

Abstract
Severe pulmonary hypertension is a lethal group of disorders which preferentially afflicts women. It appears that in recent years the patient profile has shifted towards older, obese, and postmenopausal women, suggesting that endocrine factors may be important. Several studies have revealed an increased prevalence of thyroid disease in these patients, but no studies have evaluated for a coexistence of endocrine factors. In particular, no studies have attempted to evaluate for concurrent thyroid disease, obesity and long-term estrogen exposure in patients.
88 patients attending the Pulmonary Hypertension Association 8th International meeting completed a questionnaire and were interviewed. Information was collected regarding reproductive history, height, weight, and previous diagnosis of thyroid disease.
46% met criteria for obesity. 41% reported a diagnosis of thyroid disease. 81% of women reported prior use of hormone therapy. 70% reported greater than 10 years of exogenous hormone use. 74% of female patients reported two or more of potentially disease modifying endocrine factors (obesity, thyroid disease or estrogen therapy).
The coexistent high prevalence in our cohort of exogenous estrogen exposure, thyroid disease and obesity suggests that an interaction of multiple endocrine factors might contribute to the pathogenesis of pulmonary hypertension and may represent epigenetic modifiers in genetically-susceptible individuals.
Keywords: Severe pulmonary hypertension, Estrogen, Menopause, Thyroid Disease, Autoimmunity, Obesity
Introduction
Severe pulmonary arterial hypertension continues to be categorized as idiopathic pulmonary arterial hypertension (IPAH) or secondary pulmonary hypertension associated with various disease states (APAH). The WHO pulmonary hypertension classification system includes categories of pulmonary arterial hypertension (PAH), pulmonary hypertension attributable to chronic left heart, lung or thromboembolic disease and "miscellaneous" forms [1]. Whereas thirty years ago idiopathic or then called "primary" pulmonary hypertension was recognized as a disease of young women [2] and the NIH-sponsored pulmonary hypertension registry reported a female to male ratio of 1.7: 1 [3], it appears that in recent years the patient profile has changed. The average patient diagnosed with IPAH is now older and frequently, a postmenopausal woman. The overall number of female patients also appears to have increased or is potentially overrepresented in clinical trials [4]. Data from the 2007 REVEAL registry report that 78% of 1226 patients with PAH were women with a median age of 53 years [5] and a large referral center reported on 657 patients with PAH, of which 77% were women and the mean age was 54 years [6]. In 2006, Taraseviciute published a retrospective medical record analysis of 1367 patients with severe PAH which revealed that 541 of these patients were postmenopausal women and that 217 of the women had a BMI > 30 kg/m2 [7]. The discovery of the familial IPAH gene, BMPRII [8] and of the association of ALK-1 gene mutations in patients with hereditary hemorrhagic telangiectasia (HHT) and PAH [9] provide the solid foundation for the concept of a genetic PAH predisposition. It has, however, been recognized that only about 50% of patients with germ-line BMPRII mutation develop severe PAH [10] and that the incidence of PAH in groups of patients with acknowledged risk factors, such as HIV infection or anorexigen drug intake, is low [11,12]. Remarkably, even in patients with significant intra-cardiac shunt, the fraction of patients who develop PAH is small. This raises the question whether there are additional previously unrecognized factors which, in concert with known trigger factors, can modify the risk of PAH development.
The shift in the patient profile towards older, obese, and postmenopausal women led us to postulate that there may be two previously unrecognized endocrine factors, which, when added to the genetic PAH susceptibility, may not only increase the risk of PAH development but also may be pathobiologically important. These factors include obesity, now recognized as a state of systemic inflammation [13] and exposure to potentially angiogenic estrogens in the form of hormone therapy for contraception or management of menopause. The concept of endocrine modulators in PAH is not entirely new. For example, several studies have reported an increased prevalence of autoimmune thyroid disease in PAH patients as compared to population based studies [14]. As autoimmunity, inflammation and angiogenesis [15,16] may each contribute to the pathogenesis of severe PAH, we developed a questionnaire and conducted an interview in order to obtain information regarding these putative endocrine risk factors in patient volunteers assembled at the 2008 biannual meeting of the Pulmonary Hypertension Association. In particular, information regarding reproductive history, thyroid disease, height and weight was obtained. In the design of the questionnaire we were guided by epidemiological studies in hormone responsive cancers conceived to shed light on possible associations between reproductive history and cancer development [17]. Here we present data which suggest that obesity, hormone therapy and thyroid disease are frequent findings in this group of patients with severe PAH.
Methods
Study population
Patients attending the Pulmonary Hypertension Association 8th International Pulmonary Hypertension Conference and Scientific Sessions (Houston, TX) were recruited for enrollment in the study. This conference is a forum for interactions between PH patients, motivated to learn about their disease, and PH clinicians and researchers. In recent years the conference has provided a "research room" for the purposes of recruitment of PH patients and to facilitate the collection of blood and clinical samples. Patients were considered eligible for our study if they reported a diagnosis of pulmonary hypertension, were currently receiving medical treatment for PAH and were able to give informed consent. IRB approval was obtained prior to the initiation of this study.
Medical history
Questions regarding the medical history included: current age, height, weight, age at diagnosis of PH, and current medications (in particular the use of prostacyclin or prostacyclin analogues). Patients were also asked about the "likely cause" of their pulmonary hypertension as it had been explained to them by their physician. Patients were also asked whether they had known family members with PH and to report the gender of these pulmonary hypertension patient relatives.
Baseline survey
Informed consent was obtained from each participant before study enrollment. A self-administered questionnaire which included the pertinent history regarding diagnosis and treatment of PH, current medications, family history of PH, reproductive history and history of thyroid disease was distributed to all consented patients. Completed questionnaires were collected within 2 subsequent days from 89 patients.
Patient interview
A standardized interview was conducted by a board-certified endocrinologist for the purpose of allowing the patients to ask questions and for further explanation of their written responses. All interviews were conducted by L.B.S.; and the average length of the interview was 10 minutes.
Reproductive factors and hormone use
Questions regarding reproductive factors included age at menarche, menstruation status, regularity of menstruation, length of menstruation, history of hysterectomy/oopherectomy, history of infertility (inability to become pregnant despite attempts greater than 1 year), history of polycystic ovarian syndrome (PCOS), history of pregnancy and live birth, history and length of breastfeeding, type of menopause (natural or induced) and history of exogenous estrogen exposure (female hormone treatment and ingestion of phyto-estrogens). Information collected on hormone treatment included current or past use of contraceptive agents (oral, transdermal, injection, and IUD) and agents initiated for menopause symptoms (oral preparation, transdermal preparation, vaginal estrogen cream). No questions were asked regarding dose or composition (estrogen/progestin) of hormone therapy given the anticipated difficulty in patient recall for these details [18]. Patients were instructed, however, to estimate the total duration of therapy for each agent. Patients were also asked about their personal history of hormone responsive cancers (breast or ovarian cancer) and history of anti-estrogen therapy.
Lifelong estrogen exposure
There is growing evidence to support the role of endogenous estrogens in the development of certain hormone responsive cancers [19]. Greater lifelong exposure to ovarian estrogen as occurs with early menarche, null parity and late menopause is associated with increased risk for the development of breast, endometrial and ovarian cancer in premenopausal women [20]. In postmenopausal women higher circulating estrogen levels are observed in obesity and these factors may act synergistically to confer increased risk in the development of hormone responsive cancer [21]. Although the risk of breast cancer associated with exogenous estrogen is somewhat controversial, current evidence suggests that prolonged estrogen therapy increases the risk of breast cancer, especially among younger women (age < 25 years) [22].
There has been recent interest in the relationship between hormone responsive cancer and soy-derived isoflavone ingestion. Soyfoods (isoflavones) have estrogen-like properties under certain conditions and may stimulate the growth of estrogen sensitive breast tumors [23]. Many women incorporate soyfoods into their diets to attenuate vasomotor symptoms of menopause [24].
In an effort to quantify cumulative lifelong endogenous and exogenous estrogen exposure, we evaluated subjects according to several major epidemiologic risk factors which have been previously identified in studies of hormone responsive cancers. These include obesity, parity, total years of menstruation, use exogenous female hormones, history of ovarian or breast cancer, and ingestion of soy isoflavones. Based on these factors we generated an arbitrary scoring system to characterize the extent and contribution of each element to the overall long-term estrogen exposure. Table 1 shows the elements of this scoring system. The total number of possible points for each subject was 12.
Table 1.
| Estrogen Exposure Factor | Points |
|---|---|
| BME | |
| > 25 kg/m2 | +1 |
| > 30 kg/m2 | +2 |
| Nuliiparous | +2 |
| Menstruation | |
| > 30 yrs | +1 |
| > 35 yrs | +2 |
| Exogenous hormones | |
| > 10 yrs | +1 |
| > 15 yrs | +2 |
| > 20 yrs | +3 |
| History of breast cancer | +1 |
| History of ovarian cancer | +1 |
| History of breastfeeding | |
| > 24 months | -1 |
| Isoflavone ingestion | |
| > 5 serving per week | +1 |
Thyroid disease
Participant PH patients were asked whether they had ever been diagnosed with thyroid disease and whether they had ever been on thyroid replacement therapy. During the patient interview, the interviewer asked further questions to clarify the type of thyroid disease (hypothyroidism, hyperthyroidism, thyroid cancer) reported by participants and to clarify whether the disorder required medical therapy (drug therapy, surgery or radioactive iodine). The form of thyroid disease and the treatment, if any, was recorded.
Obesity
Participants were asked to record their height and weight as measured most recently at their physician's office. BMI was calculated from self-report of height and weight.
Role of the funding source
This work has been supported by the following research grants to L.B.S.: Thomas F. Jeffress and Kate Miller Jeffress Memorial Trust research grant and VCU Health System BIRCWH award. Neither funding source was involved in the study design, collection or analysis of data, manuscript preparation or decision to submit the manuscript for publication.
Results
Characteristics of the cohort
Table 2 shows the baseline information and distribution of characteristics in the cohort. The total number of subjects was 88. 89% of subjects were female. The mean age of the cohort was 50.4 years. The mean age at diagnosis of PH was 44.2 years. A high rate of obesity was observed as the mean BMI was 31.7 kg/m2. 56% of participants reported having primary (familial or idiopathic) pulmonary hypertension. Individuals reporting PH associated with various diseases included the following: 21 patients with collagen vascular disease (RA, scleroderma, CREST), 5 patients with OSA, 6 patients with congenital or acquired cardiac disease (systemic to pulmonary shunt, valvular disease), 1 patient with portal hypertension, 1 patient with COPD, 1 patient with HIV and 1 patient with chronic thromboembolic disease. Lastly, 3 individuals reported anorexigen related PH. PH treatment included prostacyclin therapy in 37, endothelin receptor antagonists in 39, and PDE-5 inhibitors in 46 and calcium channel blockers in 10 patients. 48 subjects were on multiple drug therapy.
Table 2.
Baseline Characteristics
| No. of subjects | 88 |
| Males | 10 |
| Females | 78 |
| BMI > 30 kg/m2 | 22 |
| Mean Age (years) | 50.4 |
| Mean Age at diagnosis of PH (years) | 44.2 |
| Mean BMI (kg/m2) | 31.7 |
| Etiology of PH | |
| Primary (Famililal or Idipathic) | 49 |
| Associated forms | 39 |
| Collagen vascular disease | 21 |
| Chronic cardiac disease | 6 |
| Portal hypertension | 1 |
| Chronic lung disease | 1 |
| Obstructive sleep apnea | 5 |
| HIV | 1 |
| Thromboembolic disease | 1 |
| Anorexigen | 3 |
| Drug therapy for PH | |
| Prostacyclin/Analog | 37 |
| Endothelin receptor antagonist | 39 |
| PDE-5 inhibitor | 46 |
| Calcium channel blocker | 10 |
| Multiple drug therapy | 48 |
Reproductive factors
For decades there has been considerable interest in the female predominance of pulmonary hypertension. This has generated important questions regarding the potential for endogenous and exogenous estrogens to underlie the observed sex differences. Table 3 shows selective reproductive history characteristics of the female patients in the cohort.
Table 3.
Reproductive Characteristics of Female PAH Patients
| Premenopausal | Postmenopausal | |
|---|---|---|
| No. of subjects | 42 | 36 |
| BMI > 30 kg/m2 | 22 | 22 |
| History of fewer than 8 periods/year | 12 | 9 |
| History of infertility | 6 | 0 |
| history of acne | 3 | 3 |
| history of excess hair | 3 | 6 |
| history of PCOS | 1 | 2 |
| Years of Menstruation | ||
| < 15 | 3 | 0 |
| 15-29 | 30 | 12 |
| 30-35 | 6 | 7 |
| > 35 | 3 | 17 |
| Menopause status | ||
| Natural menopause | 17 | |
| Induced menopause | 19 | |
| Oopherectomy | 8 | |
| Parity | ||
| Parous | 37 | 30 |
| Nulliparous | 5 | 6 |
| No. of births | ||
| 1 | 3 | 5 |
| 2 | 22 | 14 |
| 3 | 8 | 6 |
| 4 | 4 | 3 |
| 5 | 0 | 2 |
| Use of exogenous female hormones | ||
| Never | 13 | 2 |
| Past | 29 | 34 |
| Oral contraceptive pills | 29 | 33 |
| Depo-Provera | 2 | 1 |
| HRT (oral) | 24 | |
| HRT (transdermal) | 2 | |
| Vaginal estrogen cream | 1 | |
| Current | ||
| Oral contraceptive pills | 1 | |
| HRT (oral) | 3 | |
| Hormone use (years) | ||
| > 10 | 19 | 20 |
| > 15 | 1 | 2 |
| > 20 | 2 | 7 |
| Use of SERMs (EVISTA) | 2 | |
| History of IUD | 3 | 9 |
| History of breast cancer | 0 | 6 |
| Use of tamoxifen | 6 | |
| History of ovarian cancer | 0 | 1 |
| History of breastfeeding | 7 | 26 |
| > 24 months | 10 | |
| History of DVT | 0 | 3 |
Exogenous estrogen exposure
A significant proportion of female PH patients reported prior use of exogenous estrogens. 69% of premenopausal females reported prior use of oral contraceptive pills (OCP), and 1 subject reported current use. 92% of postmenopausal women reported prior use of OCP and 67% reported prior use of estrogens in the form of hormone replacement therapy (HRT) following spontaneous or induced menopause. 3 subjects reported current use of HRT. These rates exceed estimates of OCP and HRT usage as compared to data derived from NHANES [25,26]. In the premenopausal subgroup, 45% the reported less than 10 yrs of OCP use, whereas 56% of postmenopausal women reported greater than 10 years of exogenous estrogen use. 2 subjects reported greater than 15 years and 7 subjects reported greater than 20 years of estrogen exposure. While specific data was not collected regarding the timing of estrogen therapy relative to diagnosis of PH, the mean age of the entire cohort was 50.4 years and the mean age at diagnosis was 44.2 years. As a majority of subjects in the postmenopausal group reported greater than 10 years of estrogen exposure, it is very likely that exogenous estrogen exposure occurred prior to diagnosis of pulmonary hypertension in this group. Among females reporting diagnosis of PH prior to 40 years of age, 75% also reported prior use of OCP.
Lifelong estrogen exposure
In an effort to roughly quantify lifelong estrogen exposure (load) we evaluated female PAH patients with regards to several major epidemiologic risk factors (some conferring increased risk and others conferring protection) which have been identified in studies of hormone responsive cancers. These included obesity, parity, total years of menstruation, exogenous hormone usage, history of ovarian or breast cancer, history of prolonged breast feeding, and ingestion of soy isoflavones. Collectively these factors were used to generate point system which was applied to each subject. Figure 1 depicts the proportion of risk factors points reported by subjects according to their menopause status. This assessment of reproductive history revealed that only 16% of premenopausal females reported none of these reproductive risk factors. A large proportion of these patients had either familial PAH or PAH associated with collagen vascular disease. With regards to the existence of multiple risk factor points, 29% of the premenopausal and 46% of postmenopausal women reported greater than 4 risk factors points. Regarding estrogen responsive cancers in the subgroup, 5 women reported breast cancer treated with anti-estrogen therapy. Of 3 subjects reporting DVT, all had used OCP and one individual had a concurrent history of breast cancer.
Figure 1.
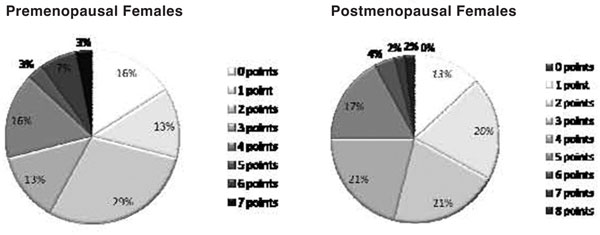
Reproductive risk factor point distribution for the cohort is shown according to menopause status.
Prevalence of thyroid disease
For the purposes of our study, thyroid disease was defined as "overt" hypo or hyperthyroidism requiring therapy or thyroid cancer. Several subjects reported a history of benign nodular thyroid disease or subclinical hypothyroidism, but these were not included for analysis. Table 2 shows the distribution of thyroid disease per self-report. The prevalence of self-reported thyroid disease in our cohort was 41%. While previous studies have reported as much as three-fold increase in prevalence of thyroid disease in PAH patients [27], our cohort reported thyroid disease at a rate six times that of large population based estimates of thyroid disease [28].
The percentage of subjects reporting thyroid disease was as follows: 84% of patients with thyroid disease reported having hypothyroidism, 8% reported having hyperthyroidism and 8% reported a history of thyroid cancer. The highest prevalence of thyroid disease was reported by patients aged 60-69 years and the lowest prevalence was reported by those aged 20-29 years. Although no discrete pattern emerged, there was a trend towards an age related increase in prevalence of thyroid disease. Controversy currently exists regarding the potential for prostacyclin therapy to induce or "unmask" thyroid disease [29]. The largest proportion of patients reporting both thyroid disease and current prostacyclin therapy occurred in the 30-39 year old group. This may explain the higher than expected prevalence of thyroid disease in this relatively young subgroup.
Thyroid disease according to PH subtype
41% of patients with "secondary" PH reported thyroid disease as compared to 38% of patients with primary disease. Nearly 50% of patients with "secondary" PAH and thyroid disease had collagen vascular disease (CREST syndrome, RA, scleroderma) which might reflect enhanced susceptibility to autoimmune disorders in these individuals. Several studies have reported associations between subclinical or overt thyroid disease and sleep disordered breathing [30]. Among individuals in our cohort with pulmonary hypertension secondary to OSA, 75% reported thyroid disease. The mean BMI of this group was 45.9 kg/m2. All individuals with OSA reported a diagnosis of hypothyroidism and current treatment with thyroid replacement hormone.
Prevalence of obesity
The mean BMI of the entire cohort was 31.7 kg/m2. 46% of patients met criteria for obesity as defined by a BMI > 30 kg/m2 (Figure 2). The prevalence of obesity was the most pronounced in the 50-59 year old subgroup, followed closely by the 40-49 year old subgroup. The rate of obesity in our cohort exceeds the prevalence of obesity as suggested by NHANES data [31]. 13 subjects were currently on glucocorticoid therapy.
Figure 2.
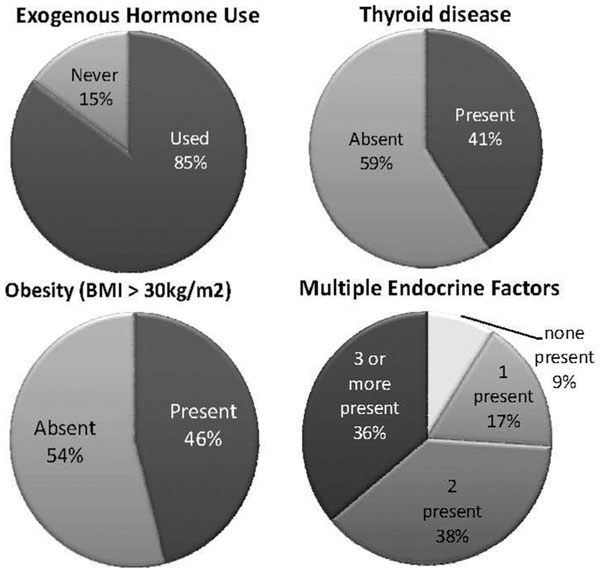
Distribution of these risk factors and the combination of risk factors.
Prevalence of multiple endocrine risk factors
We analyzed all females in the cohort for the presence of multiple endocrine risk factors to include BMI > 30 kg/m2, previous use of exogenous estrogens and thyroid disease requiring medical therapy. Figure 3 depicts the results. Nine percent of females had no risk factor points and 17% had one risk factor point. 74% of patients had multiple endocrine factors points and this group was comprised mostly of postmenopausal females (Figure 2).
Figure 3.
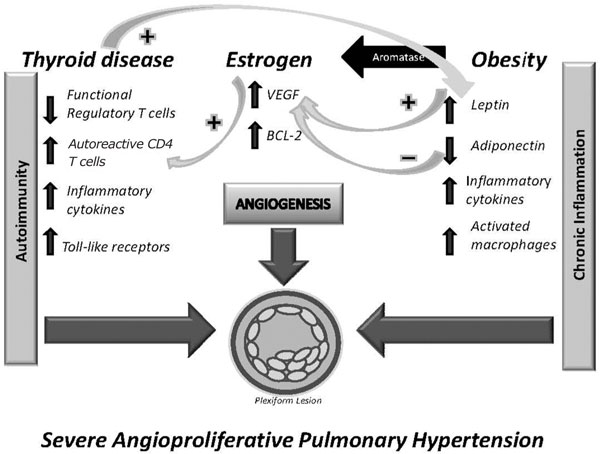
Schematic illustrating the potential interactions of pathobiologically relevant risk factos which may contribute to severe angioproliferative pulmonary arterial hypertension. Hypothetically, autoimmunity and chronic inflammation interact via thyroid disease, estrogen and obesity. VEGF = vascular endothelial growth factor, Bcl-2 = antiapoptotic B-cell lymphoma protein.
Discussion
Apparently during the last 20 years more patients in the U.S.A. have received the diagnosis of pulmonary hypertension than in preceding decades [32]. Overall, severe PAH is a group of rare diseases, either idiopathic or associated with known causes. Whereas progress has been made to elucidate the gene mutations which underlie the susceptibility for PAH development in some groups of patients [33] little is known about modifiers of disease or epigenetically important factors and conditions. One of several disease modifiers may be female gender as a greater prevalence of women in all forms of PAH has been recognized for decades [2]. It is remarkable that the fraction of women, and in particular older women, in most groups of PAH patients has increased in recent years and also that no hypothesis for this gender preference has been developed. Although a causative or permissive role of angiogenic and perhaps proinflammatory estrogen metabolites [34] is now somewhat intuitive, there are no data on cumulative life-long estrogen exposure in PH patients. Another risk factor for development of PAH is autoimmunity [35]. All forms of autoimmune disorders are more prevalent in women and hormonal factors have been considered to explain this prevalence. Increased rates of autoimmune thyroid disease have been observed in patients with IPAH and many disease-associated forms of pulmonary hypertension [36]. Lastly obesity, now recognized as a complex endocrinopathy with a systemic inflammatory component and a risk factor for hormone-sensitive cancers [37], has recently been reported in women with PAH [7].
In order to survey the prevalence of these individual factors and conditions in a cohort of patients with the diagnosis of severe PAH, both a focused endocrine disorder questionnaire was administered and an interview was conducted. Here we report a high prevalence of long-term estrogen exposure, thyroid disorder and obesity in this cohort of PAH patients. As the overwhelming number of patients in this cohort were women, we focus the following discussion on the female patients' characterization.
Long-term estrogen exposure
Early and long term estrogen exposure has been correlated with an increased risk for the development of hormone responsive cancers. There is also accumulating evidence that estrogen is a key regulator of endothelial cell proliferation and function in vivo [38]. Similar mechanisms which underlie both carcinogenesis and endothelial cell proliferation include estrogen induced VEGF expression, decreased synthesis of proteins required for apoptosis and up-regulation of protective genes in cell death pathways [39]. Given our current understanding of the pathology of PAH which includes the phenotype switch of pulmonary vascular endothelium into a highly proliferative, apoptosis-resistant phenotype [40], we have a growing interest in estrogen exposure in the context of PAH. As such, we were particularly impressed with the rates of self-reported hormone therapy in our cohort. Indeed 71% of the premenopausal and 86% of the postmenopausal women reported previous or current use of hormone therapy. In the postmenopausal subgroup, more than 50% of patients reported greater than 10 years of hormone therapy, which was most commonly a prolonged use of oral contraceptive pills. While we intentionally avoided questions regarding hormone dose and estrogen/progestin composition, the OCP preparations commonly prescribed to women of a similar age as our post-menopausal subgroup consisted of higher dose estrogen than is commonly prescribed today [41]. This prominent use of hormone therapy was reflected in our scoring of lifelong estrogen exposure in our cohort. In fact, in postmenopausal patients, obesity and prolonged hormone therapy were the most significant contributors to our putative risk factor score. Of interest, is the observation that only a small proportion of women had no risk factors. These were young women (average age < 22 years) with either familial IPAH or PAH associated with CREST syndrome. Perhaps in these individual women, genetic or disease associated factors are prominent enough to promote the development of PAH, and the contribution of a "second hit" is less important. It may also be of interest that the use of phytoestrogens was common in this patient cohort.
Autoimmune thyroid disease
It has been established that a higher prevalence of autoimmune thyroid disease exists in PH patients, and that anti-thyroid antibodies can be detected in patients without overt thyroid dysfunction at rates higher than the general population [40]. Thyrotoxicosis has also been considered a risk factor for pulmonary arterial hypertension, and treatment with thionamides in this population can reverse PAH [43,44]. We found a high proportion of patients in our cohort reporting a history of thyroid disease and current use of thyroid hormone supplementation. Indeed thyroid disease among our patients was prevalent at a rate nearly double that recently published by Burger and colleagues at Mayo Clinic [45]. In their retrospective study thyroid disease was defined as abnormal TSH, history of thyroid disease on replacement or elevated TPO antibody. While we did rely on self-report of thyroid disease in our study, we defined individuals as having thyroid dysfunction only if they required medical therapy. Of course we do not have information on the severity of thyroid gland dysfunction at the time of initiation of therapy, so it is possible that some patients were currently on therapy for subclinical thyroid disease.
The majority of patients reporting thyroid disease were hypothyroid and a significant proportion also reported current therapy with prostacyclin or prostacyclin analogs. There has been a longstanding interest in the potential of prostacyclin to induce thyroid disease in PAH patients [46]. Indeed thyroid tissue has high and low affinity receptors for prostacyclin and alterations in receptor density and kinetics have been observed in Hashimoto's, Reidel thyroiditis, autonomous thyroid nodules and thyroid cancer [47]. Prostacyclin infusion can also enhance TSH secretion and promote peripheral conversion of T4 to T3 [48]. Currently there is no consensus regarding the importance of establishing baseline thyroid function in patients with PAH, nor in monitoring thyroid function prior to or after the initiation of prostacyclin therapy. Additionally, there have been no studies to date which have evaluated the treatment of subclinical thyroid dysfunction in this population. Treatment of thyroid dysfunction in PAH may have important pathobiological consequences. While the effects of thyroid hormone on lung ventilation/oxygenation and peripheral vascular resistance [49] have been characterized for some time, new evidence suggests that thyroid hormone through non-genomic signaling may induce proliferation and promote migration of human arterial smooth muscle cells [50].
Obesity
The high prevalence of obesity in our cohort echoes recent findings from larger PH surveys and may be important in the pathobiology of PH. It is now recognized that obesity is a systemic process characterized by chronic inflammation, pro-angiogenesis and impaired immune function [51]. Adipose tissue is a dynamic organ capable of synthesis and secretion of proinflammatory cytokines, hormones (adipokines) and potent growth factors which have local paracrine and systemic effects [52]. The accumulation of intra-abdominal fat leads to impaired insulin signaling, and decreased intracellular glucose transport [53]. These signaling defects result in impaired endothelium-dependent vasodilation [54]. Additionally, obesity is associated with characteristic changes in the proportion of adipose derived hormones. Fat-pad production of adiponectin and ghrelin is decreased, and the production of leptin is increased with caloric excess [55]. While these hormones have well established roles in energy homeostasis, current research also suggests that they may be important regulators of angiogenesis [56]. Adiponectin has been shown to inhibit tumor cell proliferation, microvessel angiogenesis and VEGF-induced migration of coronary artery endothelial cells [57]. Conversely, leptin is mitogenic to vascular endothelium and and coronary artery smooth muscle cells, likely through upregulation of VEFGR-1 [58].
Leptin also induces matrix remodeling through upregulation of matrix metalloproteinases [59]. Our understanding of the relationship between obesity and innate immunity now extends beyond the concept of chronic inflammation resulting in immune dysfunction (Figure 3). The fat pad is a reservoir for macrophages which are functionally similar to "M1" or classically activated macrophages [60], and the immunodeficiency of obesity involves defective T-cell to mitogen response, and decreased NK cell activity [61]. There are also overlapping defects in key signaling pathways in obesity and PH. It is intriguing that TGF-β signaling is impaired in PH and that proper signaling through this pathway is key to the regulation of satiety in humans (through DAF-7) [62]. This has interesting implications regarding potential therapies targeted to both PH and obesity.
As investigators are beginning to consider PAH disease modifiers, each of these putative factors or conditions present in our cohort could indeed represent an initiating or disease modifying factor. Although the rationale for hypothetically linking long-term estrogen exposure, autoimmune thyroid disease and obesity with the development of PAH (Figure 3) is attractive, our approach to data gathering and data analysis can provide circumstantial evidence at best. Neither long term estrogen exposure nor obesity have previously been considered important risk or trigger factors for PAH. Abehaim et al. [63] in their prospective case control study of anorexigen induced PAH could not establish a statistically significant link between obesity and PAH, however the cohort studied was European and obesity is still less prevalent in Europe as compared to the US population. Overall larger multicenter cohorts have not been evaluated for the presence or prevalence of these putative risk factors and the concept of potentially important epigenetic factors in the development of PAH are not generally accepted. It is evident that hypothesis-driven mining of databases requires the inclusion of data which are pertinent to the subject under investigation and it is therefore regrettable that the recently established REVEAL pulmonary hypertension data bank does not contain elements of the reproductive history of women. As there is presently no general acceptance of a concept of epigenetic modifiers, there is certainly no appreciation for a potential confluence or interaction of multiple (at present putative) factors. Indeed the data derived from our questionnaire and interview document that the majority of the PAH patients presented with more than one of these putative risk factors, in fact, nearly three-quarters of these patients presented with more than one of these factors.
Limitations
Clearly, like any relatively small cohort study, ours has a number of limitations and the results are in some danger of over-interpretation. We chose to investigate patients attending the 2008 International Pulmonary Hypertension Association meeting and these individuals represent a subset highly motivated to participate in clinical studies. As such, our findings might not be generalizable to larger groups of PAH patients. Additionally, many patients were from specialized PAH treatment centers in the US. It could be argued that this group likely has enhanced access to medical care such that surveillance for and diagnosis of thyroid disease could have been increased. Second, our study design included a self-administered questionnaire and personal interview and relied largely upon recall of clinical data, and as such, was subject to recall bias. Previous studies which have compared recalled and validated elements of the reproductive history have shown fair to excellent agreement in personal recall of onset and duration of menses, parity, and use of hormone therapy. Less agreement has been observed in recall of hormone preparation, duration of hormone therapy and onset of menopause [64,65]. Also, as we relied upon self-report of most recent height and weight when calculating BMI, our results likely represent an underestimation of true BMI. Several studies suggest that self-reported weight can vary by as much as 3 kilograms from measured weight and that in most cases it is underestimated [66]. Lastly, as some of our questions could be considered sensitive, and the patient interview was conducted by a single board-certified endocrinologist, the potential exists for both interviewer and respondent bias to have influenced our results. Furthermore, the questionnaire used here has not been previously validated in the form presented to these PAH patients, although the composition of the questions is similar to those commonly used in the field of hormone-sensitive cancer epidemiology. Our decision to use an arbitrary point score system in order to attempt a more quantitative assessment of estrogen exposure was entirely ours and may be subject to severe criticism. Finally, we need to emphasize that our process of data gathering amounts to little more than a first survey of a potentially very complex interplay between genetic and endocrine risk factors in female patients with PAH. Nevertheless, the results of our survey can be seen as hypothesis-generating and may provide the basis for the design of a prospective multi-center study of women with PAH which would focus on these putative risk factors. Such a study would rely upon a questionnaire similar to ours and would for its analysis not rely upon comparative data derived from existing databases.
Authors' contributions
I declare that I have participated in study design, collection and interpretation of data, writing of the manuscript, and the decision to submit the manuscript for publication. I have no conflicts of interest with regards to this research. LBS, M.D.
I declare that I have participated in study design, interpretation of data, writing of the manuscript and the decision to submit the manuscript for publication. I have no conflicts of interest with regards to this research. NFV, M.D. (Corresponding author)
Acknowledgements
This work has been supported by the following research grants to L.B.S.: Thomas F. Jeffress and Kate Miller Jeffress Memorial Trust research grant and VCU Health System BIRCWH award. The authors wish to thank the Pulmonary Hypertension Association (PHA) leadership, in particular Dr. Greg Elliott (Intermountain Medical Center University of Utah School of Medicine) and the pulmonary hypertension patients who consented to participate in this survey. We also wish to thank Dr. John Nestler (Virginia Commonwealth University School of Medicine) for his critical reading of this manuscript.
References
- Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G. et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- Voelkel NF, Reeves JT. Primary Pulmonary Hypertension in Pulmonary Vascular Diseases. New York: Marcel Dekker; 1979. Lung Biology in Health and Disease; pp. 573–628. [Google Scholar]
- Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM. et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216–23. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- Hill NS, Preston IR, Roberts KE. Patients with Pulmonary Arterial Hypertension in Clinical Trials. Who Are They? Proc Am Thorac Soci. 2008;5:603–9. doi: 10.1513/pats.200803-032SK. [DOI] [PubMed] [Google Scholar]
- Badesch DB, Benza RL, Krichman AM, Raskob GE, Giles S. Reveal registry: baseline characteristics of the first 1,226 enrolled patients. Chest Meeting Abstracts. 2007;132(4):473b–474. [Google Scholar]
- Maradit-Kremers H, Golbin JM, Slusser JP, Scott CG, Kane GC, McGoon MD. Treatment patterns and predictors of drug therapy in pulmonary arterial hypertension (PAH) between 1995 and 2005. Mayo Clinic, Rochester, MN, USA; 2008. [Google Scholar]
- Taraseviciute A, Voelkel NF. Severe pulmonary hypertension in postmenopausal obese women. Eur J Med Res. 2006;11(5):198–202. [PubMed] [Google Scholar]
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G. et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD. et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40(12):865–71. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S. et al. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol. 2004;43(12 Suppl S):33S–9S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Sitbon O, Lascoux-Combe C, Delfraissy JF, Yeni PG, Raffi F, De ZD. et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177(1):108–13. doi: 10.1164/rccm.200704-541OC. [DOI] [PubMed] [Google Scholar]
- Galie N, Manes A, Palazzini M, Negro L, Marinelli A, Gambetti S. et al. Management of pulmonary arterial hypertension associated with congenital systemic-to-pulmonary shunts and Eisenmenger's syndrome. Drugs. 2008;68(8):1049–66. doi: 10.2165/00003495-200868080-00004. [DOI] [PubMed] [Google Scholar]
- Wassink AM, Olijhoek JK, Visseren FL. The metabolic syndrome: metabolic changes with vascular consequences. Eur J Clin Invest. 2007;37(1):8–17. doi: 10.1111/j.1365-2362.2007.01755.x. [DOI] [PubMed] [Google Scholar]
- Ferris A, Jacobs T, Widlitz A, Barst RJ, Morse JH. Pulmonary arterial hypertension and thyroid disease. Chest. 2001;119(6):1980–1. doi: 10.1378/chest.119.6.1980. [DOI] [PubMed] [Google Scholar]
- Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26(6):1110–8. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM. et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- La VC, Negri E, Franceschi S, Parazzini F. Long-term impact of reproductive factors on cancer risk. Int J Cancer. 1993;53(2):215–9. doi: 10.1002/ijc.2910530207. [DOI] [PubMed] [Google Scholar]
- Ko KP, Park SK, Kim Y, Bae J, Jun JK, Gwack J. et al. [Reliability of a questionnaire for women's reproductive history] J Prev Med Public Health. 2008;41(3):181–5. doi: 10.3961/jpmph.2008.41.3.181. [DOI] [PubMed] [Google Scholar]
- Layde PM, Webster LA, Baughman AL, Wingo PA, Rubin GL, Ory HW. The independent associations of parity, age at first full term pregnancy, and duration of breast-feeding with the risk of breast cancer. Cancer and Steroid Hormone Study Group. J Clin Epidemiol. 1989;42(10):963–73. doi: 10.1016/0895-4356(89)90161-3. [DOI] [PubMed] [Google Scholar]
- Eliassen AH, Hankinson SE. Endogenous hormone levels and risk of breast, endometrial and ovarian cancers: prospective studies. Adv Exp Med Biol. 2008;630:148–65. doi: 10.1007/978-0-387-78818-0_10. [DOI] [PubMed] [Google Scholar]
- Vona-Davis L, Howard-McNatt M, Rose DP. Adiposity, type 2 diabetes and the metabolic syndrome in breast cancer. Obes Rev. 2007;8(5):395–408. doi: 10.1111/j.1467-789X.2007.00396.x. [DOI] [PubMed] [Google Scholar]
- Breast cancer and combined oral contraceptives: results from a mulit-national study: WHO collaborative study of neoplasia and steroid contraceptives. Br J Cancer. 1990;61(1):110–9. doi: 10.1038/bjc.1990.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina MJ, Loprinzi CL. Soy for breast cancer survivors: a critical review of the literature. J Nutr. 2001;131(11 Suppl):3095S–108S. doi: 10.1093/jn/131.11.3095S. [DOI] [PubMed] [Google Scholar]
- Maskarinec G, Singh S, Meng L, Franke AA. Dietary soy intake and urinary isoflavone excretion among women from a multiethnic population. Cancer Epidemiol Biomarkers Prev. 1998;7(7):613–9. [PubMed] [Google Scholar]
- Crespo CJ, Smit E, Snelling A, Sempos CT, Andersen RE. Hormone replacement therapy and its relationship to lipid and glucose metabolism in diabetic and nondiabetic postmenopausal women: results from the Third National Health and Nutrition Examination Survey (NHANES III) Diabetes Care. 2002;25(10):1675–80. doi: 10.2337/diacare.25.10.1675. [DOI] [PubMed] [Google Scholar]
- Taichman LS, Eklund SA. Oral contraceptives and periodontal diseases: rethinking the association based upon analysis of National Health and Nutrition Examination Survey data. J Periodontol. 2005;76(8):1374–85. doi: 10.1902/jop.2005.76.8.1374. [DOI] [PubMed] [Google Scholar]
- Li JH, Safford RE, Aduen JF, Heckman MG, Crook JE, Burger CD. Pulmonary hypertension and thyroid disease. Chest. 2007;132(3):793–7. doi: 10.1378/chest.07-0366. [DOI] [PubMed] [Google Scholar]
- Aoki Y, Belin RM, Clickner R, Jeffries R, Phillips L, Mahaffey KR. Serum TSH and total T4 in the United States population and their association with participant characteristics: National Health and Nutrition Examination Survey (NHANES 1999-2002) Thyroid. 2007;17(12):1211–23. doi: 10.1089/thy.2006.0235. [DOI] [PubMed] [Google Scholar]
- Arroliga AC, Dweik RA, Rafanan AL. Primary pulmonary hypertension and thyroid disease. Chest. 2000;118(4):1224–5. doi: 10.1378/chest.118.4.1224. [DOI] [PubMed] [Google Scholar]
- Resta O, Pannacciulli N, Di GG, Stefano A, Barbaro MP, De PG. High prevalence of previously unknown subclinical hypothyroidism in obese patients referred to a sleep clinic for sleep disordered breathing. Nutr Metab Cardiovasc Dis. 2004;14(5):248–53. doi: 10.1016/S0939-4753(04)80051-6. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999-2000. JAMA. 2002;288(14):1723–7. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- Hyduk A, Croft JB, Ayala C, Zheng K, Zheng ZJ, Mensah GA. Pulmonary hypertension surveillance--United States, 1980-2002. MMWR Surveill Summ. 2005;54(5):1–28. [PubMed] [Google Scholar]
- Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S. et al. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol. 2004;43(12 Suppl S):33S–9S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354(3):270–82. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- Badesch DB, Wynne KM, Bonvallet S, Voelkel NF, Ridgway C, Groves BM. Hypothyroidism and primary pulmonary hypertension: an autoimmune pathogenetic link? Ann Intern Med. 1993;119(1):44–6. doi: 10.7326/0003-4819-119-1-199307010-00008. [DOI] [PubMed] [Google Scholar]
- Arroliga AC, Dweik RA, Rafanan AL. Primary pulmonary hypertension and thyroid disease. Chest. 2000;118(4):1224–5. doi: 10.1378/chest.118.4.1224. [DOI] [PubMed] [Google Scholar]
- Rinaldi S, Key TJ, Peeters PH, Lahmann PH, Lukanova A, Dossus L. et al. Anthropometric measures, endogenous sex steroids and breast cancer risk in postmenopausal women: a study within the EPIC cohort. Int J Cancer. 2006;118(11):2832–9. doi: 10.1002/ijc.21730. [DOI] [PubMed] [Google Scholar]
- Applanat MP, Buteau-Lozano H, Herve MA, Corpet A. Vascular endothelial growth factor is a target gene for estrogen receptor and contributes to breast cancer progression. Adv Exp Med Biol. 2008;617:437–44. doi: 10.1007/978-0-387-69080-3_42. [DOI] [PubMed] [Google Scholar]
- Lee MY, Jung SC, Lee JH, Han HJ. Estradiol-17beta protects against hypoxia-induced hepatocyte injury through ER-mediated upregulation of Bcl-2 as well as ER-independent antioxidant effects. Cell Res. 2008;18(4):491–9. doi: 10.1038/cr.2008.42. [DOI] [PubMed] [Google Scholar]
- Rai PR, Cool CD, King JAC, Stevens T, Burns N, Winn RA. et al. The Cancer Paradigm of Severe Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2008;178(6):558–64. doi: 10.1164/rccm.200709-1369PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey PM, Cerhan JR, Pruthi S. Oral contraceptive use and risk of breast cancer. Mayo Clin Proc. 2008;83(1):86–90. doi: 10.4065/83.1.86. [DOI] [PubMed] [Google Scholar]
- Arroliga AC, Dweik RA, Rafanan AL. Primary pulmonary hypertension and thyroid disease. Chest. 2000;118(4):1224–5. doi: 10.1378/chest.118.4.1224. [DOI] [PubMed] [Google Scholar]
- Ma RC, Cheng AY, So WY, Hui DS, Tong PC, Chow CC. Thyrotoxicosis and pulmonary hypertension. Am J Med. 2005;118(8):927–8. doi: 10.1016/j.amjmed.2005.03.038. [DOI] [PubMed] [Google Scholar]
- Siu CW, Zhang XH, Yung C, Kung AW, Lau CP, Tse HF. Hemodynamic changes in hyperthyroidism-related pulmonary hypertension: a prospective echocardiographic study. J Clin Endocrinol Metab. 2007;92(5):1736–42. doi: 10.1210/jc.2006-1877. [DOI] [PubMed] [Google Scholar]
- Li JH, Safford RE, Aduen JF, Heckman MG, Crook JE, Burger CD. Pulmonary hypertension and thyroid disease. Chest. 2007;132(3):793–7. doi: 10.1378/chest.07-0366. [DOI] [PubMed] [Google Scholar]
- Ferris AM, Morse JH, Barst RJ. Thyroid disease and pulmonary arterial hypertension (PAH) in patients treated with prostacylin. Circulation. 2000;102[18](suppl II):426. Ref Type: Abstract. [Google Scholar]
- Virgolini I, Steurer G, Keminger K, Sinzinger H, Kraupp O. Evaluation of prostaglandin receptors in the human thyroid gland. Prog Clin Biol Res. 1987;242:35–42. [PubMed] [Google Scholar]
- Mitsuma T, Nogimori T, De HS, Chaya M. Effect of prostaglandin A1 and A2 on thyrotropin secretion in rats. Endocrinol Exp. 1986;20(4):371–7. [PubMed] [Google Scholar]
- Vinzio S, Morel O, Schlienger JL, Goichot B. Cellular mechanisms of thyroid hormone action. Presse Med. 2005;34(16 Pt 1):1147–52. doi: 10.1016/s0755-4982(05)84141-7. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Tsunekawa K, Seki K, Mori M, Murakami M. Regulation of iodothyronine deiodinase and roles of thyroid hormones in human coronary artery smooth muscle cells. Atherosclerosis. 2006;186(1):207–14. doi: 10.1016/j.atherosclerosis.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Karagiannides I, Pothoulakis C. Obesity, innate immunity and gut inflammation. Curr Opin Gastroenterol. 2007;23(6):661–6. doi: 10.1097/MOG.0b013e3282c8c8d3. [DOI] [PubMed] [Google Scholar]
- Bakhai A. Adipokines--targeting a root cause of cardiometabolic risk. QJM. 2008. [DOI] [PubMed]
- Despres JP. Health consequences of visceral obesity. Ann Med. 2001;33(8):534–41. doi: 10.3109/07853890108995963. [DOI] [PubMed] [Google Scholar]
- Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103(5):398–406. doi: 10.1007/s00395-008-0733-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays HE, Gonzalez-Campoy JM, Bray GA, Kitabchi AE, Bergman DA, Schorr AB. et al. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev Cardiovasc Ther. 2008;6(3):343–68. doi: 10.1586/14779072.6.3.343. [DOI] [PubMed] [Google Scholar]
- Hausman GJ, Richardson RL. Adipose tissue angiogenesis. J Anim Sci. 2004;82(3):925–34. doi: 10.2527/2004.823925x. [DOI] [PubMed] [Google Scholar]
- Kelesidis I, Kelesidis T, Mantzoros CS. Adiponectin and cancer: a systematic review. Br J Cancer. 2006;94(9):1221–5. doi: 10.1038/sj.bjc.6603051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Brakenhielm E, Wahlestedt C, Thyberg J, Cao Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc Natl Acad Sci USA. 2001;98(11):6390–5. doi: 10.1073/pnas.101564798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE. et al. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. 2001;33(2):95–102. doi: 10.1038/emm.2001.17. [DOI] [PubMed] [Google Scholar]
- Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14(12):1225–30. doi: 10.2174/138161208784246153. [DOI] [PubMed] [Google Scholar]
- Nieman DC, Henson DA, Nehlsen-Cannarella SL, Ekkens M, Utter AC, Butterworth DE. et al. Influence of obesity on immune function. J Am Diet Assoc. 1999;99(3):294–9. doi: 10.1016/S0002-8223(99)00077-2. [DOI] [PubMed] [Google Scholar]
- You YJ, Kim J, Raizen DM, Avery L. Insulin, cGMP, and TGF-beta signals regulate food intake and quiescence in C. elegans: a model for satiety. Cell Metab. 2008;7(3):249–57. doi: 10.1016/j.cmet.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich S, Rubin L, Walker AM, Schneeweiss S, Abenhaim L. Anorexigens and pulmonary hypertension in the United States: results from the surveillance of North American pulmonary hypertension. Chest. 2000;117(3):870–4. doi: 10.1378/chest.117.3.870. [DOI] [PubMed] [Google Scholar]
- Bosetti C, Tavani A, Negri E, Trichopoulos D, La VC. Reliability of data on medical conditions, menstrual and reproductive history provided by hospital controls. J Clin Epidemiol. 2001;54(9):902–6. doi: 10.1016/S0895-4356(01)00362-6. [DOI] [PubMed] [Google Scholar]
- Nischan P, Ebeling K, Thomas DB, Hirsch U. Comparison of recalled and validated oral contraceptive histories. Am J Epidemiol. 1993;138(9):697–703. doi: 10.1093/oxfordjournals.aje.a116907. [DOI] [PubMed] [Google Scholar]
- Shields M, Gorber SC, Tremblay MS. Estimates of obesity based on self-report versus direct measures. Health Rep. 2008;19(2):61–76. [PubMed] [Google Scholar]