. Epub 2008 Jul 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerer T, Weiss C, Hammes HP, Braun C, Hesse A, Alken P, Knoll T. Evaluation of urolithiasis: a link between stone formation and diabetes mellitus? Urol Int. 2009;82:350–5. doi: 10.1159/000209371. [DOI] [PubMed] [Google Scholar]
- Miller NL, Gillen DL, Williams JC Jr, Evan AP, Bledsoe SB, Coe FL, Worcester EM, Matlaga BR, Munch LC, Lingeman JE. A formal test of the hypothesis that idiopathic calcium oxalate stones grow on Randall's plaque. BJU Int. 2009;103:966–71. doi: 10.1111/j.1464-410X.2008.08193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkema KY, Velic A, Dijkman HB, Verkaart S, van der Kemp AW, Nowik M, Timmermans K, Doucet A, Wagner CA, Bindels RJ, Hoenderop JG. The Calcium-Sensing Receptor Promotes Urinary Acidification to Prevent Nephrolithiasis. J Am Soc Nephrol. 2009. in press . [DOI] [PMC free article] [PubMed]