

Abstract
Many neurodegenerative disorders, although related by their destruction of brain function, display remarkable cellular and/or regional pathogenic specificity likely due to a deregulated functionality of the mutant protein. However, neurodegenerative disease genes, for example huntingtin (HTT), the ataxins, the presenilins (PSEN1/PSEN2) are not simply localized to neurons but are ubiquitously expressed throughout peripheral tissues; it is therefore paramount to properly understand the earliest precipitating events leading to neuronal pathogenesis to develop effective long-term therapies. This means, in no unequivocal terms, it is crucial to understand the gene's normal function. Unfortunately, many genes are often essential for embryogenesis which precludes their study in whole organisms. This is true for HTT, the β-amyloid precursor protein (APP) and presenilins, responsible for early onset Alzheimer's disease (AD). To better understand neurological disease in humans, many lower and higher eukaryotic models have been established. So the question arises: how reasonable is the use of organisms to study neurological disorders when the model of choice does not contain neurons? Here we will review the surprising, and novel emerging use of the model organism Dictyostelium discoideum, a species of soil-living amoeba, as a valuable biomedical tool to study the normal function of neurodegenerative genes. Historically, the evidence on the usefulness of simple organisms to understand the etiology of cellular pathology cannot be denied. But using an organism without a central nervous system to understand diseases of the brain? We will first introduce the life cycle of Dictyostelium, the presence of many disease genes in the genome and how it has provided unique opportunities to identify mechanisms of disease involving actin pathologies, mitochondrial disease, human lysosomal and trafficking disorders and host-pathogen interactions. Secondly, I will highlight recent studies on the function of HTT, presenilin γ-secretase and Hirano bodies conducted in Dictyostelium. I will then outline the limitations and future directions in using Dictyostelium to study disease, and finally conclude that given the evolutionary conservation of genes between Dictyostelium and humans and the organisms' genetic tractability, that this system provides a fertile environment for discovering normal gene function related to neurodegeneration and will permit translational studies in higher systems.
Keywords: Dictyostelium discoideum, Model organism, Neurodegeneration, Huntingtin, Presenilin, γ-secretase, Hirano bodies, Neurotransmitter homologues
Review
Dictyostelium - emergence of a classic model organism for the study of neurodegenerative disease
Dictyostelium discoideum is a species of soil-living amoeba belonging to the Kingdom Amoebozoa and phylum Mycetozoa. Dictyostelium, or social amoeba, is a eukaryote that when starved, transitions from individual, self-sustainable professional phagocytes into a highly coordinated developmental program (Figure 1a). During this period of multicellular development, the cells execute a series of morphological changes that proceed in defined stages over a 24 h period to form a bona fide multicellular organism. During the earliest developmental stage, cells secrete, and undergo chemotaxis toward cyclic adenosine monophosphate (cAMP) to form aggregation territories. The secretion of cAMP promotes a G protein-coupled receptor signaling cascade that results in the formation of discrete mounds containing as many as 100,000 cells [1,2]. Cells within the mound remain motile and are directed to differentiate into either prestalk or prespore cells, leading to morphogenetic changes yielding a multicellular stalk, supporting a ball of encapsulated dormant spores [3]. The entire process is clearly depicted in Figure 1b by scanning electron microscopy and although the life cycle appears simplistic, the entire process is complex with still many unresolved questions including how do cells secrete cAMP, regulate organism size, and initiate cell-fate choices to name but a few. Nevertheless, it is becoming clearer that Dictyostelium possesses signal-transduction pathways that are closely related to metazoans. Yet, with at least one major and exploitable fundamental difference: when animals undergo embryogenesis, they develop through coordinated cell division, morphogenetic movements and differentiation followed by growth of the organism. In contrast, Dictyostelium growth precedes development. This simplifies developmental studies and provides a novel route to examine with exceptional cellular clarity biological functions including cytokinesis, endocytosis, secretion, protein trafficking, intra- and extracellular signaling, gene expression, cell-cell communication, adhesion, differentiation and many biochemical aspects of cell motility. With the sequencing of the genome complete [4] we are entering a renaissance in the use of this organism as a biomedical research tool. Bacteria, yeasts and several other invertebrate or vertebrate model systems are known for their contribution to our understanding of basic biology and human disease, but Dictyostelium, unfortunately is rarely included among this list. Upon publication of the genome, public availability of RNA seq and DNA microarray data, research using Dictyostelium has revealed many common cellular features shared across various phyla, and that many genes encode for proteins more similar to their human counterparts than are those of yeasts (Saccharomyces cerevisiae) [4,5]. Yeasts are single, free-living cells that reproduce by budding, whose genome consists of 6,000 genes. Similar to Dictyostelium, experiments can be performed over hours, days or weeks whereas in mice these experiments might take years, if at all possible. Moreover, unlike yeasts, the 34 Mb, Dictyostelium genome is densely populated with 12, 646 genes and is surprisingly close in size to the 13,676 genes found in the Drosophila genome [6]. The fly, unlike Dictyostelium, is a multicellular animal with more complex behaviors and nervous system with a lifespan of 2-3 months and generational time of 10 days. Worms (Caenorhabditis elegans), also multicellular contains more genes (19,099) than Dictyostelium or the fly, a generation time of 3 days and lifespan of 2-3 weeks. Despite their obvious differences, relatively low cost compared to mice, and way of life, importantly, all of these model organisms make proteins that carry out the same core functions as in humans and thus might offer clues to pathogenic processes. Whereas higher eukaryotes often express a number of similar genes with redundant functions, Dictyostelium, due to its haploid genome often carries only a single homologous gene making genetic manipulation by targeted gene deletion simple, quick and at a fraction of the cost of higher eukaryotes. A quick scan of the genome reveals many genes that cause neurological disease including, but not limited to adrenoleukodystrophy (ABCD1), amyotrophic lateral sclerosis (SOD1), Miller-Dieker lissencephaly (LIS1), Parkinson's disease (UCHL1) and Neuronal ceroid lipofuscinosis (PPT1, CLN2, CLN3, CLN5) [4]. A conservative search (threshold value of e ≤ 10-40), detected 64 proteins involved in human diseases [4]. However, searches using advanced programs reveal many orthologous proteins that cause disease (e.g., PSEN1, PSEN2, HTT, Neurofibromatosis 1, LRRK2) such that the number of predicted human disease genes in Dictyostelium is greater than previously thought [7-11]. What I hope to convince you of in this short review is the importance of using all available model systems, and posit that, despite its' evolutionary distance, reverse genetics in Dictyostelium offers great promise for translational studies aimed at understanding the earliest events leading to neurodegeneration.
Figure 1.
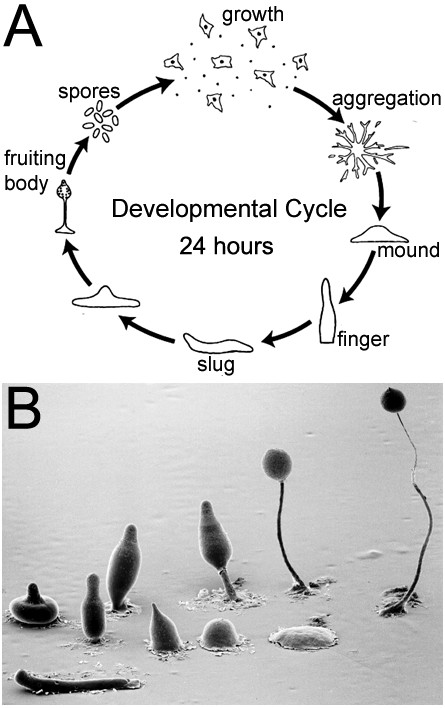
The life cycle of Dictyostelium discoideum. (A) Most of its life exists in its growth phase, as a haploid social amoeba preying upon bacteria in the soil and dividing by mitosis. Once the food source is depleted cells enter into a 24 h multicellular developmental program. During this transition, amoebae aggregate towards secreted cAMP by chemotaxis in the thousands to form a tight mound and then enter a stage where cells begin to differentiate. Cells within the mound remain motile and are directed to differentiate, by secreted morphogens, into either prestalk or prespore cells, culminating to form a fruiting body comprised of a multicellular stalk that supports a ball of encapsulated dormant spores. (B) Scanning electron microscopy showing the various structures formed during development. Permissions: CC Creative Commons Attribution - Share Alike 3.0, David Brown & Joan E. Strassmann. SEM courtesy of MJ Grimson & RL Blanton, Biological Sciences Electron Microscopy, Texas Tech University.
Alzheimer's disease: The amyloid precursor protein and discovery of presenilin-dependent γ-secretase activity
Alzheimer's disease (AD) is the most common cause of dementia, estimated to contribute to about 60 to 70% of cases [12]. AD is a terminal disease that culminates in death. Impaired recent memory usually is an initial symptom of AD, but other cognitive deficits (e.g., changes in attention, problem-solving abilities) may also be present. Amyloid plaques and neurofibrillary tangles, the characteristic pathological hallmarks of AD, are believed to be causative and accrue in the years preceding clinical symptoms. Amyloid plaques are accumulations of insoluble protein aggregates in the extracellular space of the brain and the primary component of plaques is the Aβ peptide, a 38- to 43-amino acid peptide derived from the larger pre-cursor protein APP [13,14]. Mutations in APP [15,16] and two other genes, presenilin-1 (PSEN1) [17] and presenilin-2 (PSEN2), have been identified that result in autosomal dominant forms of early-onset FAD [18]. The intimate relationship between APP and the presenilins in the production of Aβ is beyond the scope of this review but is detailed elsewhere [19]. The presenilins are homologous, ubiquitously expressed multiple transmembrane domain proteins and many seminal reports provide compelling evidence that presenilins provide the catalytic core of γ-secretase [20-23]. The protein composition of the γ-secretase complexes have been resolved and each consist of four proteins that are all required for proteolytic activity [24-26]: APH-1, PEN2, and nicastrin [25,27]. The normal biological function of the presenilins has been difficult to determine largely due to the fact that it is essential and knockout mice die in utero. To fully understand and appreciate the normal in vivo function of the presenilins they must be studied in the context of organismal models including Dictyostelium.
The identification of presenilins and γ-secretase activity in Dictyostelium
A recent development in Dictyostelium biomedical research was the identification of homologous genes to presenilin 1, presenilin 2, nicastrin, aph-1 and pen-2, the core components that constitute the γ-secretase complex [8]. In humans, mutations in the presenilins are intimately involved in the aberrant proteolytic processing of APP in a pathway leading to FAD. How the mutations cause disease, or more importantly the time course or precipitating events that lead to the pathogenesis of AD are not known, and as such, highlight the importance of uncovering the normal function of the γ-secretase complex. In this respect, Dictyostelium represents a novel organismal model, to aid in further understanding the normal physiological role of the presenilins and γ-secretase from a conserved and evolutionary point of view in a system that has no Notch or APP equivalent [4]. To study the γ-secretase in Dictyostelium, strains were created containing single and/or double gene disruptions for ps1-, ps2-, aph1-, ncst- and ps1/ps2-, aph1/ps2-, ncst/ps2-, and aph/ncst- nulls. It is important to note that mutants of these genes in Dictyostelium are viable, unlike the embryonic lethality seen in mammalian systems, and that mutants display a variety of phenotypic defects during specific periods of development. Moreover, phylogenetic sequence analysis reveals that the Dictyostelium presenilins are structurally similar to human presenilin rather than the related signal peptidyl proteases (Figure 2A-D), and a majority of mutations in human PSEN causing early-onset FAD [28] are conserved in Dictyostelium PSEN proteins [8].
Figure 2.
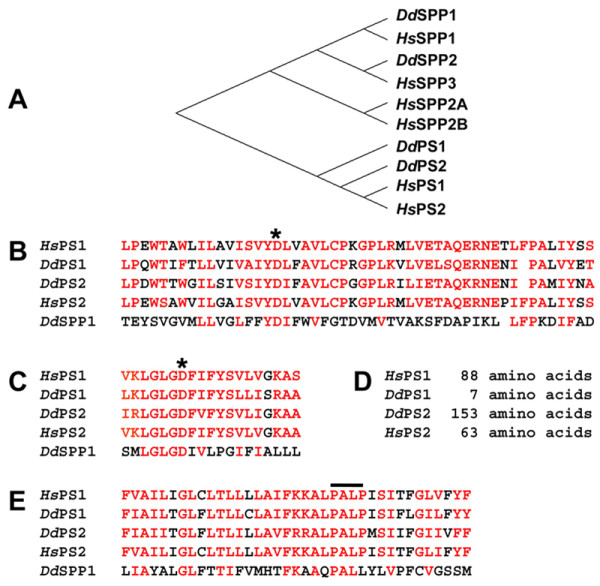
Dictyostelium PS proteins align with human presenilins. (A) Phylogenic comparison of amino acid sequences of Dictyostelium and the human PS and signal peptidyl proteases (SPP) proteins. The Dictyostelium PS proteins cluster closely with human PS. (B-E) Sequence alignments of functional domains of PS and SPP proteins. Similar amino acid residues are indicated in red. Residue alignment surrounding the N-terminal enzymatic aspartate (*) is shown. Residue alignment surrounding the C-terminal enzymatic aspartate (*) is shown. Residue alignment within the conserved, overlined PALP domain region. Permissions: McMains, V.C. et al. (2010). Dictyostelium possesses highly diverged presenilin/γ-secretase that regulates growth and cell-fate specification and can accurately process human APP: a system for functional studies of the presenilin/γ-secretase complex. Dis. Model. Mech. 3, pp. 581-594.
The presenilin γ-secretase complexes regulate cell differentiation and phagocytosis in Dictyostelium
Cells deficient for ps1 did not show post-aggregative developmental defects compared to cells deficient for ps2 suggesting that in Dictyostelium, PSEN1 γ-secretase complexes may carry out different functional activities than PSEN2 γ-secretase complexes during specific phases of the life cycle. This observation fits well with the suggestion that that there may be several distinct presenilin complexes that have different biological activities [29]. Wild type and ps1- cells are able to complete development within 24 h, whereas ps2-, aph1- and ncst- mutants not only form fewer fruiting bodies, but predominantly arrest as abnormal intermediate structures within the same time frame [8]. The authors also show that in Dictyostelium γ-secretase complexes function to regulate cell differentiation as measured by cell-type specific gene expression and firmly establish that presenilin γ-secretase activity is required in a cell-autonomous pathway that determines cell fate during development [8]. Once again, these in vivo observations support a large body of suggestive evidence for presenilin γ-secretase playing an essential role in cellular and neuronal differentiation during neural development and adult neurogenesis in mouse and zebrafish models [30,31]. Cells deficient for γ-secretase also display a reduced ability for phagocytosis. Quantification of phagocytic rates for the various mutant strains revealed that cells lacking PSEN1 had highly reduced rates of phagocytosis, but this was not the case for ps2- cells. The described phagocytosis defect supports observations that mammalian presenilins are present in lysosomal membranes, but also strengthens the argument that distinct presenilin complexes exist and have different biological activities. Further to this, when wild type amoeba were made to express a variant of human APP, the cells were clearly able to process APP in a manner identical to mammalian cells resulting in the differential production of C-terminal fragments and Aβ40/Aβ42, the toxic fragment believed to be the causative agent in AD (Figure 3).
Figure 3.
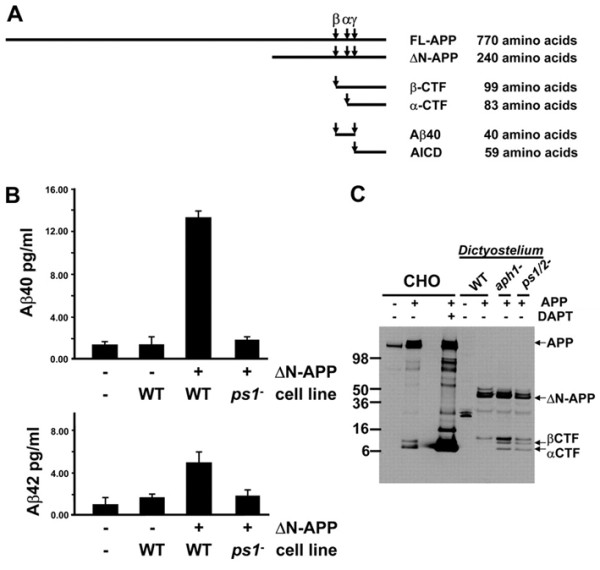
Dictyostelium have PS-dependent γ-secretase activity that processes human APP to release Aβ peptides. (A) Schematic showing various APP derived products and their respective sizes. (B) ΔN-APP, a truncated human APP expressed in transformed wild type (WT) and ps1-null cells were analyzed for secreted levels of Aβ40 and Aβ42 peptides by quantitative ELISA. Fresh media and media conditioned by native WT cells were used a negative controls. Bars indicate standard errors derived from two independent experiments, each with two replicates. (C) Protein samples were collected from native and APP-expressing CHO cells untreated or treated with DAPT, from growing WT Dictyostelium or WT, aph1-null and ps1/2-null Dictyostelium that express ΔN-APP. APP expression and processing was determined by immunoblot assay using anti-APP C-terminus antibodies. Permissions: McMains, V.C. et al. (2010). Dictyostelium possesses highly diverged presenilin/γ-secretase that regulates growth and cell-fate specification and can accurately process human APP: a system for functional studies of the presenilin/γ-secretase complex. Dis. Model. Mech. 3, pp. 581-594.
This work demonstrates the evolutionary conservation of this key regulatory enzyme. The most obvious value stemming from this work for the study of presenilin function are the possibilities for elucidating presenilin-independent function(s) as wild-type presenilins are required for proper protein degradation through the autophagosome-lysosome system thereby implicating a role for these proteins in autophagy [32]; secondly, the exciting prospects surrounding the finding that proteolytic processing of APP occurs in Dictyostelium exactly the same way as has been shown animal cells. This finding is important not only because it provides an excellent system in which to test therapeutic agents that target Aβ production in vivo, but it is remarkable that this enzyme, given the evolutionary distance between Dictyostelium and humans, is capable of cleaving a non-endogenous substrate to produce C-terminal fragments and ratios of secreted Aβ40/Aβ42 identical to mammalian systems. Knowledge gained about human diseases including cancers from the use of yeast cannot be denied, and with Dictyostelium, we also have an equally genetically tractable organism that not only contains genes uncannily similar to humans, but as a single cell behaves much like an animal cell and further offers the ability to study gene function under conditions of multicellularity all in the same model. Even with the evolutionary distance between humans and Dictyostelium one might expect genes to play different roles, but with regard to amyloidogenic processing of APP, this does not appear to be the case. It should be mentioned that a survey of the Dictyostelium genome does not readily identify or predict the presence of either the known α-secretase or β-secretase [4] and suggests the presence of yet to be identified first-cleavage sheddases either unique to Dictyostelium or potentially conserved across metazoan species. Moreover, this model offers a biochemical clarity not offered by any other system to date to study γ-secretase function directly, free from the multiple secondary effects that can arise from aberrant proteolytic processing of more than 60 identified substrates [33]. This study provides strong in vivo evidence for presenilin function in regulating phagocytosis, only previously suspected in animal cell culture [34]. Lastly, with the availability of Dictyostelium strains carrying multiple loss-of-function mutations to the components of the γ-secretase, this model system allows for the rapid systematic analysis of how specific mutations to human presenilins might result in gain-of-function or loss-of-function effects that can be readily assessed in higher models. Further studies will be required to determine whether the mammalian PSEN gene can rescue Dictyostelium ps1-null phenotypes and proteolytic processing of human APP.
Higher organism and cellular models of Huntington's disease
Huntington's disease (HD) is a fatal neurodegenerative disease [35] also known as Huntington's chorea, that affects muscle coordination from preferential loss of efferent medium spiny neurons in the striatum of the basal ganglia, leads to cognitive decline, dementia and eventually death [36-38]. Genetic evidence suggests the disease is caused by an unstable CAG trinucleotide repeat within the coding region of the htt gene leading to expansion (> 35) of glutamine (Q) residues near the amino-terminus of the protein [35]. Mutant HTT confers an unknown gain-of-function property to the protein, but might also impair normal protein function [39]. Huntingtin is a large (350 kDa) alpha-solenoid HEAT (huntingtin, elongation factor 3, the A subunit of protein phosphatase 2A, and TOR1) repeat protein [40] with little resemblance to any known protein. The normal function of HTT, the nature of the polyQ mutation which is expressed in both neuronal and non-neuronal cells and how it contributes to pathogenesis still remains unclear [41]. Although genetically precise mouse models for HD have contributed significantly towards understanding HTT normal function, these mice are expensive to create, to maintain and therefore difficult to recapitulate results. Thus, defining the disease-producing 'gain-of-function' - either a polyglutamine-length dependent increase or deregulation of a normal huntingtin activity or the introduction of a novel polyglutamine-length dependent activity, will require an understanding of the protein's normal function(s).
A wide range of HD animal models are currently available and have been used to investigate pathological pathways, molecular targets, and potential therapeutics. Invertebrates such as Drosophila melanogaster, non-mammalian species (e.g., Danio rerio) and mammals, including mice, rats, sheep and non-human primates, have been genetically engineered to model the HD mutation. Although these systems are extremely important tools to understand HD, a large majority of studies focus upon the late end-stage disease symptoms and thus offer minimal insight into early manifestation including HTT normal function. The existence of HD cell lines has been, to a finite degree, useful for the dissection of huntingtin function and assessment of potential therapeutic compounds [42,43]. Further, in vitro cell lines often do not show overt defects suggesting they are unable to reproduce the pathophysiological mechanisms induced by the mutant gene.
Many established model systems are based upon an amino-terminal fragment that results in perturbation of many cellular processes but these remove the polyglutamine tract from its normal protein context. Therefore, one cannot understate that strategies are badly needed to reveal full length huntingtin-dependent biochemical processes that contribute to HD pathology. Because of the evolutionary conservation of huntingtin, lower organism systems offer an attractive and affordable route that has yet to be exploited to understand huntingtin function.
The identification of Dictyostelium huntingtin
The Dictyostelium genome was screened and found to contain a single gene with sequence homology to human huntingtin. Bioinformatic and phylogenetic analysis of the primary amino acid sequence placed the protein firmly within the huntingtin family, including size, and the presence of numerous HEAT and HEAT-like repeats. Dictyostelium mutants' deficient for HTT cells are viable, unlike the embryonic lethality seen in higher eukaryotes, but are fragile in that they display a number of subtle phenotypes suggesting that HTT is involved in a number of cellular processes [9]. We show that huntingtin deficiency impacts upon osmoregulation, cation homeostasis, cell motility, cell shape, chemotactic cAMP relay, homotypic cell-cell adhesion, cell fate determination and cytokinesis in Dictyostelium [9,44]. Importantly, these phenotypic deficits are greatly dependent upon the environment in which the cells are placed which suggests that HTT aids in translating extracellular signals into cellular processes. These findings support the large body of evidence that suggest in metazoa, huntingtin is a multifunctional protein with roles in embryogenesis, cell fate, cytoskeleton, apoptosis, vesicular and mitochondrial transport, iron homeostasis, autophagy, energy metabolism and transcriptional regulation [39,41,45,46]. A major issue of concern is which, if any, of these functions directly involve HTT or are secondary effects to HTT deficiency or mutant HTT activity. The power behind the Dictyostelium system is that it allows for the identification of in vivo HTT functions at the level of both single cells and a multi-cellular environment all in the same organism with a biochemical and cellular clarity to identify functions that directly depend on HTT.
Dictyostelium cells deficient for huntingtin display cell shape and cytoskeletal defects
When htt- cells are placed under low ionic strength phosphate buffer, the cells elicit a round phenotype without membrane extensions, a reduction of F-actin at the cortex of the cell and under hypoosmotic conditions rapidly swell and undergo complete lysis within 5-6 h [9]. This fits well with reports that mutant HTT in cells from HD patients or when normal huntingtin levels are reduced, cells display defective actin-remodeling under conditions of stress [47] and consistent with mammalian studies that huntingtin regulates neurological processes including actin-rich dendritic spine formation and membrane branching [48,49]. In vitro binding of the HTT N-terminus to F-actin has been reported [50] and may indicate a normal, perhaps transient, function for HTT in the regulation of the actin cytoskeleton. Dictyostelium provides a valuable tool to uncover mechanisms by which HTT regulates actin-cytoskeleton responses.
Huntingtin modulates chemotaxis in Dictyostelium
The failure of htt- cells to osmoregulate and sensitivity to extreme hypoosmotic shock secondarily affects intracellular ion homeostasis. As a consequence, unless specific cations (Ca2+ or Mg2+) are provided exogenously htt- cells appear unable to initiate cAMP-induced Ca2+-transients that may act in a feedback loop to positively reinforce cAMP relay which impairs chemotaxis during Dictyostelium development. Although the a role for HTT in chemotaxis has not been established in mammalian systems, a consensus is gradually emerging that the dyshomeostasis of Ca2+ is an important factor in the linkage of the HTT mutation to the onset and progression of the disease [51] and together suggest abnormal Ca2+ signaling and other Ca2+ signaling proteins should be explored further as an evolutionary conserved role for HTT.
Huntingtin regulates cell fate during development in Dictyostelium
It was shown that HTT modulates cell fate in Dictyostelium (Figure 4). Using chimeras where cells are marked with GFP, htt- cells in the presence of wild type cells fail to populate the prespore region of the slug, and as a consequence do not become spores [9]. Cell fate defects in huntingtin deficient cells have also been observed in vertebrates. In zebrafish, reduced expression of huntingtin differentially targets development of telencephalic neurons compared to mid- and hind-brain [52]. In mouse chimeras, Hdh-/- cells preferentially colonize the hypothalamus, midbrain, and hindbrain relative to the telencephalon and the thalamus during early development [53]. Thus, like these latter neuronal populations, Dictyostelium cells require huntingtin for the proper development of viable spores. Not surprisingly, the data thus far do not provide a simple definition for a single normal function for huntingtin, but, as huntingtin deficiency in Dictyostelium produces pleiotropic effects, the findings are consistent with the consensus from mammalian studies that huntingtin is a multifunctional protein that impacts upon many biochemical processes. Exciting new avenues of research have emerged in delineating which of these functions are conserved in mammals. Ultimately, determining to what degree these functions are altered by expansion of the polyglutamine tract in human huntingtin will provide much needed insights into the mechanism by which mutant huntingtin triggers HD pathogenesis.
Figure 4.
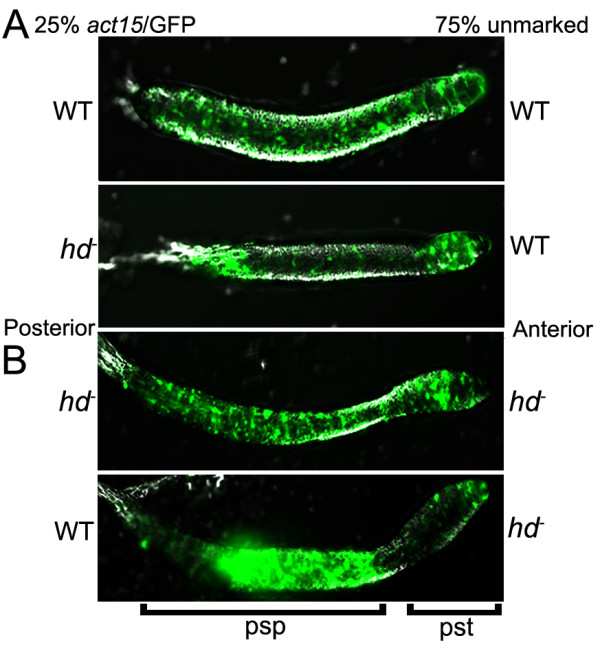
Huntingtin regulates prespore/spore differentiation cell-autonomously. (A) GFP was expressed in htt- cells and their position in chimeras with unlabelled wild type cells was monitored. GFP:htt- cells fail to populate the prespore domain. (B) Reverse experiment in which GFP: WT cells in chimeras with unlabelled htt- cells occupy the prespore domain. The prestalk (pst) and prespore (psp) domains are underlined. Cell types within each region are shown and slugs are arranged with the front facing towards the right. Permission: Myre M.A. et al. (2009). Deficiency of huntingtin has pleiotropic effects in the social amoeba Dictyostelium discoideum. PLoS Genetics, 7(4), pp. e1002052.
Insight into Hirano bodies - Dictyostelium discoideum
Hirano bodies, described about 40 years ago, are bright eosinophilic intracytoplasmic inclusions/protein aggregates with crystalloid fine rod structures that occur preferentially in the neuronal processes of individuals with neurodegenerative diseases including Alzheimer's disease, Creutzfeldt-Jacob disease, amyotrophic lateral sclerosis, parkinsonism-dementia and Pick's disease [54,55]. Hirano bodies are largely considered a pathological hallmark of postmortem brain samples from patients suffering from neurodegenerative disorders representing "tombstones" of neuronal cell death. Hirano bodies have been reported in glial cells, in peripheral nerve axons and in extraocular muscles of eyes [56] suggesting they are more than products of cell death. It should be noted that Hirano bodies, as a function of age, appear in individuals without neurodegeneration [57]. Hirano bodies are complex inclusions comprised of many proteins including actin and actin-associated proteins (cofillin and alpha-actinin), tau, a C-terminal fragment of APP; microtubule associated bundling proteins, and neurofilaments [55]. Moreover, they have been described in various experimental animals. Understanding the underlying pathological molecular mechanism responsible for their formation has been precluded by the lack of either an in vitro or in vivo experimental model system.
Research using Dictyostelium revealed that formation of Hirano bodies is a pathological event [58]. The expression in Dictyostelium of the CT fragment (amino acids 124-295) of the 34 kDa protein, the 34 kDa protein is one of 11 actin crosslinking proteins present in Dictyostelium, a protein that exhibits activated actin binding and calcium-insensitive actin filament cross-linking activity induced cells to form ellipsoidal paracrystalline inclusions that contain ordered assemblies of F-actin, CT-myc, myosin II, cofilin and alpha-actinin that remarkably resembled the structure of Hirano bodies. They extended their findings to mammalian murine cells and discovered that F-actin rearrangements caused by a reduction in stress fibers accumulate as numerous punctate foci and large aggregates: Hirano bodies [58]. So the failure to regulate either the activity and/or affinity of an actin cross-linking protein provides a signal that forms Hirano bodies. Whereas wild type cells complete development in 24 h, CT-myc cells contained significantly higher amounts of F-actin, formed fruiting bodies with delayed developmental timing by ~6 h. The accumulation of F-actin was also shown to occur in murine cells expressing CT-myc. This report established for the first time that Hirano bodies are not restricted to mammalian cells or nerve cells occur because of aberrant function of the actin cytoskeleton and that Hirano bodies can exert defects on normal cell function. Because of using Dictyostelium, Hirano bodies and induction of F-actin rearrangements in mammalian cell cultures by expression of the CT 34-kDa actin-bundling protein was found to occur in HEK 293, HeLa, Cos7 cells, neuroblastoma and astrocytic cells, and in primary neurons [59].
Conclusion
Several Dictyostelium genes are homologous or retain a significant amount of structural conservation to human genes making the organism a useful biomedical model system. With the entire genome sequenced and publicly available in a model organism database called dictyBase, it allows molecular and cellular biologists to examine with more clarity the complex multifunctional aspects of gene function. However, one limitation of using unicellular organisms (e.g., yeast) as models for assessing the function of genes involved in disease is that pathologies which affect specific organs cannot be as readily assessed as it might be in the relevant organism. This limitation may also apply to genes that are present in unicellular organisms but are required in a more stringent manner in specialized tissues, or expressed as a variety of different isoforms in specific cell types. However, this limitation has not precluded the use of lower organisms to understand cell division and cancers in many different organs. Although Dictyostelium develops into bona fide multicellular organism, it does present a limitation in the number of cell types that can be analyzed and therefore discoveries made using this organism must be rigorously assessed in the relevant mammalian cell type. Coupled with translational experiments and validation in higher eukaryotes, research using Dictyostelium will expedite and contribute to our understanding of human neurological diseases. Individual cell behavior accounts for the many phases of health and pathogenic mechanisms that initiate disease. This has been elegantly portrayed in Dictyostelium with respect to the precise production of Aβ from the heterologous expression of human APP. Chemotaxis is a critical cellular function with implications in embryogenesis. Dictyostelium cells deficient for HTT show highly defective chemotactic behavior coupled with cytoskeletal/membrane deficits under conditions of low ionic conditions that may reveal conserved roles for this protein in axon guidance, neuritic extensions and embryogenesis. Future endeavors that increases the amount of translational research conducted in higher organisms or neuronal cell models will be beneficial for understanding both Dictyostelium cellular physiology and, more importantly, the cellular mechanisms of human disease. I wish to posit that multiple model systems can and should be employed in the cross-genomic analysis of human neurodegenerative disease genes to address multiple basic eukaryotic cellular functions (e.g., Dictyostelium), to their assembly into various types of more complex molecular pathways (e.g., flies and worms), and then validated and assessed in models of human neurodegenerative disease (e.g., mice).
Abbreviations
HTT: Huntingin; PSEN1: Presenilin 1; PSEN2: Presenilin 2; APP: Amyloid precursor protein; AD: Alzheimer's disease; HD: Huntington's disease; cAMP: Cyclic adenosine monophosphate; FAD: Familial Alzheimer's disease; HEAT: Huntingtin: elongation factor 3: the A subunit of protein phosphatase 2A: and TOR1.
Competing interests
The author declares that they have no competing interests.
Authors' contributions
MAM contributed to the conception, design and drafting of this manuscript.
Acknowledgements
I wish to thank Marcy E. MacDonald and James F. Gusella for their support, foresight in using this particular model organism and numerous insightful, scientific discussions.
MM was supported by grants NINDS NS032765, Massachusetts HD Center Without Walls NS016367, Huntington's Disease Society of America Coalition for the Cure, and the CHDI Foundation.
References
- Parent CA, Devreotes P. Molecular genetics of signal transduction in Dictyostelium. Annu Rev Biochem. 1996;65:411–440. doi: 10.1146/annurev.bi.65.070196.002211. [DOI] [PubMed] [Google Scholar]
- Soderbom F, Loomis B. Cell-cell signaling during Dictyostelium development. Trends Microbiol. 1998;6(10):402–406. doi: 10.1016/S0966-842X(98)01348-1. [DOI] [PubMed] [Google Scholar]
- Williams JG, Duffy KT, Lane DP, McRobbie SJ, Harwood AJ, Traynor D, Kay RR, Jermyn KA. Origins of the prestalk-prespore pattern in Dictyostelium development. Cell. 1989;59(6):1157–1163. doi: 10.1016/0092-8674(89)90771-X. [DOI] [PubMed] [Google Scholar]
- Eichinger L, Pachebat JA, Glöckner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, Tunggal B, Kummerfeld S. et al. The genome of the social amoeba Dictyostelium discoideum. Nature. 2005;435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh A, Miranda ER, Katoh-Kurasawa M, Fuller D, Rot G, Zagar L, Curk T, Sucgang R, Chen R, Zupan B, Loomis WF, Kuspa A, Shaulsky G. Conserved developmental transcriptomes in evolutionarily divergent species. Genome Biol. 2010;11(3):R35. doi: 10.1186/gb-2010-11-3-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE. et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- McMains VC, Myre MA, Kreel L, Kimmel AR. Dictyostelium possesses highly diverged presenilin/gamma-secretase that regulates growth and cell-fate specification and can accurately process human A: a system for functional studies of the presenilin/γ-secretase complex. Dis Models Mech. 2010;3(9-10):581–594. doi: 10.1242/dmm.004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myre MA, Lumsden AL, Thompson MN, Wasco W, Macdonald ME, Gusella JF. Deficiency of huntingtin has pleiotropic effects in the social amoeba Dictyostelium discoideum. PLoS Genet. 2011;7(4):e1002052. doi: 10.1371/journal.pgen.1002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Steimle PA, Ren Y, Ross CA, Robinson DN, Egelhoff TT, Sesaki H. Iijima: Dictyostelium huntingtin controls chemotaxis and cytokinesis through the regulation of myosin II phosphorylation. M Mol Biol Cell. 2011;22(13):2270–2281. doi: 10.1091/mbc.E10-11-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Charest PG, Firtel RA. Spatiotemporal regulation of Ras activity provides directional sensing. Curr Biol. 2008;18(20):1587–1593. doi: 10.1016/j.cub.2008.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, Waters C, Jimison P, Shepherd E, Sevush S, Graff-Radford N, Newland D. et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16(4):203–212. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta. 2000;1502(1):172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1995;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Ying-Hui F, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, TSuda T, Mar L. et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer's disease. Mol Brain. 2011;4:3. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398(6727):522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38(1):9–12. doi: 10.1016/S0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci USA. 2003;100(11):6382–6387. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L. et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and Aβ processing. Nature. 2000;407(6800):48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- Kim SD, Kim J. Sequence analyses of presenilin mutations linked to familial Alzheimer's disease. Cell Stress Chaperones. 2008;13:401–412. doi: 10.1007/s12192-008-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Sanjo N, Chen F, Hasegawa H, Petit A, Ruan X, Li W, Shier C, Kawarai T, Schmitt-Ulms G, Westaway D, St. George-Hyslop P, Fraser PE. The presenilin proteins are components of multiple membrane-bound complexes that have different biological activities. J Biol Chem. 2004;279(30):31329–31336. doi: 10.1074/jbc.M401548200. [DOI] [PubMed] [Google Scholar]
- Baumeister R. The physiological role of presenilins in cellular differentiation: lessons from model organisms. Eur Arch Psychiatry Clin Neurosci. 1999;249(6):280–287. doi: 10.1007/s004060050100. [DOI] [PubMed] [Google Scholar]
- van Tijn P, Kamphuis W, Marlatt MW, Hol EM, Lucassen PJ. Presenilin mouse and zebrafish models for dementia: focus on neurogenesis. Prog Neurobiol. 2011;93(2):149–164. doi: 10.1016/j.pneurobio.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Neely KM, Green KN, LaFerla FM. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a γ-secretase-independent manner. J Neurosci. 2011;31(8):2781–2791. doi: 10.1523/JNEUROSCI.5156-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JV, Twomey C, Wujek P. Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell Mol Life Sci. 2009;66(9):1534–1555. doi: 10.1007/s00018-009-8435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras I, Laplante A, Boulais J, Brunet S, Thinakaran G, Desjardins M. γ-secretase is a functional component of phagosomes. J Biol Chem. 2005;280(43):36310–36317. doi: 10.1074/jbc.M504069200. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's Disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1998;85(15):5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60(10):1615–1620. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65(5):745–747. doi: 10.1212/01.wnl.0000174432.87383.87. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: An alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6(12):919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Andrade MA, Bork P. HEAT repeats in the huntingtin protein. Nat Genet. 1995;11(2):115–116. doi: 10.1038/ng1095-115. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90(3):905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Bolognini D, Mutti C, Zuccato C, Tartari M, Sola F, Valenza M, Kazantsev AG, Cattaneo E. Loss of huntingtin function complemented by small molecules acting as repressor element 1/neuron restrictive silencer element silencer modulators. J Biol Chem. 2007;282(34):24554–24562. doi: 10.1074/jbc.M609885200. [DOI] [PubMed] [Google Scholar]
- Varma H, Voisine C, DeMarco CT, Cattaneo E, Lo DC, Hart AC, Stockwell BR. Selective inhibitors of death in mutant huntingtin cells. Nat Chem Biol. 2007;3(2):99–100. doi: 10.1038/nchembio852. [DOI] [PubMed] [Google Scholar]
- Wang Y, Steimle PA, Ren Y, Ross CA, Robinson DN, Egelhoff TT, Sesaki H, Iijima. Dictyostelium huntingtin controls chemotaxis and cytokinesis through the regulation of myosin II phosphorylation. M Mol Biol Cell. 2011;22(13):2270–2281. doi: 10.1091/mbc.E10-11-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden AL, Henshall TL, Dayan S, Lardelli MT, Richards RI. Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum Mol Genet. 2007;16(16):1905–1920. doi: 10.1093/hmg/ddm138. [DOI] [PubMed] [Google Scholar]
- Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR, Rubinsztein DC. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008;412(2):191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- Munsie L, Caron N, Atwal RS, Marsden I, Wild EJ, Bamburg JR, Tabrizi SJ, Truant R. Mutant huntingtin causes defective actin remodeling during stress: defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet. 2011;20(10):1937–1951. doi: 10.1093/hmg/ddr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Merriam EB, Hu X. The dynamic cytoskeleton: backbone of dendritic spine plasticity. Curr Opin Neurobiol. 2011;21(1):175–181. doi: 10.1016/j.conb.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP. Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;12:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli S, Shao J, Diamond MI. F-actin binding regions on the androgen receptor and huntingtin increase aggregation and alter aggregate characteristics. PLoS One. 2010;5(2):e9053. doi: 10.1371/journal.pone.0009053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I. Inositol 1,4,5-tripshosphate receptor, calcium signalling and Huntington's disease. Subcell Biochem. 2007;45:323–335. doi: 10.1007/978-1-4020-6191-2_11. [DOI] [PubMed] [Google Scholar]
- Henshall TL, Tucker B, Lumsden AL, Nornes S, Lardelli MT. Selective neuronal requirement for huntingtin in the developing zebrafish. Hum Mol Genet. 2009;18(24):4830–4842. doi: 10.1093/hmg/ddp455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Del Mar N, Meade CA, Yang H, Dragatsis I. Neurons lacking huntingtin differentially colonize brain and survive in chimeric mice. J Neurosci. 2001;21(19):7608–7619. doi: 10.1523/JNEUROSCI.21-19-07608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier L, Galvez S, Gajdusek DC. Familial clustering of the ataxic form of Creutzfeldt-Jacob disease with Hirano bodies. J Neurol Neurosurg Psychiatry. 1985;48(3):234–238. doi: 10.1136/jnnp.48.3.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A. Hirano bodies and related neuronal inclusions. Neuropath Appl Neurobiol. 1994;20:3–11. doi: 10.1111/j.1365-2990.1994.tb00951.x. [DOI] [PubMed] [Google Scholar]
- Tomanaga M. Hirano body in extraocular muscle. Acta Neuropathol. 1983;60:309–313. doi: 10.1007/BF00691883. [DOI] [PubMed] [Google Scholar]
- Gibson PH, Tomlinson BE. Numbers of Hirano bodies in the hippocampus of normal and demented people with Alzheimer's disease. J Neurol Sci. 1997;33:199–206. doi: 10.1016/0022-510x(77)90193-9. [DOI] [PubMed] [Google Scholar]
- Maselli AG, Davis R, Furukawa R, Fechheimer M. Formation of Hirano bodies in Dictyostelium and mammalian cells induced by expression of a modified form of an actin-crosslinking protein. J Cell Sci. 2002;115:(Pt 9):1939–1949. doi: 10.1242/jcs.115.9.1939. [DOI] [PubMed] [Google Scholar]
- Davis RC, Furukawa R, Fechheimer M. A cell culture model for investigation of Hirano bodies. Acta Neuropathol. 2008;115(2):205–217. doi: 10.1007/s00401-007-0275-9. [DOI] [PubMed] [Google Scholar]