

Abstract
The folate enzyme, FDH (10-formyltetrahydrofolate dehydrogenase, ALDH1L1), a metabolic regulator of proliferation, activates p53-dependent G1 arrest and apoptosis in A549 cells. In the present study, we have demonstrated that FDH-induced apoptosis is abrogated upon siRNA knockdown of the p53 downstream target PUMA. Conversely, siRNA knockdown of p21 eliminated FDH-dependent G1 arrest and resulted in an early apoptosis onset. The acceleration of FDH-dependent apoptosis was even more profound in another cell line, HCT116, in which the p21 gene was silenced through homologous recombination (p21−/− cells). In contrast to A549 cells, FDH caused G2 instead of G1 arrest in HCT116 p21+/+ cells; such an arrest was not seen in p21-deficient (HCT116 p21−/−) cells. In agreement with the cell cycle regulatory function of p21, its strong accumulation in nuclei was seen upon FDH expression. Interestingly, our study did not reveal DNA damage upon FDH elevation in either cell line, as judged by comet assay and the evaluation of histone H2AX phosphorylation. In both A549 and HCT116 cell lines, FDH induced a strong decrease in the intracellular ATP pool (2-fold and 30-fold, respectively), an indication of a decrease in de novo purine biosynthesis as we previously reported. The underlying mechanism for the drop in ATP was the strong decrease in intracellular 10-formyltetrahydrofolate, a substrate in two reactions of the de novo purine pathway. Overall, we have demonstrated that p21 can activate G1 or G2 arrest in the absence of DNA damage as a response to metabolite deprivation. In the case of FDH-related metabolic alterations, this response delays apoptosis but is not sufficient to prevent cell death.
Keywords: ALDH1L1, apoptosis, cell cycle arrest, folate metabolism, p21, PUMA
Introduction
The p21 (WAF1/Cip1) protein is an inhibitor of cyclin-dependent kinases1-3 and a predominant member of the Cip/Kip family of Cdk inhibitors.4,5 As a Cdk inhibitor, p21 controls cell cycle progression and negatively regulates cellular proliferation.2,3 In support of the role of p21 in controlling the cell cycle, p21 knockout mice, while undergoing normal development, lack the ability to arrest in G1 in response to DNA damage or nucleotide pool perturbation.6 The p21 gene is also one of the main transcriptional targets of the p53 tumor suppressor7 and is required for p53-dependent G1 and G2 cell cycle arrest.8-10 For example, DNA damage produced by ionizing radiation or by treatment with drugs such as adriamycin induces p53 and p21, leading to G1 and/or G2 arrest.10-12
The cell cycle arrest is a common cellular response to DNA damage and is viewed as a delay period in DNA replication during which the cell can attempt to repair the damage. If this attempt fails, cell death pathways will be activated. Based on this paradigm, the p21-induced cell cycle arrest is considered as an antiapoptotic response.13 Numerous studies support this notion, indicating that inhibition of cell cycle progression by elevated levels of p21 prevents apoptosis.13-17 Likewise, the loss of p21 often enhances apoptosis induced by certain stimuli.18 Contrary to such studies, some reports suggested that p21 could also have a proapoptotic role. For example, ectopic expression of p21 in the p53-defective human ovarian adenocarcinoma cell line SKOV3 not only makes the cells more susceptible to apoptosis but also enhances the cytotoxic effect of cisplatin.19 It has been also reported that p21 is required for sodium butyrate–induced apoptosis in MCF-7 cells.20 In line with this observation, it has been demonstrated that elevated p21 increases expression of proapoptotic protein Bax and accelerates apoptosis in C6-ceramide–treated Hep3B cells.21 Of note, p21 can directly regulate apoptosis independently of its function in the cell cycle control.16,17 Overall, p21 perhaps can evoke either proapoptotic or antiapoptotic responses, depending on the cell type, stress stimuli, and accompanying signaling events.13,15,22
In cell culture experiments, p21 is often up-regulated in response to treatment with anticancer drugs.22 Among these drugs are antifolates, a class of antimetabolites used in the treatment of cancer since the late 1940s.23 Several studies have demonstrated that MTX (inhibitor of dihydrofolate reductase), AG2034 (inhibitor of glycinamid ribonucleotide formyltransferase), and pemetrexed (inhibits both of the above enzymes plus thymidylate synthase, TS) all result in the increase of p21 levels.24-27 These drugs target folate metabolism, ultimately inhibiting de novo purine and TMP biosynthesis. It has been also reported that sensitivity to 5-FU, another TS inhibitor, was increased in cells upon elevation of p21 levels.28 This effect was associated with decreased expression of TS due to transcriptional regulation of the TS gene through the p21/CDK axis.29 A connection between folate metabolism and p21 was also demonstrated in the study by Crott et al., in which a decrease of media folate was accompanied by a concomitant increase in the p21 transcript levels.30
Elevation of p21 was also observed in studies of FDH (10-formyltetrahydrofolate dehydrogenase, ALDH1L1), a major folate enzyme controlling the overall flux of one-carbon groups through the folate pool.31 The enzyme functions as a metabolic regulator, which removes carbon groups, in the form of CO2, from the reduced folate pool, thus diverting them from biosynthetic pathways.32,33 This function results from FDH-catalyzed reaction of the conversion of 10-formyltetrahydrofolate (10-fTHF) to tetrahydrofolate (THF) and CO2 in a NADP+-dependent manner.33 FDH, being abundant in normal tissues, is commonly down-regulated or silenced in cancers.34 One of the main mechanisms of such regulation is FDH gene (ALDH1L1) promoter methylation.35 In our previous experiments, the expression of the enzyme in FDH-deficient A549 cells resulted in G1 cell cycle arrest and p53-dependent apoptosis.31 These effects are likely the result of a strong depletion of intracellular purine pools associated with the FDH enzymatic activity.36 In the present study, we have demonstrated metabolic alterations induced by this enzyme and evaluated the role of p21 in cytotoxic effects of FDH on cancer cells.
Results
Knockdown of p21 in Tet-On A549/FDH cells
We have previously shown that FDH induces expression of p21 in a p53-dependent manner in A549 cells.31 The overall response to FDH in p53-positive cells was G1 arrest and apoptosis. To determine whether p21 is acting as an antiapoptotic or proapoptotic signal in response to FDH, we first investigated the effect of FDH expression in Tet-On A549/FDH cells31 with knocked-down p21. The p21 knockdown was achieved using a siRNA approach with p21-specific short-chain oligonucleotides (Table 1). We simultaneously transfected Tet-On A549/FDH cells with p21-targeted siRNA and induced FDH expression with doxycycline.31 In agreement with our published studies,31 in control nontransfected cells or cells transfected with scrambled siRNA, we observed the appearance of p21 protein 24 hours after FDH induction, with its further graduate elevation between 24 and 72 hours after FDH induction (Fig. 1). In contrast, transfection of FDH-expressing cells with p21-targeting siRNA resulted in a significant decrease in p21 mRNA and protein levels, observed as early as 24 hours after transfection, with the indication of a more complete p21 knockdown at 48 and 72 hours (Fig. 1A and 1B). Of note, equal levels of FDH expression in control, scrambled, and siRNA transfected cells were demonstrated in these experiments (Fig. 1B).
Table 1.
Oligonucleotides Used for siRNA and PCR Experiments
| Target | siRNA oligonucleotides | PCR primers |
|---|---|---|
| CDKN1A/p21 | 5′-GAACUUCGACUUUGUCACCGAGACA-3′ | Forward 5′-gcgccatgtcagaaccgtcag-3′ |
| 5′-UGUCUCGGUGACAAAGUCGAAGUUC-3′ | Reverse 5′-GGGCTTCCTCTTGGAGAAGATC-3′ | |
| BBC3/PUMA | 5′-UUGUACAGGACCCUCCAGGGUGAGG-3′ | Forward 5′-TCCTCAGCCCTCGCTCTCGC-3′ |
| 5′-CCUCACCCUGGAGGGUCCUGUACAA-3′ | Reverse 5′-CCGATGCTGAGTCCATCAGC-3′ |
Figure 1.

Silencing of p21 prevents FDH-induced G1 arrest and accelerates apoptosis in A549 cells. (A) Levels of p21 mRNA in A549 cells after transfection with p21-specific siRNA and FDH induction. (B) Levels of FDH and p21 proteins (Western blot) at different times of FDH induction in untreated (control), treated with scrambled siRNA (Scr), and treated with p21-specific siRNA (siRNA). Actin is shown as loading control. (C) Distribution of cells between cell cycle phases in p21-proficient cells without FDH (top), p21-proficient cells after FDH induction (middle), and p21-silenced cells after FDH induction (bottom). Assay was carried out at 48 hours after FDH induction. (D) Apoptosis evaluated by annexin V/PI staining in p21-proficient (scrambled) and silenced (siRNA) cells in the presence (+) and absence (–) of FDH. (E) Calculation of apoptotic cells (% of the total cells) from D. Error bars represent ±SD (n = 3).
As expected, p21 silencing prevented G1 arrest in response to FDH (Fig. 1C), confirming that FDH-induced folate stress leads to the arrest through p21 as a downstream target. In contrast, apoptosis was seen in p21-deficient as well as p21-proficient cells (Fig. 1D). Calculation of the number of apoptotic cells revealed that, in response to FDH, p21-deficient cells exhibited enhanced apoptosis at the earlier time point compared to p21-expressing cells, with about 1.5-fold increase in apoptotic cells at 48 hours after FDH induction (Fig. 1E). Interestingly, this difference became much smaller at 72 hours after the FDH induction (about 1.2-fold increase in p21-deficient cells), indicating a strong apoptotic response in both p21-proficient and p21-deficient cells (Fig. 1D and 1E).
Effects of FDH expression on p21-deficient HCT116 cells
To further investigate the effects of FDH in p21-deficient cells, we transiently expressed FDH in HCT116 and HCT116 p21−/− cells. Expression of FDH induced the expression of the p21 protein in HCT116 cells, whereas HCT116 p21−/− cells lack such capabilities (Fig. 2A). Analysis of the apoptotic cell population by annexin V/PI staining (Fig. 2B) demonstrated that FDH expression results in a significant increase in apoptotic cells at 24 and 48 hours after FDH transfection in HCT116 p21−/− cells as compared to p21-positive HCT116 cells (Fig. 2C). Remarkably, by 72 hours after FDH transfection, HCT116 p21+/+ and HCT116 p21−/− cells show little difference in the number of apoptotic cells (Fig. 2C).
Figure 2.

FDH-induced antiproliferative effects are different in HCT116 p21+/+ and p21−/− cells. (A) Levels of FDH and p21 proteins (Western blot) at different times after FDH transfection. Actin is shown as loading control. (B) Apoptosis evaluated by annexin V/PI staining in p21+/+ and p21−/− cells in the presence (+) and absence (–) of FDH. Time after FDH transfection is indicated. (C) Calculation of apoptotic cells (% of the total cells) from B. (D) Distribution of HCT116 p21+/+ and p21−/− cells between cell cycle phases at 48 hours after FDH transfection. Error bars represent ±SD. For statistical analysis, a Student t test was performed (*P < 0.05, n = 3).
We have previously reported that FDH induces G1 arrest in A549 cells, which we attributed to the p21 activation.31 In the present study, such an arrest was not seen in p21 knocked-down cells (above), which provides direct evidence for the p21-dependent G1 arrest in FDH-expressing cells. To confirm this functional association between FDH and p21, we evaluated the distribution of HCT116 cells (p21+/+ and p21−/−) between cell cycle phases in the absence and presence of FDH. Surprisingly, we have observed that in HCT116 p21+/+ cells, FDH induced strong G2 arrest instead of G1 arrest seen in A549 cells (Fig. 2D). Furthermore, this arrest was not seen in p21-deficient (HCT116 p21−/−) cells (Fig. 2D).
siRNA-mediated knockdown of PUMA prevents FDH-induced apoptosis
The above data suggested that p21 functions to prevent FDH-induced apoptosis without direct participation in apoptotic signaling. To confirm these findings, we studied whether the p53 downstream target PUMA, which is also up-regulated in response to FDH, is solely responsible for p53-dependent FDH-induced apoptosis. Toward this end, we investigated the effects of PUMA knockdown on FDH-induced apoptosis in Tet-On A549/FDH cells. Utilizing a PUMA-specific siRNA (Table 1), we were able to abolish the expression of PUMA mRNA and protein in response to FDH expression at 48 and 72 hours after siRNA transfection (Fig. 3A). We further observed that cells with knocked-down PUMA were not sensitive to FDH: FDH expression in cells transfected with scrambled siRNA caused approximately 50% of the cell population to stain positive for annexin V 48 hours after doxycycline addition, whereas cells transfected with PUMA siRNA showed very little annexin V staining (Fig. 3B and 3C). While p21 only partially and temporarily protects cells against FDH-induced apoptosis, the PUMA knockdown provides complete and permanent protection. Thus, these data highlight PUMA as a main downstream effector of FDH-induced apoptosis in p53-proficient cells.
Figure 3.

PUMA knockdown protects A549 cells from FDHinduced apoptosis. (A) Levels of PUMA mRNA (left) and protein (right) at different times after FDH induction. (B) Apoptosis evaluated by annexin V/PI staining. (C) Calculation of apoptotic cells (%) from B. Error bars represent ±SD. For statistical analysis, a Student t test was performed (**P < 0.005, n = 3).
Effect of FDH on DNA damage and ATP levels
G1 or G2 arrest is a common cellular response to DNA damage.37 These arrests can be also activated by nucleotide pool deprivation.38-40 We have evaluated DNA damage in A549 and HCT116 cells upon FDH expression by assessing the levels of histone H2AX phosphorylation. These experiments indicated that DNA damage does not take place in response to FDH elevation (Fig. 4A). In contrast, the ATP pool was decreased about 2- and 30-fold after FDH induction in A549 and HCT116 cells, respectively (Fig. 4B). This was in agreement with our previous report36 and with the fact that FDH should deplete 10-formyltetrahydrofolate, a substrate for two reactions of the de novo purine biosynthesis. To further confirm that FDH does not cause DNA damage, we also carried out a comet assay41 in A549 cells with and without FDH induction. This assay evaluates single- and double-stranded DNA breaks based upon the ability of denatured, cleaved DNA fragments to migrate out of the nucleoid under the influence of an electric field. No DNA damage upon FDH expression was observed in these experiments compared to cells positive for comet tails (Fig. 5).
Figure 4.

Effects of FDH expression on DNA damage and intracellular ATP pool. (A) DNA damage was evaluated in FDH-expressing HCT116 cells (36 hours posttransfection) and A549 cells (48 hours postinduction). Etoposide (5 µM for 4 hours) was used as a positive control. (B) ATP levels were measured in A549 (24 hours postinduction) and HCT116 (24 hours posttransfection). Error bars represent ± SD (n = 3).
Figure 5.

DNA damage assessed by comet assay (representative micrographs of fluorescent DNA stain of A549 cells are shown). (A) Untreated cells with undamaged DNA (negative control). (B) Cells exposed to 100 mM of H2O2 for 20 minutes at 4°C (positive control; denatured DNA fragments migrate out from the nucleoid in a different length of comet tail). (C, D) FDH-expressed cells have undamaged supercoiled DNA (it remains inside of the nucleoid). Cells were examined at different time points (C: 24 hours; D: 48 hours) of FDH induction.
Levels of reduced folate pools
A strong decrease in 10-fTHF was observed in both cell lines upon FDH expression. While in A549 cells this decrease was 4-fold, in HCT116 cells, 10-fTHF was depleted to the undetectable levels upon FDH expression (Fig. 6). Of note, the levels of 10-fTHF in HCT116 cells were initially much lower (about 30-fold) than in A549 cells (Fig. 6). Alongside with 10-fTHF, another folate pool, 5-mTHF, was also significantly decreased in response to FDH (2-fold and 6.5-fold, respectively, for A549 and HCT116 cells) (Fig. 6A and 6B). This is in agreement with the notion that FDH decreases folate-bound one-carbon groups (Fig. 6C).32 Changes in the THF/5,10-methylene-THF pool were insignificant in both cell lines (Fig. 6). Overall, while the levels of reduced folate pools are different in A549 and HCT116 cells (with the former having almost 2.5-fold higher total folate), changes in intracellular folates in response to FDH showed a similar trend in both cell lines including the decrease of the total folate pool.
Figure 6.

Intracellular levels of reduced folate pools in A549 (A) and HCT116 (B) cells. THF = tetrahydrofolate; 5,10-mTHF = methylenetetrahydrofolate; 5-mTHF = methyltetrahydrofolate; 10-fTHF = 10-formyltetrahydrofolate. Error bars represent ±SD (n = 4). (C) Schematic depicting a simplified version of pathways of the reduced folate coenzyme conversion. The biosynthetic outcome of folate metabolism (production of SAM, purines, and TMP) is indicated. FDH catalyzes the conversion of 10-fTHF to THF and CO2, thus depleting folate-bound one-carbon groups (these groups leave the folate pool as CO2) and inhibiting folate-dependent biosynthetic reactions. Most directly, the FDH action affects de novo purine biosynthesis.
Subcellular distribution of p21
We have investigated subcellular localization of p21 in response to FDH by confocal microscopy. In both A549 and HCT116 cell lines, strong nuclear accumulation of p21 was observed (Fig. 7). Such an accumulation is in agreement with the function of p21 in the regulation of the cell cycle.42 These experiments indicated that in both cell lines, upon FDH expression, p21 accumulated in nucleoli. This observation, which is in line with earlier reports,43,44 was further confirmed by double labeling of cells for p21 and the nucleolar protein, fibrillarin.44 Of note, much less profound accumulation of p21 was also seen in the cytoplasm (Fig. 7).
Figure 7.

Subcellular distribution of p21 in FDH-deficient and FDH-expressing A549 and HCT116 cells assessed by confocal microscopy. (A) Green: p21 protein stained with specific antibody and secondary antibody conjugated with Alexa Fluor 488; red: nuclei stained with TO-PRO-3 iodide dye; yellow: colocalization. (B) Green: p21 protein stained with specific antibody and secondary antibody conjugated with Alexa Fluor 488; red: fibrillarin stained with specific antibody and secondary antibody conjugated with Alexa Fluor 555; yellow: colocalization.
Discussion
Our early studies have shown that FDH activates p53-dependent G1 arrest and apoptosis in rapidly proliferating cancer cells,31,36 which perhaps explains down-regulation of the protein in many cancers.34 FDH elevation is accompanied by the increased levels of p21 and PUMA.31,36 PUMA, a proapoptotic BH3-only member of Bcl2 family proteins, is considered as one of the main downstream effectors of p53 in apoptosis induction.45,46 In turn, p21 is a well-known downstream target of p53 in G1 arrest.15 In addition, numerous reports imply a role of p21 as a proapoptotic or antiapoptotic effector.13,15-17 While mechanisms by which p21 can promote apoptosis are not clear, the inhibition of apoptosis by p21 can be either direct (through the binding to key apoptotic molecules) or indirect (in cases when progression through the cell cycle is required for apoptosis).16,17 Thus, the function of p21 in apoptosis may or may not be related to its role in the cell cycle control.16,17 In the present study, we have addressed the role of p21 in FDH antiproliferative effects.
If p21 would play a proapoptotic role, direct or indirect, in the cellular response to FDH, its knockdown could be expected to inhibit FDH-induced apoptosis, which was not the case in our experiments. Instead, A549 cells become more sensitive to FDH upon treatment with p21-specific siRNA. This was reflected in an early apoptosis onset apparently due to the lack of the G1 arrest–dependent delay of the cellular response in p21-deficient cells. In addition, p21 could potentially slow down cellular response to FDH by inhibiting procaspase-347 or SAPK,48,49 FDH downstream effectors in apoptosis induction.31,50 In contrast, the silencing of PUMA by siRNA completely protected A549 cells from FDH-induced apoptosis, indicating that this protein is a key effector in this process. The effect of p21 deletion was even more profound in another cell line, HCT116. Thus, at early time points (24 and 48 hours after FDH transfection), two-fold more p21-deficient cells undergo apoptosis than in the case of p21-positive cells. The more profound effect in HCT116 cells could be attributed to the fact that in HCT116 p21−/− cells, the p21 gene was deleted through homologous recombination, which ensures the complete knockout.8
The enhanced apoptotic response to FDH in HCT116 p21−/− compared to p21+/+ cells could be also explained by the fact that in the HCT116 cell line, FDH induces G2 but not G1 arrest as in A549 cells. As an inhibitor of Cdk1 phosphorylation, p21 can be involved in both G1 and G2 cell cycle arrest.15 Together with GADD45 and 14-3-3s, the p21 protein is responsible for the maintenance of G2 arrest through effects on the cyclin B1-Cdk1 complex.51 Moreover, it has been suggested that G2 arrest and the induction of apoptosis are two parallel processes directed by the levels of p21.17,18 Of note, levels of p21 in response to FDH were lower in HCT116 p21+/+ than in A549 cells. Such levels might be insufficient to induce G1 arrest. While the role of p21 in G2 arrest is well appreciated in the literature, this arrest can be also p21 independent. Thus, in response to the proteasome inhibitor MG132, HCT116 cells undergo G2 arrest independent of p53 or p21 status.52 However, although cells lacking p53 or p21 can still undergo G2 arrest in response to DNA damage, the p21 protein (activated by p53) is essential in sustaining the G2 arrest.10 In support of the role of p21 in FDH-induced G2 arrest in HCT116 cells, the isogenic cell line, HCT116 p21−/−, did not undergo arrest upon FDH elevation.
In a stress response, the proportion of cells that arrest at G1-S or G2-M depends on cell type, growth conditions, and the checkpoint controls operative in the cell.10 For example, paclitaxel induces p53 and p21 in A549 cells, causing G1 and G2 arrest, but HCT116 cells do not up-regulate p53 in response to this drug.53 Paclitaxel, however, is not a DNA-damaging agent, while the best-known example of G2 checkpoint activation is by DNA damage.51 Folate deficiency, on the other hand, is known to induce DNA damage.54 The underlying mechanism is an increased misincorporation of uracil into DNA at conditions of insufficient TMP biosynthesis, a folate-dependent reaction.55 Of note, the elevation of FDH could produce, to some extent, similar effects on the intracellularly reduced folate pool as folate deficiency. This phenomenon is associated with the fact that the enzyme depletes folate-bound one-carbon groups (Fig. 6C), while folate deficiency decreases the group carriers. The outcome toward folate-dependent biosynthetic reactions could be similar with both insults. Our study, however, demonstrated that the FDH induction does not lead to DNA damage. Instead, it severely depletes intracellular purine pools,36 which in turn should inhibit DNA and RNA synthesis. Of note, depletion of intracellular ribonucleotides, but not deoxyribonucleotides, has been shown to reversibly activate p53,39 the effect also seen in FDH-expressing cells.36 Changes in reduced folate pools provide an explanation for the lack of DNA damage in response to FDH elevation: the 10-fTHF pool responsible for purine biosynthesis was dramatically depleted, but the 5,10-methylene-THF/THF pool, part of which is responsible for TMP biosynthesis, stays at about the same level (Fig. 6A and 6C).
Interestingly, while the purine depletion in response to FDH was profound (as judged by ATP levels) in both A549 and HCT116 cell lines, the cell cycle arrest took place at different checkpoints, G1 and G2, respectively. The p21-dependent G1 arrest in response to nucleotide deprivation is a common phenomenon to prevent entering S phase, in which DNA biosynthesis requires an abundant supply of nucleotides. It is not completely clear at present why FDH induces p21-dependent G2 arrest in HCT116 cells. In this regard, it is known that G2/M cyclines are subject to inhibition by growth-restricting conditions.51 Therefore, withdrawal of crucial nutrients, such as folate, could be expected to result in this type of arrest. In support of this view, G2 arrest in HepG2 cells in response to folate withdrawal has been demonstrated.56 In line with this mechanism, the present study has demonstrated the overall decrease in intracellularly reduced folate pools in response to FDH expression. The activation of different checkpoints in two cell lines could be associated with the fact that HCT116 cells are mismatch repair (MMR) deficient.57 In this regard, different effects of folate depletion on MMR-proficient and -deficient cells have been reported, with MMR-deficient cells being resistant to apoptosis induced by folate withdrawal.58 This mechanism, however, might not be relevant to FDH effects because strong apoptosis was observed in FDH-expressing HCT116 cells (Fig. 2).
The cell cycle regulatory function of p21 is associated with its nuclear localization, but the protein can also localize in the cytoplasm.5 While p21 does not have a nuclear export signal, the phosphorylation near its nuclear localization signal (NLS) retains the protein in the cytoplasm.59 Another mechanism to retain p21 in the cytoplasm, which is the truncation of the NLS-containing C-terminus by specific proteases, has also been reported.60 In the nucleus, in addition to the regulation of the cell cycle progression, p21 is also involved in a variety of transcriptional responses.44 In the cytoplasm, p21 can initiate antiapoptotic responses by inhibiting proapoptotic kinase ASK142 or by binding to procaspase 3, thus preventing its proteolytic activation.47 In a more general sense, it has been suggested that the subcellular localization of p21 defines its function as a tumor suppressor (nuclear localization) or oncoprotein (cytoplasmic localization).44 Indeed, some human cancers display elevated levels of cytoplasmic p21, which is associated with poor prognosis.61-65 In response to FDH, p21 accumulates in the nuclei of both A549 and HCT116 cell lines, with strongest accumulation seen in nucleoli (Fig. 7), which is in agreement with the cell cycle arrest observed in our studies.42-44 While some accumulation in the cytoplasm was also seen, the role of cytoplasmic p21 in FDH effects is not clear. Interestingly, we have recently shown that FDH inhibits motility by affecting the cofilin/actin pathway.66 In this regard, effects of cytoplasmic p21 on cellular motility, through inhibition of ROCK kinase (an upstream effector in the LIMK/cofilin/actin pathway), were reported.67,68 Whether the cytoplasmic accumulation of p21 is a cellular attempt to counteract effects of FDH on motility is a subject for future studies.
Overall, our data suggest that activation of p21-dependent G1 or G2 cell cycle arrest counteracts the FDH-induced apoptotic response (Fig. 8). In support of this mechanism, the lack of p21 sensitizes cells to FDH, resulting in an earlier onset of the apoptosis. This conclusion is in line with the observation that p21 attenuates apoptotic signals in methotrexate-treated cells.69 Of note, both methotrexate and FDH work in the same direction with regard to folate metabolism, inhibiting folate-dependent biosynthetic reactions. The activation of p21, however, is not sufficient to rescue cells from FDH-induced apoptosis.
Figure 8.
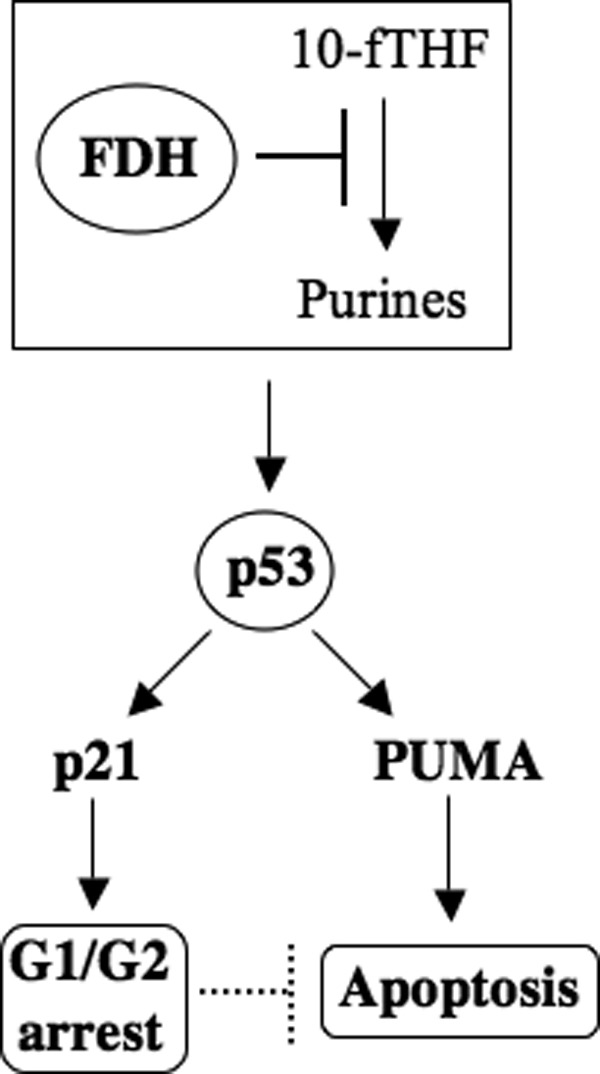
Schematic depicting the FDH anti-proliferative cascade. FDH depletes the 10-fTHF pool, leading to inhibition of the de novo purine biosynthesis and depletion of purines in the cell. This activates the p53 protein, which in turn induces transcriptional activation of p21 and proapoptotic PUMA. Accumulated p21 protein activates either G1 or G2 arrest, both of which delay apoptosis.
Materials and Methods
Cell culture
Cell culture media were purchased from Mediatech (Manassas, VA), and fetal bovine serum was purchased from Atlanta Biologicals (Lawrenceville, GA). Tet-certified fetal bovine serum was obtained from Clontech (Mountain View, CA). Tet-On A549/FDH cells were generated in our previous studies.31 HCT116 and HCT116 p21−/− cell lines, a gift from Dr. Bert Vogelstein (The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine), were grown in RPMI 1640 culture media supplemented with 10% (v/v) fetal bovine serum at 37°C under humidified air containing 5% CO2.
Transient transfection
HCT116 cells (~1.0 × 106) were transfected with 2 µg pcDNA3.1 or pcDNA3.1/FDH plasmid using Amaxa nucleofector kit “V” (Lonza, Basel, Switzerland) according to the manufacturer’s protocol.
siRNA
Tet-On A549/FDH cells (1.2 × 105) were transfected with 25 nmol of Stealth RNAi (Invitrogen, Carlsbad, CA) (Table 1) using 5 to 10 µL Lipofectamine 2000 (Invitrogen). Scrambled Stealth RNAi (Invitrogen) was used as a negative control. Transfection was performed following the manufacturer’s protocols. Total RNA was purified with RNA Easy Mini Kit (Qiagen, Hilden, Germany). Reverse transcriptase reaction was performed with an Oligo (dT)18 primer using Advantage RT-for-PCR Kit (Clontech). Conventional PCR was carried out using specific primers (Table 1) and Pfu-turbo DNA polymerase (Stratagene, La Jolla, CA).
Immunoblot assays
Cell lysates were prepared in buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 1 mM PMSF, and mammalian protease inhibitor cocktail (Sigma, St. Louis, MO). Lysates were subjected to SDS-PAGE followed by immunoblot with specified antibodies. Expression of FDH protein was verified with an in-house FDH-specific polyclonal antibody (1:10,000).31,34,36 p21 was detected using a p21-specific monoclonal antibody (5 µg/mL) (Sigma). A polyclonal PUMA antibody was used to detect PUMA (1:1,000) (Cell Signaling Technology, Danvers, MA). Actin was detected using a monoclonal antibody from Calbiochem (1:8,000) (La Jolla, CA).
Cell cycle and apoptosis assays
Cell cycle analysis was carried out using propidium iodide staining/FACS. Cells (1.0 × 106) were washed once with cold PBS and centrifuged at 1,000 rpm for 5 minutes. Pellets were resuspended in 200 μL of cold PBS, fixed by the addition of 4 mL of 70% ethanol, and incubated overnight at −20°C. Following centrifugation, samples were resuspended in 500 μL of PBS containing 40 μg/mL propidium iodide and 100 μg/mL Rnase (both from Roche, Basel, Switzerland), incubated for 30 minutes at 37°C, and analyzed by FACS. Apoptotic cells were detected by annexin V and PI labeling using Annexin V-FLUOS staining kit (Roche). All cells (floating and attached) were used in these experiments. Samples were analyzed at the flow cytometry core facility at Hollings Cancer Center.
DNA damage assay
DNA damage assays were performed using the EpiQuik In Situ DNA Damage Assay Kit (Epigentek, Farmingdale, NY). This assay measures the phosphorylation at serine 139 of histone H2AX, which is one of the earliest chromatin modification events in DNA damage response. All experiments were performed following the manufacturer’s protocols.
Comet assay
DNA strand breaks were assessed using Alkaline Comet Assay kit (Trevigen, Gaithersburg, MD). All experiments were preformed following the manufacturer’s protocols. Images were captured using a Zeiss Axio Observer D1 multichannel fluorescent microscope (Oberkochen, Germany).
ATP assay
ATP levels were measured using ATP Bioluminescence Assay Kit CLS II (Roche) following the manufacturer’s protocols as we previously described.36
Assays of reduced folate pools
Approximately 5 × 106 cells were collected and rapidly washed 3 times with ice-cold PBS. The cell pellet was resuspended in 50 mM Tris-HCl buffer, pH 7.4, containing 50 mM sodium ascorbate. Cells were lysed by heating for 3 minutes in a boiling water bath. Cell lysates were chilled on ice and centrifuged for 5 minutes at 17,000g at 4°C. Folate pools were measured in cell lysates by the ternary complex assay method70 as we previously described.71 Folate levels were calculated per milligram of cellular protein measured by the Bradford assay.
Confocal microscopy
Cells seeded in Lab-Tek II Chamber (Nalge Nunc International, Penfield, NY) were fixed with 3.7% of methanol-free formaldehyde for 10 minutes and permeabilized with 0.1% Triton X-100 for 5 minutes. After blocking with 10% preimmune goat serum in PBS for 45 minutes, slides were incubated with p21-specific monoclonal antibody (1:200) (Pharmingen, San Diego, CA) for 1 hour at room temperature. This was followed by incubation with secondary goat anti-mouse antibody labeled with Alexa Fluor 488 (Life Technologies, Grand Island, NY) in a dark chamber for 45 minutes at room temperature. Nuclei were counterstained using To-Pro3 (Life Technologies, Grand Island, NY). Nucleoli were visualized by staining with fibrillarin polyclonal antibody (1:100) (Abcam, Cambridge, UK) and secondary antibody conjugated with Alexa Fluor 555 (Molecular Bioprobes). Images were captured and processed using a Leica TCS SP2 AOBS scanning confocal microscope (Wetzlar, Germany) at the imaging core facility at Hollings Cancer Center.
Acknowledgments
The authors thank Dr. Bert Vogelstein for the kind gift of HCT116 p21−/− cells.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported in part by the National Institutes of Health [grant numbers DK54388 and CA095030]. Flow cytometry and imaging core facilities were supported in part by the Cancer Center [grant number P30 CA138313] to the Hollings Cancer Center, Medical University of South Carolina. L.A.X. was supported by an Abney Foundation scholarship.
References
- 1. Gu Y, Turck CW, Morgan DO. Inhibition of CDK2 activity in vivo by an associated 20K regulatory subunit. Nature. 1993;366:707-10 [DOI] [PubMed] [Google Scholar]
- 2. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805-16 [DOI] [PubMed] [Google Scholar]
- 3. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701-4 [DOI] [PubMed] [Google Scholar]
- 4. Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004;18:851-5 [DOI] [PubMed] [Google Scholar]
- 5. Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65-70 [DOI] [PubMed] [Google Scholar]
- 6. Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675-84 [DOI] [PubMed] [Google Scholar]
- 7. el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817-25 [DOI] [PubMed] [Google Scholar]
- 8. Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187-90 [PubMed] [Google Scholar]
- 9. Wu H, Wade M, Krall L, Grisham J, Xiong Y, Van Dyke T. Targeted in vivo expression of the cyclin-dependent kinase inhibitor p21 halts hepatocyte cell-cycle progression, postnatal liver development and regeneration. Genes Dev. 1996;10:245-60 [DOI] [PubMed] [Google Scholar]
- 10. Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497-501 [DOI] [PubMed] [Google Scholar]
- 11. Dulic V, Kaufmann WK, Wilson SJ, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013-23 [DOI] [PubMed] [Google Scholar]
- 12. Ji C, Marnett LJ, Pietenpol JA. Cell cycle re-entry following chemically-induced cell cycle synchronization leads to elevated p53 and p21 protein levels. Oncogene. 1997;15:2749-53 [DOI] [PubMed] [Google Scholar]
- 13. Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639-49 [PubMed] [Google Scholar]
- 14. Garner E, Raj K. Protective mechanisms of p53-p21-pRb proteins against DNA damage-induced cell death. Cell Cycle. 2008;7:277-82 [DOI] [PubMed] [Google Scholar]
- 15. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43-56 [DOI] [PubMed] [Google Scholar]
- 17. Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res. 2010;704:12-20 [DOI] [PubMed] [Google Scholar]
- 18. Wendt J, Radetzki S, von Haefen C, et al. Induction of p21CIP/WAF-1 and G2 arrest by ionizing irradiation impedes caspase-3-mediated apoptosis in human carcinoma cells. Oncogene. 2006;25:972-80 [DOI] [PubMed] [Google Scholar]
- 19. Lincet H, Poulain L, Remy JS, et al. The p21(cip1/waf1) cyclin-dependent kinase inhibitor enhances the cytotoxic effect of cisplatin in human ovarian carcinoma cells. Cancer Lett. 2000;161:17-26 [DOI] [PubMed] [Google Scholar]
- 20. Chopin V, Toillon RA, Jouy N, Le Bourhis X. P21(WAF1/CIP1) is dispensable for G1 arrest, but indispensable for apoptosis induced by sodium butyrate in MCF-7 breast cancer cells. Oncogene. 2004;23:21-9 [DOI] [PubMed] [Google Scholar]
- 21. Kang KH, Kim WH, Choi KH. p21 promotes ceramide-induced apoptosis and antagonizes the antideath effect of Bcl-2 in human hepatocarcinoma cells. Exp Cell Res. 1999;253:403-12 [DOI] [PubMed] [Google Scholar]
- 22. Liu S, Bishop WR, Liu M. Differential effects of cell cycle regulatory protein p21(WAF1/Cip1) on apoptosis and sensitivity to cancer chemotherapy. Drug Resist Updat. 2003;6:183-95 [DOI] [PubMed] [Google Scholar]
- 23. Goldman ID, Chattopadhyay S, Zhao R, Moran R. The antifolates: evolution, new agents in the clinic, and how targeting delivery via specific membrane transporters is driving the development of a next generation of folate analogs. Curr Opin Investig Drugs. 2010;11:1409-23 [PubMed] [Google Scholar]
- 24. Dabrowska M, Mosieniak G, Skierski J, Sikora E, Rode W. Methotrexate-induced senescence in human adenocarcinoma cells is accompanied by induction of p21(waf1/cip1) expression and lack of polyploidy. Cancer Lett. 2009;284:95-101 [DOI] [PubMed] [Google Scholar]
- 25. Huang WY, Yang PM, Chang YF, Marquez VE, Chen CC. Methotrexate induces apoptosis through p53/p21-dependent pathway and increases E-cadherin expression through downregulation of HDAC/EZH2. Biochem Pharmacol. 2011;81:510-7 [DOI] [PubMed] [Google Scholar]
- 26. Wu MF, Hsiao YM, Huang CF, et al. Genetic determinants of pemetrexed responsiveness and nonresponsiveness in non-small cell lung cancer cells. J Thorac Oncol. 2010;5:1143-51 [DOI] [PubMed] [Google Scholar]
- 27. Obajimi O, Keen JC, Melera PW. Inhibition of de novo purine synthesis in human prostate cells results in ATP depletion, AMPK activation and induces senescence. Prostate. 2009;69:1206-21 [DOI] [PubMed] [Google Scholar]
- 28. Noro R, Miyanaga A, Minegishi Y, et al. Histone deacetylase inhibitor enhances sensitivity of non-small-cell lung cancer cells to 5-FU/S-1 via down-regulation of thymidylate synthase expression and up-regulation of p21(waf1/cip1) expression. Cancer Sci. 2010;101:1424-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takagi K, Sowa Y, Cevik OM, Nakanishi R, Sakai T. CDK inhibitor enhances the sensitivity to 5-fluorouracil in colorectal cancer cells. Int J Oncol. 2008;32:1105-10 [PubMed] [Google Scholar]
- 30. Crott JW, Liu Z, Choi SW, Mason JB. Folate depletion in human lymphocytes up-regulates p53 expression despite marked induction of strand breaks in exons 5-8 of the gene. Mutat Res. 2007;626:171-9 [DOI] [PubMed] [Google Scholar]
- 31. Oleinik NV, Krupenko SA. Ectopic expression of 10-formyltetrahydrofolate dehydrogenase in a549 cells induces g(1) cell cycle arrest and apoptosis. Mol Cancer Res. 2003;1:577-88 [PubMed] [Google Scholar]
- 32. Anguera MC, Field MS, Perry C, et al. Regulation of folate-mediated one-carbon metabolism by 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 2006;281:18335-42 [DOI] [PubMed] [Google Scholar]
- 33. Krupenko SA. FDH: an aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem Biol Interact. 2009;178:84-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krupenko SA, Oleinik NV. 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ. 2002;13:227-36 [PubMed] [Google Scholar]
- 35. Oleinik NV, Krupenko NI, Krupenko SA. Epigenetic silencing of ALDH1L1, a metabolic regulator of cellular proliferation, in cancers. Genes Cancer. 2011;2:130-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oleinik NV, Krupenko NI, Priest DG, Krupenko SA. Cancer cells activate p53 in response to 10-formyltetrahydrofolate dehydrogenase expression. Biochem J. 2005;391:503-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med. 2006;7:165-72 [DOI] [PubMed] [Google Scholar]
- 38. Speevak MD, Chevrette M. Identification of chromosomes implicated in suppression of apoptosis in somatic cell hybrids. Biochem Cell Biol. 1994;72:655-62 [DOI] [PubMed] [Google Scholar]
- 39. Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM. A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev. 1996;10:934-47 [DOI] [PubMed] [Google Scholar]
- 40. Agarwal ML, Agarwal A, Taylor WR, Chernova O, Sharma Y, Stark GR. A p53-dependent S-phase checkpoint helps to protect cells from DNA damage in response to starvation for pyrimidine nucleotides. Proc Natl Acad Sci U S A. 1998;95:14775-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McArt DG, McKerr G, Howard CV, Saetzler K, Wasson GR. Modelling the comet assay. Biochem Soc Trans. 2009;37:914-7 [DOI] [PubMed] [Google Scholar]
- 42. Asada M, Yamada T, Ichijo H, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cazzalini O, Perucca P, Valsecchi F, et al. Intracellular localization of the cyclin-dependent kinase inhibitor p21CDKN1A-GFP fusion protein during cell cycle arrest. Histochem Cell Biol. 2004;121:377-81 [DOI] [PubMed] [Google Scholar]
- 44. Abella N, Brun S, Calvo M, et al. Nucleolar disruption ensures nuclear accumulation of p21 upon DNA damage. Traffic. 2010;11:743-55 [DOI] [PubMed] [Google Scholar]
- 45. Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27(Suppl 1):S71-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chipuk JE, Green DR. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle. 2009;8:2692-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suzuki A, Tsutomi Y, Akahane K, Araki T, Miura M. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17:931-9 [DOI] [PubMed] [Google Scholar]
- 48. Shim J, Lee H, Park J, Kim H, Choi EJ. A non-enzymatic p21 protein inhibitor of stress-activated protein kinases. Nature. 1996;381:804-6 [DOI] [PubMed] [Google Scholar]
- 49. Xue Y, Ramaswamy NT, Hong X, Pelling JC. Association of JNK1 with p21waf1 and p53: modulation of JNK1 activity. Mol Carcinog. 2003;36:38-44 [DOI] [PubMed] [Google Scholar]
- 50. Oleinik NV, Krupenko NI, Krupenko SA. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene. 2007;26:7222-30 [DOI] [PubMed] [Google Scholar]
- 51. Foijer F, te Riele H. Check, double check: the G2 barrier to cancer. Cell Cycle. 2006;5:831-6 [DOI] [PubMed] [Google Scholar]
- 52. Kim OH, Lim JH, Woo KJ, et al. Influence of p53 and p21Waf1 expression on G2/M phase arrest of colorectal carcinoma HCT116 cells to proteasome inhibitors. Int J Oncol. 2004;24:935-41 [PubMed] [Google Scholar]
- 53. Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001;20:3806-13 [DOI] [PubMed] [Google Scholar]
- 54. Ames BN. Micronutrient deficiencies: a major cause of DNA damage. Ann N Y Acad Sci. 1999;889:87-106 [DOI] [PubMed] [Google Scholar]
- 55. Blount BC, Mack MM, Wehr CM, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94:3290-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang RF, Ho YH, Lin HL, Wei JS, Liu TZ. Folate deficiency induces a cell cycle-specific apoptosis in HepG2 cells. J Nutr. 1999;129:25-31 [DOI] [PubMed] [Google Scholar]
- 57. Parsons R, Li GM, Longley MJ, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227-36 [DOI] [PubMed] [Google Scholar]
- 58. Gu L, Wu J, Qiu L, Jennings CD, Li GM. Involvement of DNA mismatch repair in folate deficiency-induced apoptosis small star, filled. J Nutr Biochem. 2002;13:355-63 [DOI] [PubMed] [Google Scholar]
- 59. Child ES, Mann DJ. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5:1313-9 [DOI] [PubMed] [Google Scholar]
- 60. Levkau B, Koyama H, Raines EW, et al. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a caspase cascade. Mol Cell. 1998;1:553-63 [DOI] [PubMed] [Google Scholar]
- 61. Coppola D, Lu L, Fruehauf JP, et al. Analysis of p53, p21WAF1, and TGF-beta1 in human ductal adenocarcinoma of the pancreas: TGF-beta1 protein expression predicts longer survival. Am J Clin Pathol. 1998;110:16-23 [DOI] [PubMed] [Google Scholar]
- 62. Bales ES, Dietrich C, Bandyopadhyay D, et al. High levels of expression of p27KIP1 and cyclin E in invasive primary malignant melanomas. J Invest Dermatol. 1999;113:1039-46 [DOI] [PubMed] [Google Scholar]
- 63. Winters ZE, Hunt NC, Bradburn MJ, et al. Subcellular localisation of cyclin B, Cdc2 and p21(WAF1/CIP1) in breast cancer: association with prognosis. Eur J Cancer. 2001;37:2405-12 [DOI] [PubMed] [Google Scholar]
- 64. Winters ZE, Leek RD, Bradburn MJ, Norbury CJ, Harris AL. Cytoplasmic p21WAF1/CIP1 expression is correlated with HER-2/ neu in breast cancer and is an independent predictor of prognosis. Breast Cancer Res. 2003;5:R242-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Koster R, di Pietro A, Timmer-Bosscha H, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 2010;120:3594-605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Oleinik NV, Krupenko NI, Krupenko SA. ALDH1L1 inhibits cell motility via dephosphorylation of cofilin by PP1 and PP2A. Oncogene. 2010;29:6233-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tanaka H, Yamashita T, Asada M, Mizutani S, Yoshikawa H, Tohyama M. Cytoplasmic p21(Cip1/WAF1) regulates neurite remodeling by inhibiting Rho-kinase activity. J Cell Biol. 2002;158:321-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee S, Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J Biol Chem. 2004;279:1885-91 [DOI] [PubMed] [Google Scholar]
- 69. Kraljevic Pavelic S, Cacev T, Kralj M. A dual role of p21waf1/cip1 gene in apoptosis of HEp-2 treated with cisplatin or methotrexate. Cancer Gene Ther. 2008;15:576-90 [DOI] [PubMed] [Google Scholar]
- 70. Priest DG, Schmitz JC, Bunni MA, Stuart RK. Pharmacokinetics of leucovorin metabolites in human plasma as a function of dose administered orally and intravenously. J Natl Cancer Inst. 1991;83:1806-12 [DOI] [PubMed] [Google Scholar]
- 71. Oleinik NV, Krupenko NI, Reuland SN, Krupenko SA. Leucovorin-induced resistance against FDH growth suppressor effects occurs through DHFR up-regulation. Biochem Pharmacol. 2006;72:256-66 [DOI] [PubMed] [Google Scholar]