

Abstract
Anaplasma phagocytophilum is an emerging tick-borne pathogen that infects humans, domestic animals and wildlife throughout the Holarctic. In the far-western United States, multiple rodent species have been implicated as natural reservoirs for A. phagocytophilum. However, the presence of multiple A. phagocytophilum strains has made it difficult to determine which reservoir hosts pose the greatest risk to humans and domestic animals. Here we characterized three genetic markers (23S–5S rRNA intergenic spacer, ank and groESL) from 73 real-time TaqMan PCR-positive A. phagocytophilum strains infecting multiple rodent and reptile species, as well as a dog and a horse, from California. Bayesian and maximum-likelihood phylogenetic analyses of all three genetic markers consistently identified two major clades, one of which consisted of A. phagocytophilum strains infecting woodrats and the other consisting of strains infecting sciurids (chipmunks and squirrels) as well as the dog and horse strains. In addition, analysis of the 23S–5S rRNA spacer region identified two unique and highly dissimilar clades of A. phagocytophilum strains infecting several lizard species. Our findings indicate that multiple unique strains of A. phagocytophilum with distinct host tropisms exist in California. Future epidemiological studies evaluating human and domestic animal risk should incorporate these distinctions.
Introduction
Anaplasma phagocytophilum is a tick-transmitted rickettsial parasite of humans and other animals. Since tick-borne fever was first described and attributed to ‘Rickettsia phagocytophila’ in small ruminants in Great Britain in the early 1900s (Macleod & Gordon, 1933; Foggie, 1949, 1951), related diseases were identified in voles (Microtus pennsylvanicus) in 1938 (Tyzzer, 1938; Foggie, 1962), horses in the 1950s in California (Stannard et al., 1969), dogs in California in 1982 (Madewell & Gribble, 1982) and human beings in the upper Midwest in 1993 (Chen et al., 1994). In 2001, Ehrlichia equi, Ehrlichia phagocytophila and the agent of human granulocytic ehrlichiosis were synonymized as A. phagocytophilum on the basis of 16S rRNA and groEL genes and morphological and phenotypic characteristics (Dumler et al., 2001).
Nevertheless, phenotypic and genetic data support the presence of distinct strains. The North American strain Ap-Variant 1, which has distinctive 16S rRNA, msp4, msp2 and ank genes, occurs in ticks and deer (Massung et al., 2003); deer, goats and tick-derived cell lines can be infected with Ap-Variant 1, but rodents cannot (Massung et al., 2007). In England, two genetically distinct subpopulations of A. phagocytophilum coexist in separate enzootic cycles, one involving deer and Ixodes ricinus ticks and the other involving field voles (Microtus agrestis) and Ixodes trianguliceps (Bown et al. 2009). Strains in western North America show at least two phenotypes based on pathogenicity for horses. The equine-origin MRK strain, as well as strains from chipmunks (Tamias species) and tree squirrels (Sciurus and Tamiasciurus species), reproducibly induce severe disease in horses (Pusterla et al., 1999; Foley et al., 2009b) consistent with other data showing that squirrels and chipmunks are reservoir-competent for A. phagocytophilum (Nieto & Foley, 2008, 2009). In contrast, woodrat-origin strains are minimally infectious and apathogenic in horses (Foley et al., 2009b).
The ecological and epidemiological significance of genetic and phenotypic variants of A. phagocytophilum is poorly understood. The goal of the present study was to analyse A. phagocytophilum strains from a diversity of wild and domestic animal hosts in order to determine whether genotypes in the western US cluster according to host species or geographical location. These data are critical for interpreting surveillance and human risk and will contribute important baseline information for understanding the ecology of this emerging zoonotic disease.
Methods
Sample collection and preparation.
DNA samples for molecular characterization were obtained from small mammals including chipmunks, dusky-footed woodrats, eastern grey squirrels (Sciurus carolinensis) and western grey squirrels (Sciurus griseus) between October 2005 and May 2008. Animals were sampled in central and northern California at the following sites: Big Basin State Park (BB, Santa Cruz County; 37° 10.621′, 122° 12.328′), Henry Cowell State Park (HC, Santa Cruz County; 37° 08.705′, 122° 11.12′), Samuel P. Taylor State Park (SPT, Marin County; 38° 01.232′, 122° 40.774′), Hendy Woods State Park (HW, Mendocino County; 39° 04.25′, 123° 28.238′) and Hoopa Valley Indian Reservation (HV, Humboldt County; 41° 16.65′, 123° 70.12′). Several lizard species including northern alligator lizards (Elgaria coerulea), sagebrush lizards (Sceloporus graciosus) and western fence lizards (Sceloporus occidentalis) were also sampled in HV during May and June 2006. Blood samples from small mammals and lizards were collected as described previously (Foley et al., 2009a; Nieto et al., 2009). DNA from all blood samples was extracted using a Qiagen blood and tissue kit as per the manufacturer’s instructions. A number of sampled animals harboured ticks, results of which have been described separately (Foley et al., 2011; Rejmanek et al., 2011). Three additional A. phagocytophilum strains used in this study included HZ (the fully sequenced human-origin strain from New York state) (Dunning Hotopp et al., 2006), MRK (originally isolated from an infected horse in Shasta County, CA, and maintained in infected leukocytes obtained by horse passage) and Dog_CA, a strain isolated from an infected dog from Tuolumne County, CA, that was a patient at the UC Davis Veterinary Medicine Teaching Hospital (VMTH) and which presented with high fever, lethargy, thrombocytopenia and A. phagocytophilum inclusions in approximately 100 % of its neutrophils (courtesy of Steven Dumler, John Hopkins). This strain has been maintained in a human promyelocytic leukaemia (HL-60) cell line. Locations of sampling sites and California origin strains (MRK and Dog_CA) are illustrated in Fig. 1.
Fig. 1.
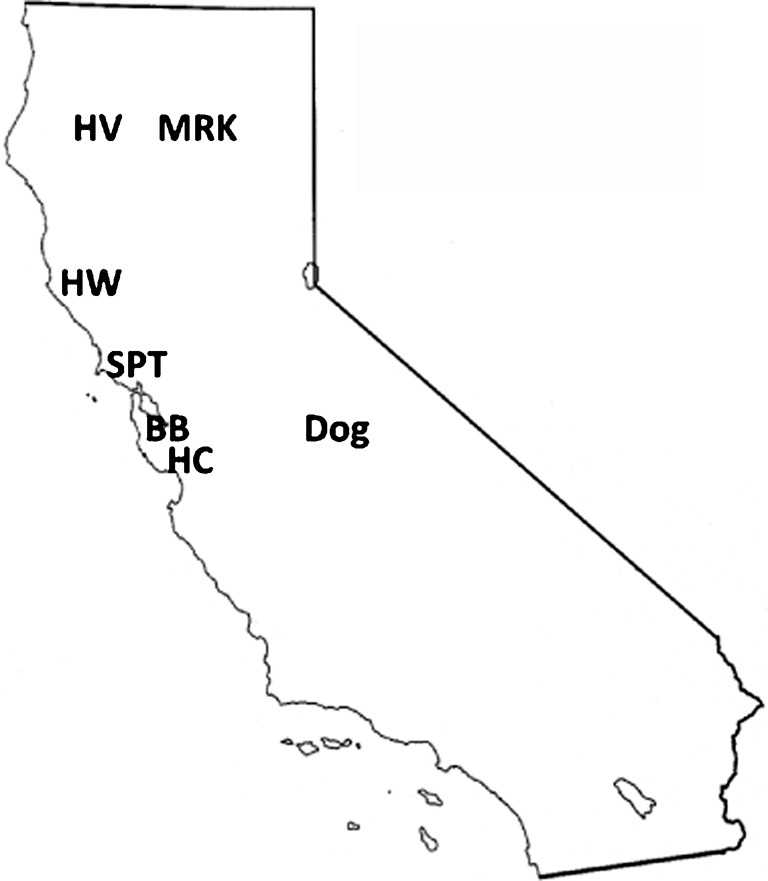
Small mammal sampling sites and locations of A. phagocytophilum-infected dog and horse (MRK). HV, Hoopa Valley State Park; HW, Hendy Woods State Park; SPT, Samuel P. Taylor State Park; BB, Big Basin State Park; HC, Henry Cowell State Park.
PCR analysis and cloning.
All DNA samples were initially screened for the presence of A. phagocytophilum DNA using a highly sensitive real-time TaqMan PCR assay targeting the msp2/p44 gene (Drazenovich et al., 2006). This assay targets a conserved region of the msp2/p44 gene present in ~100 copies throughout the genome and it has been used to successfully amplify DNA from a wide variety of strains from within the US and other parts of world, including China, Guatemala and Germany (Drazenovich et al., 2006). Results of real-time PCR were considered positive if they had a cycle threshold (CT) value <40 and a characteristic amplification curve. Nested conventional PCR assays targeting sections of the ank gene and the groESL heat shock operon, as well as the 23S–5S rRNA (rrl–rrs) intergenic spacer region, were then performed on all positive samples. Amplification of the ank gene was performed as described by Massung et al. (2007) using external primers ANK-F1 (5′-GAAGAAATTACAACTCCTGAAG-3′) and ANK-R1 (5′-CAGCCAGATGCAGTAACGTG-3′), followed by internal primers ANK-F2 (5′-TTGACCGCTGAAGCACTAAC-3′) and ANK-R2 (5′-ACCATTTGCTTCTTGAGGAG-3′), yielding an approximately 600 bp product. A segment of the groESL operon was amplified using external primers HS1a (5′-AITGGGCTGGTAITGAAAT-3′) and HS6a (5′-CCICCIGGIACIAIACCTTC-3′), followed by internal primers HS43 [5′-AT(A/T)GC(A/T)AA(G/A)GAAGCATAGTC-3] and HSVR (5′-CTCAACAGCAGCTCTAGTAGC-3′), yielding an approximately 1200 bp product as described by Liz et al. (2002). The 23S–5S rRNA spacer region was amplified with newly designed external primers ITS2F (5′-AGGATCTGACTCTAGTACGAG-3′) and ITS2R (5′-CTCCCATGTCTTAAGACAAAG-3′), and internal primers ITS2iF (5′-ATACCTCTGGTGTACCAGTTG-3′) and ITS2iR (5′-TTAACTTCCGGGTTCGGAATG-3′), using the following thermocycler conditions: 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 1 min, followed by a 5 min extension at 72 °C. This PCR assay was designed so that both sets of forward primers are embedded within the conserved 23S rRNA gene and both reverse primers are within the 5S rRNA gene. The nested amplicon spans a 300 bp region covering the entire non-coding (less conserved) spacer region. Amplified DNA was visualized on a 1 % agarose gel stained with GelStar nucleic acid stain (Lonza). Bands of the expected size were excised and cleaned with a Qiagen gel extraction kit. For sequencing of the ank gene, gel-extracted amplicons were cloned using the pGEM-T easy vector system (Promega) prior to sequencing.
DNA sequencing and alignment.
Gel-extracted or cloned PCR products were sequenced in both forward and reverse directions on an ABI 3730 sequencer (Davis Sequencing). Sequences initially were aligned using the clustal_x sequence alignment program (Larkin et al., 2007) and trimmed to a final length of 291 bp (23S–5S rRNA), 567 bp (ank) and 1164 bp (groESL). To minimize the potential for incorporating base-pair substitution errors introduced during PCR, cloning or DNA sequencing into the final consensus sequence, any samples exhibiting a unique sequence that differed from all other sequences were reamplified and resequenced. GenBank accession numbers for all unique sequences are presented in Table 1.
Table 1. Description of A. phagocytophilum strains and PCR results with corresponding GenBank accession numbers for individual genes analysed in this study.
| Strain ID | Host species | Origin | PCR results (accession no. shown if successful*) | ||
| 23S–5S rRNA | groESL | ank | |||
| Scg_752_HV | Sceloporus graciosus | Hoopa Valley | JF487929 | na | na |
| Sco_622_HV | Sceloporus occidentalis | Hoopa Valley | JF487929 | na | na |
| Sco_760_HV | Sceloporus occidentalis | Hoopa Valley | JF487930 | na | na |
| Ec_765_HV | Elgaria coeruleus | Hoopa Valley | JF487930 | na | na |
| Sg_730_HV | Sciurus griseus | Hoopa Valley | JF451142 | JF494840 | JF303732 |
| Sg_167_BB | Sciurus griseus | Big Basin | JF451139 | JF494840 | JF303732 |
| Sg_168_BB | Sciurus griseus | Big Basin | JF451139 | na | JF303733 |
| Sc_1225_BB | Sciurus carolinensis | Big Basin | na | na | JF303732 |
| To_1591_HW | Tamias ochrogenys | Hendy Woods | JF451141 | na | na |
| To_1634_HW | Tamias ochrogenys | Hendy Woods | JF451141 | na | na |
| To_1755_HW | Tamias ochrogenys | Hendy Woods | JF451139 | na | na |
| Ts_219_SPT | Tamias sonomae | Samuel P. Taylor | JF451139 | JF494840 | JF303733 |
| Ts_279_SPT | Tamias sonomae | Samuel P. Taylor | JF451139 | na | JF303733 |
| Nf_1026_HW | Neotoma fuscipes | Hendy Woods | JF451141 | JF494841 | JF303734 |
| Nf_1217_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | JF494838 | JF303737 |
| Nf_1218_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | JF494838 | JF303737 |
| Nf_1220_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | JF494837 | JF303738 |
| Nf_1222_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | JF494837 | JF303738 |
| Nf_1443_HW | Neotoma fuscipes | Hendy Woods | JF451141 | na | JF303735 |
| Nf_1496_HW | Neotoma fuscipes | Hendy Woods | JF451141 | na | JF303739 |
| Nf_1503_HW | Neotoma fuscipes | Hendy Woods | JF451141 | na | JF303736 |
| Nf_1603_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | JF494836 | JF776828 |
| Nf_1605_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | JF494837 | JF303738 |
| Nf_1606_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | JF494837 | JF303737 |
| Nf_1607_HV | Neotoma fuscipes | Hoopa Valley | JF451141 | na | na |
| Nf_1619_HC | Neotoma fuscipes | Henry Cowell | JF451141 | JF494834 | JF303740 |
| Nf_1629_HC | Neotoma fuscipes | Henry Cowell | JF451141 | JF494835 | JF303741 |
| Nf_1692_HC | Neotoma fuscipes | Henry Cowell | JF451141 | na | na |
| Nf_1707_HC | Neotoma fuscipes | Henry Cowell | JF451141 | na | na |
| Nf_1712_HC | Neotoma fuscipes | Henry Cowell | JF451141 | na | na |
| Nf_DU1_HW | Neotoma fuscipes | Hendy Woods | JF451141 | JF494841 | JF303739 |
| Nf_13.2_HW | Neotoma fuscipes | Hendy Woods | JF451141 | JF494841 | JF303739 |
| Dog_CA | Canis lupus familiaris | Tuolumne County | JF451139 | JF494833 | JF303732 |
| MRK_CA | Equus caballus | Shasta County | JF451139 | JF494840 | JF303732 |
| HZ_NY | Homo sapiens | New York State | JF451140 | JF494839 | JF303731 |
Identical sequences are represented by a single accession number.
Phylogenetic analysis.
For each genetic marker, phylogenetic analyses were performed using the program Treefinder (www.treefinder.de/former_index.html), which employs a maximum-likelihood (ML) method to resolve phylogenies, and MrBayes 3.1.2 (Ronquist & Huelsenbeck, 2003), which uses a Bayesian statistical approach based on probability distributions. For ML analyses, the optimal nucleotide substitution model was determined using the program jModeltest 0.1.1 (Posada, 2008). The HKY model of substitution was used for reconstructing the groESL and ank phylogenies while the GTR model was deemed most appropriate for phylogeny reconstruction using the 23S–5S marker. Bootstrap support for the ML trees was based on 1000 pseudoreplicate datasets generated from the original sequence alignments.
For Bayesian analysis, protein-coding ank and groESL DNA sequences and non-coding 23S–5S DNA sequences were aligned using muscle v3.8 (Edgar, 2004). Coding sequences were translated to ensure that the sequences were in-frame and did not contain stop codons. Identical sequences were excluded from the analysis then reincluded for the phylogeny figures.
Preliminary analyses were performed using the GTR model of nucleotide substitution, with a discrete gamma distribution used to accommodate among-site variation in the rate of nucleotide substitution (i.e. GTR+G). We accommodated process heterogeneity within the coding sequences (ank and groESL) by partitioning these gene regions by codon position: a separate GTR+G model was applied to each subset of nucleotides comprising the three codon positions.
For each of the three separate gene regions, we approximated the joint posterior probability distribution of trees and other parameters of the GTR+G substitution model using the Metropolis-Coupled Markov Chain Monte Carlo (MC3) samplers implemented in MrBayes. Preliminary analyses of each gene region entailed four independent MC3 analyses run for 1×107 generations, each with four incrementally heated chains (with the temperature parameter set to 0.2), where the cold chain was thinned by sampling every 1×103 cycles. For the purposes of diagnosing MCMC performance (see below), we also estimated the joint prior probability density for each of the three gene regions by running the MCMC samplers in MrBayes on an ‘empty’ alignment consisting of entirely missing data (e.g. a matrix of sequences where each sequence is a string of question marks equal in length to the corresponding sequence in the original data matrix).
For each of the three gene regions, we assessed performance of the MCMC analyses by scrutinizing the samples from each of the four independent chains using Tracer 1.4 (http://beast.bio.ed.ac.uk/Tracer). We assessed convergence by comparing the marginal posterior probability density for each of the model parameters to ensure that estimates from each of the four independent chains were effectively identical. We assessed mixing of the chains over each parameter using three diagnostics: (1) the rate at which proposed updates to each parameter were accepted (with a target window of 20–70 %); (2) by examining the form of the marginal posterior probability densities (to ensure that the density for each parameter was smooth and unimodal); and (3) by comparing the marginal posterior probability density for each parameter to its corresponding prior probability density (to assess the degree of information in the data for each parameter).
We identified weak parameters as those with low effective sample size values (i.e. <200) and for which the inferred marginal posterior and prior probability densities were similar. We eliminated any weak parameters by collapsing to submodels of the GTR family as necessary, and then running new analyses under these simplified models (using the same details as described above). Specifically, this led to simplifying the models for protein-coding sequences by collapsing from three partitions for each gene (corresponding to the three codon positions) to two partitions (with first and second codon positions combined into a single data partition, and third position as a single data partition) and finally to a single partition for all sites in each gene region.
During the model search, all three gene trees exhibited the MrBayes Long Tree pathology (Brown et al., 2010) so empirical branch-length priors were obtained from their corresponding ML estimates. ML estimates for each dataset were obtained using RAxML 7.0.4 (Stamatakis, 2006), performed with 30 individual replicates under the same partitioning schemes as those used in the MrBayes analyses. The tree with the highest log-likelihood was selected, and its tree length was extracted using the ape package in R (Paradis et al., 2004), and divided by (N–1), where N is the number of species in each dataset. This process resulted in the selection of the following substitution models. For ank, a separate GTR model was applied to each of three partitions (by codon position) with unlinked stationary frequencies, instantaneous rate matrices and rate priors, and an unconstrained exponential branch length prior across all partitions with a lambda of 80.447. For groESL, F81 was applied to each of two data partitions (first and second codon positions in one position, and third codon position in a second) with unlinked stationary frequencies, instantaneous rate matrices and rate priors, and an unconstrained exponential branch length prior with a lambda of 1338.250. For 23S–5S rRNA, GTR was applied to a single data partition for the alignment, with an unconstrained exponential branch length prior with a lambda of 16.202.
For the phylogenies presented below, all final MrBayes analyses were carried out with four independent runs and four chains per run. Each analysis ran for 5×107 generations, sampled every 1×103 generations, with a burn-in of 1×104 sampled generations. To summarize the posterior distribution of trees, we present only the majority-rule consensus trees for each of the gene regions (Holder et al., 2008). Alignments, trace files and tree posteriors are available upon request.
Results
PCR analysis
A total of 73 individual animals sampled from multiple sites throughout northern California between October 2005 and May 2008 were positive for infection with A. phagocytophilum, as detected by real-time PCR. They included 38 woodrats, 11 redwood chipmunks (Tamias ochrogenys), 6 Sonoma chipmunks (Tamias sonomae), 6 western grey squirrels, 3 eastern grey squirrels and 9 lizards including 7 western fence lizards, a northern alligator lizard and a sagebrush lizard. The overall infection prevalence was 9.3 % in woodrats, 6.3 % in chipmunks, 10.5 % in squirrels and 5.3 % among lizards. Successful amplification and DNA sequencing of the three genetic markers varied considerably. Amplification of the 23S–5S rRNA spacer region was the most sensitive, with 31 of the 73 real-time PCR-positive samples (42 %) amplifying successfully. The ank gene was less sensitive, with only 21 (30 %) samples amplifying successfully, while amplification of the groESL gene was the least sensitive, with only 15 (21 %) samples amplifying. The PCR results for all three genes are summarized in Table 1.
Phylogenetic analysis
Samples that amplified at the 23S–5S rRNA region included strains from 19 woodrats, 5 chipmunks, 3 western grey squirrels, 2 western fence lizards, the northern alligator lizard and the sagebrush lizard. Excluding the 4 lizards, only 3 single nucleotide polymorphisms (SNPs) were identified among all of the rodent-origin samples as well as the HZ, MRK and Dog_CA strains. All of the woodrats, which were sampled at three different sites (HC, HW and HV) separated by several hundred kilometres, shared a strain with a unique sequence with two of the redwood chipmunks from HW. This sequence varied by one SNP from the HZ strain sequence. The other three chipmunks (one from HW and two from SPT) and two of the squirrels (both from BB) had a sequence that was identical to the MRK and Dog_CA sequences, while the third squirrel sampled in HV had a unique SNP that further differentiated it from the other squirrels. In contrast, two considerably different sequences containing multiple SNPs and several insertions/deletions (indels) were detected among the four lizard-origin A. phagocytophilum strains.
The phylogenetic relationships among all of the sequences were assessed by ML and Bayesian analysis. Both types of analyses resulted in nearly identical phylograms differing only slightly in their support for particular nodes. The Bayesian tree with ML nodal support values is presented in Fig. 2. The phylogram separated the samples into three unique clades. The sagebrush lizard strain (Scg_752_HV) and one from a western fence lizard (Sco_622_HV) grouped together and strains from the other two lizards (Ec_765_HV and Sco_760_HV) formed their own distant clade. The other clearly defined clade consisted of all the mammalian-origin samples and was further partitioned based on the SNPs described above, although the posterior probabilities and bootstrap support for these partitions were weak, probably due to a low level of nucleotide variation.
Fig. 2.

Majority-rule consensus phylogram inferred from an alignment of A. phagocytophilum 23S–5S rRNA gene sequences. Bayesian analyses of phylogeny were performed using MrBayes v. 3.1.2. Identical sequences were excluded from analysis and reincluded for this figure. Values of nodal support are Bayesian posterior probabilities. For nodes recovered in both Bayesian and ML analyses, ML bootstrap confidence values exceeding 50 % are also shown. Branch lengths are in units of expected number of nucleotide substitutions per site. Amarginale, A. marginale (GenBank accession no. AY048815); HZ_NY, human strain from New York state; MRK_CA, horse strain from California; Dog_CA, dog strain from California; Sg, Sciurus griseus; Ts, Tamias sonomae; To, Tamias ochrogenys; Nf, Neotoma fuscipes; Scg, Sceloporus graciosus; Sco, Sceloporous occidentalis; Ec, Elgaria coerulea; HV, Hoopa Valley; BB, Big Basin; SPT, Samuel P. Taylor; HW, Hendy Woods; HC, Henry Cowell.
Amplification of the groESL marker was successful for only 15 rodent samples including 12 woodrats, 2 western grey squirrels and 1 Sonoma chipmunk. The HZ, MRK and Dog_CA strains also amplified successfully. Among these samples, eight unique sequences were identified. The two squirrel, chipmunk and MRK strains all shared identical sequences that differed by one SNP from the HZ strain, by another SNP from the Dog_CA strain and by one or more unique SNPs from all of the woodrat strains. A phylogenetic tree based on Bayesian and ML analysis illustrates the differences among the samples (Fig. 3). Similar to the partitioning of samples based on the 23S–5S rRNA marker, analysis of the groESL sequences shows that the HZ strain is unique from all others, branching out from the group of identical sequences that includes the MRK strain along with the squirrel and chipmunk strains. The woodrat strains form their own separate group. However, unlike the lack of sequence variation among woodrats across the 23S–5S rRNA region, further resolution of these samples was observed with the groESL marker.
Fig. 3.

Majority-rule consensus phylogram inferred from an alignment of A. phagocytophilum groESL sequences. Bayesian analyses of phylogeny were performed using MrBayes v. 3.1.2. Identical sequences were excluded from analysis and reincluded for this figure. Values of nodal support are Bayesian posterior probabilities. For nodes recovered in both Bayesian and ML analyses, ML bootstrap confidence values exceeding 50 % are also shown. Branch lengths are in units of expected number of nucleotide substitutions per site. HZ_NY, human strain from New York state; MRK_CA, horse strain from California; Dog_CA, dog strain from California; Sg, Sciurus griseus; Ts, Tamias sonomae; Nf, Neotoma fuscipes; HV, Hoopa Valley; BB, Big Basin; SPT, Samuel P. Taylor; HW, Hendy Woods; HC, Henry Cowell.
Amplification of the ank gene was successful for 21 samples including strains from 15 woodrats, 3 western grey squirrels, 1 eastern grey squirrel and 2 Sonoma chipmunks. The ank gene also showed considerably more variation among samples than either groESL or the 23S–5S rRNA marker. Sequence variation was attributable to numerous SNPs throughout the entire gene segment. In addition, a single 3 nt indel, corresponding to the amino acid leucine, distinguished the woodrat strains from the other strains. As for the other two markers, the HZ strain displayed a unique sequence differentiating it from the others. The MRK and Dog_CA strains had identical sequences to those of the eastern grey squirrel and two of the western grey squirrels and they varied by 1 SNP from the other western grey squirrel and the two chipmunks. These sequences varied from all of the woodrat sequences by at least 22 unique SNPs in addition to the indel described above.
As observed with the other two markers, phylogenetic relationships inferred by Bayesian and ML analysis indicated (with strong nodal support) the existence of three main clades (Fig. 4). The HZ strain formed its own clade, the MRK and VMTH dog strains formed a clade with all of the squirrels and chipmunks, and the woodrats formed a separate highly diverse clade. Interestingly, within the woodrat clade there was strong support for further partitioning of strains based on sampling location. In fact, only two woodrats, Nf_1026_HW and Nf_1603_HV, were not grouped by location.
Fig. 4.

Majority-rule consensus phylogram inferred from an alignment of A. phagocytophilum ank sequences. Bayesian analyses of phylogeny were performed using MrBayes v. 3.1.2. Identical sequences were excluded from analysis and reincluded for this figure. Values of nodal support are Bayesian posterior probabilities. For nodes recovered in both Bayesian and ML analyses, ML bootstrap confidence values exceeding 50 % are also shown. Branch lengths are in units of expected number of nucleotide substitutions per site. HZ_NY, human strain from New York state; MRK_CA, horse strain from California; Dog_CA, dog strain from California; Sg, Sciurus griseus; Sc, Sciurus carolinensis; Ts, Tamias sonomae; Nf, Neotoma fuscipes; HV, Hoopa Valley; BB, Big Basin; SPT, Samuel P. Taylor; HW, Hendy Woods; HC, Henry Cowell.
Discussion
With multiple reservoir hosts and several tick vectors, the ecology of A. phagocytophilum in the far-western US is complex and remains poorly understood. In order to understand the risk factors associated with transmission of a tick-borne pathogen to humans and domestic animals, it is important to know whether clinically or ecologically meaningful phenotypic and genetic diversity exists in the environment and which hosts carry pathogenic strains. In an effort to characterize the strains circulating in this environment, we utilized three genetic markers with varying levels of resolution and sensitivity to compare A. phagocytophilum DNA extracted from small mammal and lizard species collected in northern California.
Woodrats, chipmunks and squirrels have all been implicated as reservoirs for A. phagocytophilum in humans, dogs and horses (Foley et al., 2008b; Nieto & Foley, 2008, 2009). Based on sequence analysis across all three genetic markers, we have shown that strains capable of causing granulocytic anaplasmosis in a horse (MRK) and a dog (Dog_CA) are genetically identical or nearly identical to strains isolated from squirrels and chipmunks from multiple sites in northern California. In contrast, all of the woodrat strains collected from many of the same locations differed from the other strains consistently across all three markers. Although local human-origin strains of A. phagocytophilum were not available for analysis, previous findings indicated that human and equine strains from northern California were genetically identical or nearly identical across multiple genetic markers (Foley et al., 2009b). It was interesting that, at least in the intergenic spacer region, the human-origin HZ strain clustered with the woodrat strains. This finding could be due to shared, derived sequence evolution, or it could be the result of other processes, such as homoplastic evolution. We considered it unlikely to be a shared, derived character given the geographical distance between this strain origin and California. The close relationship was not observed for the other genes but further work using this marker more broadly geographically is warranted. Nevertheless, our findings lend strong support to the growing body of evidence suggesting that A. phagocytophilum strains infecting woodrats in California are fundamentally distinct from other strains including those capable of causing disease in humans and domestic animals. For example, there appears to be a lack of spatial overlap between clinical human, canine and equine cases of A. phagocytophilum and infection in woodrats (Chae et al., 2000). In addition, horses experimentally inoculated with woodrat strains of A. phagocytophilum did not become infected, while the same horses did become infected when they were subsequently inoculated with an A. phagocytophilum strain of chipmunk origin (Foley et al., 2008a). Lastly, genetic characterization of MSP2/P44 expression sites showed a clear difference between A. phagocytophilum strains originating from humans and dogs with those originating from woodrats (Morissette et al., 2009). Together, these findings indicate that in northern California, the likely reservoirs for granulocytic anaplasmosis, in humans and domestic animals, are sciurids (tree-squirrels and chipmunks) and not woodrats as was previously postulated.
Although all of the woodrat samples consistently grouped together phylogenetically and nearly all the sciurid samples clustered together as well, two redwood chipmunk samples (To_1591_HW and To_1634_HW) were an exception, clustering instead with the woodrat samples according to their 23S–5S rRNA gene sequences. Unfortunately, neither the ank nor the groESL genes were successfully amplified from these samples. These data suggest that, in some instances, chipmunks may acquire A. phagocytophilum strains that are typical in woodrats. Interestingly, laboratory studies have shown that woodrats are susceptible to several strains including HZ, MRK and Dog_CA (Nieto et al., 2010). However, infections with these strains were transient, typically detectable by PCR for only several weeks. In contrast, woodrats infected with an A. phagocytophilum strain of woodrat origin exhibited PCR-detectable infections for up to 8 months (Castro et al., 2001). One factor to consider is whether reservoir hosts could be infected concurrently with more than one strain of A. phagocytophilum. Limited experimental data suggest that infection with one A. phagocytophilum strain precludes infection with a second strain (J. Foley & D. Rejmanek, unpublished data). If true in the wild, this could further help to maintain the observed host tropism among strains.
There appears to be greater strain diversity among the woodrat samples than among sciurid samples, based on variations within the groESL and ank genes, although fewer sciurid samples were collected and sequenced than from woodrats. For the ank gene in particular, there was only 1 nt that differed among the six sciurids and MRK and Dog_CA strains, even though these samples originated from diverse locations across northern California. In contrast, the woodrat samples partitioned into multiple, well-supported clades or branches, each of which varied by at least 7 SNPs. Why there would be greater diversity among woodrat strains of A. phagocytophilum as compared to sciurid strains is not clear. One possibility is that the sciurid strains detected in this study are recent descendants of a highly successful strain that may have been able to outcompete other less successful sciurid strains. Another explanation may be that woodrat strains, which induce long-lasting infections in their hosts, could be partitioned into different geographical sites or host–vector cycles. This is supported by the fact that nearly all of the unique woodrat strains collected were spatially clustered, such that no single strain was detected in more than one location. The existence of multiple strains in any one area suggests that none may have a particular fitness advantage.
Of final note, the detection of two highly dissimilar strains of A. phagocytophilum in multiple lizard species is quite intriguing. Although western fence lizards have been shown to be both PCR- and seropositive for A. phagocytophilum in the wild, the dissimilarity of the 23S–5S rRNA gene lizard sequences from all other strains tested in the current study suggests that these may be more than just different strains but potentially novel species. It is unfortunate that the lizard strains did not amplify at either of the other two genes, although, given the variation within the 23S–5S rRNA marker, it is possible that the primer sequences for these more variable genes were too dissimilar to anneal properly. Additional research will be necessary to further classify these strains based on more conserved genetic regions (i.e. 16S rRNA gene and mitochondrial DNA). A broader survey of A. phagocytophilum strains infecting lizards and their associated ticks will also help us to understand the role and extent of these strains in the environment.
In conclusion, we have shown that multiple strains of A. phagocytophilum with distinct host tropisms exist in northern California. These include several highly divergent strains detected in lizards, several closely related sciurid strains that are identical or nearly identical to a disease-causing strain isolated from dogs and horses, and a more divergent group of strains unique to woodrats. Future epidemiological studies evaluating human and domestic animal risk need to incorporate these distinctions. With increasing reports of dissimilar genotypes of A. phagocytophilum from multiple regions of the world, it is important to continue to define distinct phenotypes based on ecological data as well.
Acknowledgements
This work was supported by the UC Davis Center for Vectorborne Diseases and the National Institutes of Health Allergy and Infectious Disease Evolution of Infectious Disease program #RO1 GM081714. We thank Anthony Barbet and Patrick Foley for helpful discussion, Nate Nieto for sample collection, laboratory work and early contributions to this manuscript, and Nat Lim, Joy Worth and Susan Wang for laboratory assistance.
Abbreviations:
- BB
Big Basin State Park
- HC
Henry Cowell State Park
- HV
Hoopa Valley Indian Reservation
- HW
Hendy Woods State Park
- ML
maximum-likelihood
- SNP
single nucleotide polymorphism
- SPT
Samuel P. Taylor State Park
References
- Bown K. J., Lambin X., Ogden N. H., Begon M., Telford G., Woldehiwet Z., Birtles R. J. (2009). Delineating Anaplasma phagocytophilum ecotypes in coexisting, discrete enzootic cycles. Emerg Infect Dis 15, 1948–1954 10.3201/eid1512.090178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. M., Hedtke S. M., Lemmon A. R., Lemmon E. M. (2010). When trees grow too long: investigating the causes of highly inaccurate bayesian branch-length estimates. Syst Biol 59, 145–161 10.1093/sysbio/syp081 [DOI] [PubMed] [Google Scholar]
- Castro M. B., Nicholson W. L., Kramer V. L., Childs J. E. (2001). Persistent infection in Neotoma fuscipes (Muridae: Sigmodontinae) with Ehrlichia phagocytophila sensu lato Am J Trop Med Hyg 65, 261–267 [DOI] [PubMed] [Google Scholar]
- Chae J. S., Foley J. E., Dumler J. S., Madigan J. E. (2000). Comparison of the nucleotide sequences of 16S rRNA, 444 Ep-ank, and groESL heat shock operon genes in naturally occurring Ehrlichia equi and human granulocytic ehrlichiosis agent isolates from Northern California. J Clin Microbiol 38, 1364–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.-M., Dumler J. S., Bakken J. S., Walker D. H. (1994). Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J Clin Microbiol 32, 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazenovich N., Foley J., Brown R. N. (2006). Use of real-time quantitative PCR targeting the msp2 protein gene to identify cryptic Anaplasma phagocytophilum infections in wildlife and domestic animals. Vector Borne Zoonotic Dis 6, 83–90 10.1089/vbz.2006.6.83 [DOI] [PubMed] [Google Scholar]
- Dumler J. S., Barbet A. F., Bekker C. P. J., Dasch G. A., Palmer G. H., Ray S. C., Rikihisa Y., Rurangirwa F. R. (2001). Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol 51, 2145–2165 10.1099/00207713-51-6-2145 [DOI] [PubMed] [Google Scholar]
- Dunning Hotopp J. C., Lin M., Madupu R., Crabtree J., Angiuoli S. V., Eisen J. A., Seshadri R., Ren Q., Wu M. & other authors (2006). Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet 2, e21 10.1371/journal.pgen.0020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004). muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foggie A. (1949). Studies on tick-borne fever in sheep. J Gen Microbiol 3, v. [PubMed] [Google Scholar]
- Foggie A. (1951). Studies on the infectious agent of tick-borne fever in sheep. J Pathol Bacteriol 63, 1–15 10.1002/path.1700630103 [DOI] [PubMed] [Google Scholar]
- Foggie A. (1962). Studies on tick pyaemia and tick-borne fever. In Aspects of Disease Transmission by Ticks, pp. 51–58. London: Zoological Society of London [Google Scholar]
- Foley J., Nieto N. C., Madigan J., Sykes J. (2008a). Possible differential host tropism in Anaplasma phagocytophilum strains in the Western United States. Ann N Y Acad Sci 1149, 94–97 10.1196/annals.1428.066 [DOI] [PubMed] [Google Scholar]
- Foley J. E., Clueit S. B., Brown R. N. (2008b). Differential exposure to Anaplasma phagocytophilum in rodent species in northern California. Vector Borne Zoonotic Dis 8, 49–56 10.1089/vbz.2007.0175 [DOI] [PubMed] [Google Scholar]
- Foley J. E., Nieto N. C., Foley P. (2009a). Emergence of tick-borne granulocytic anaplasmosis associated with habitat type and forest change in northern California. Am J Trop Med Hyg 81, 1132–1140 10.4269/ajtmh.2009.09-0372 [DOI] [PubMed] [Google Scholar]
- Foley J. E., Nieto N. C., Massung R., Barbet A., Madigan J., Brown R. N. (2009b). Distinct ecologically relevant strains of Anaplasma phagocytophilum. Emerg Infect Dis 15, 842–843 10.3201/eid1505.081502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley J. E., Rejmanek D., Fleer K., Nieto N. (2011). Nidicolous ticks of small mammals in Anaplasma phagocytophilum-enzootic sites in northern California. Ticks Tick Borne Dis 2, 75–80 10.1016/j.ttbdis.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder M. T., Sukumaran J., Lewis P. O. (2008). A justification for reporting the majority-rule consensus tree in Bayesian phylogenetics. Syst Biol 57, 814–821 10.1080/10635150802422308 [DOI] [PubMed] [Google Scholar]
- Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A. & other authors (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- Liz J. S., Sumner J. W., Pfister K., Brossard M. (2002). PCR detection and serological evidence of granulocytic ehrlichial infection in roe deer (Capreolus capreolus) and chamois (Rupicapra rupicapra). J Clin Microbiol 40, 892–897 10.1128/JCM.40.3.892-897.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod J., Gordon W. (1933). Studies in tick-borne fever of sheep. I. Transmission by the tick, Ixodes ricinus, with a description of the disease produced. Parasitology 25, 273–285 10.1017/S0031182000019442 [DOI] [Google Scholar]
- Madewell B. R., Gribble D. H. (1982). Infection in two dogs with an agent resembling Ehrlichia equi. J Am Vet Med Assoc 180, 512–514 [PubMed] [Google Scholar]
- Massung R. F., Priestley R. A., Miller N. J., Mather T. N., Levin M. L. (2003). Inability of a variant strain of Anaplasma phagocytophilum to infect mice. J Infect Dis 188, 1757–1763 10.1086/379725 [DOI] [PubMed] [Google Scholar]
- Massung R. F., Levin M. L., Munderloh U. G., Silverman D. J., Lynch M. J., Gaywee J. K., Kurtti T. J. (2007). Isolation and propagation of the Ap-Variant 1 strain of Anaplasma phagocytophilum in a tick cell line. J Clin Microbiol 45, 2138–2143 10.1128/JCM.00478-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette E., Massung R. F., Foley J. E., Alleman A. R., Foley P., Barbet A. F. (2009). Diversity of Anaplasma phagocytophilum strains, USA. Emerg Infect Dis 15, 928–931 10.3201/eid1506.081610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto N. C., Foley J. E. (2008). Evaluation of squirrels (Rodentia: Sciuridae) as ecologically significant hosts for Anaplasma phagocytophilum in California. J Med Entomol 45, 763–769 10.1603/0022-2585(2008)45[763:EOSRSA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Nieto N. C., Foley J. E. (2009). Reservoir competence of the redwood chipmunk (Tamias ochrogenys) for Anaplasma phagocytophilum. Vector Borne Zoonotic Dis 9, 573–577 10.1089/vbz.2008.0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto N. C., Foley J. E., Bettaso J., Lane R. S. (2009). Reptile infection with Anaplasma phagocytophilum, the causative agent of granulocytic anaplasmosis. J Parasitol 95, 1165–1170 10.1645/GE-1983.1 [DOI] [PubMed] [Google Scholar]
- Nieto N. C., Madigan J. E., Foley J. E. (2010). The dusky-footed woodrat (Neotoma fuscipes) is susceptible to infection by Anaplasma phagocytophilum originating from woodrats, horses, and dogs. J Wildl Dis 46, 810–817 [DOI] [PubMed] [Google Scholar]
- Paradis E., Claude J., Strimmer K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 10.1093/bioinformatics/btg412 [DOI] [PubMed] [Google Scholar]
- Posada D. (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25, 1253–1256 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- Pusterla N., Pusterla J. B., Braun U., Lutz H. (1999). Experimental cross-infections with Ehrlichia phagocytophila and human granulocytic ehrlichia-like agent in cows and horses. Vet Rec 145, 311–314 10.1136/vr.145.11.311 [DOI] [PubMed] [Google Scholar]
- Rejmanek D., Nieto N. C., Barash N., Foley J. E. (2011). Temporal patterns of tick-borne granulocytic anaplasmosis in California. Ticks Tick Borne Dis 2, 81–87 10.1016/j.ttbdis.2010.12.003 [DOI] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 10.1093/bioinformatics/btg180 [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- Stannard A. A., Gribble D. H., Smith R. S. (1969). Equine ehrlichiosis: a disease with similarities to tick-borne fever and bovine petechial fever. Vet Rec 84, 149–150 10.1136/vr.84.6.149 [DOI] [PubMed] [Google Scholar]
- Tyzzer E. (1938). Cytoecetes microti, n.g. (n.sp.), a parasite developing in granulocytes and infective for small rodents. Parasitology 30, 242–257 10.1017/S0031182000025774 [DOI] [Google Scholar]