

Abstract
The environmental bacterium Listeria monocytogenes survives and replicates in a variety of diverse ecological niches that range from the soil to the cytosol of infected mammalian cells. The ability of L. monocytogenes to replicate within an infected host requires the expression of a number of secreted bacterial gene products whose expression is regulated by the transcriptional activator PrfA. PrfA becomes activated following bacterial entry into host cells; however, the mechanism by which this activation occurs remains unknown. Here we describe a novel C-terminal mutation that results in the high-level constitutive activation of PrfA and yet, in contrast with other described prfA* activation mutations, only modestly increases PrfA DNA binding affinity. L. monocytogenes strains containing the prfA P219S mutation exhibited high levels of PrfA-dependent virulence gene expression, were hyperinvasive in tissue culture models of infection, were fully motile and were hypervirulent in mice. In contrast with PrfA G145S and other mutationally activated PrfA proteins, the PrfA P219S protein readily formed homodimers and did not exhibit a dramatic increase in its DNA-binding affinity for target promoters. Interestingly, the prfA P219S mutation is located adjacent to the prfA K220 residue that has been previously reported to contribute to PrfA DNA binding activity. prfA P219S therefore appears to constitutively activate PrfA via a novel mechanism which minimally affects PrfA DNA binding in vitro.
Introduction
Listeria monocytogenes is a Gram-positive bacterial pathogen that survives in a myriad of diverse environmental conditions that range from the soil and decaying plant matter to the cytosol of mammalian host cells (Czuprynski, 2005; Freitag et al., 2009; Gandhi & Chikindas, 2007; Gray et al., 2006; Lecuit, 2007; Toledo-Arana et al., 2009). This resilient and highly adaptable bacterium can contaminate food sources and as a result has led to thousands of cases of food-borne illness as well as some of the most expensive food recalls in the USA (Centers for Disease Control and Prevention, 2004; Cossart, 2007; Gandhi & Chikindas, 2007; Lynch et al., 2006; Mead et al., 2006; Swaminathan et al., 2006; Swaminathan & Gerner-Smidt, 2007). Once ingested by a susceptible individual, L. monocytogenes translocates across the gastrointestinal barrier and disseminates, resulting in severe infections that include meningitis and bacteraemia (Cossart & Toledo-Arana, 2008; Drevets & Bronze, 2008; Ramaswamy et al., 2007; Swaminathan & Gerner-Smidt, 2007). In pregnant women, L. monocytogenes can cross the fetoplacental barrier and result in abortion, stillbirth or the premature birth of a severely infected infant (Delgado, 2008; Smith et al., 2009; Smith et al., 2008; Swaminathan & Gerner-Smidt, 2007).
The L. monocytogenes transition from life as a saprophyte to life as an intracellular pathogen requires bacterial recognition of its host environment and an associated increase in the expression of a number of genes whose products are required for cell invasion, phagosomal membrane disruption, cytosolic replication and bacterial spread to neighbouring host cells (Cossart & Toledo-Arana, 2008; Dussurget, 2008; Freitag et al., 2009; Gray et al., 2006; Kreft et al., 2002; Scortti et al., 2007; Toledo-Arana et al., 2009; Vázquez-Boland et al., 2001). The majority of L. monocytogenes gene products that are known to be required for host infection are regulated by a central virulence regulatory protein known as PrfA (Freitag, 2006; Scortti et al., 2007). PrfA exists in a low-activity form in bacteria growing outside of host cells, but becomes highly activated via an unknown mechanism following host cell entry (Freitag et al., 2009; Shetron-Rama et al., 2002, 2003). Activation of PrfA is required for L. monocytogenes pathogenesis, and the transition of PrfA from a low-activity to a high-activity form is thought to serve as a critical switch enabling L. monocytogenes to exploit its intracellular replication niche.
PrfA is a 27 kDa protein which belongs to the cAMP receptor protein (Crp)–Fnr family of transcriptional regulators (Eiting et al., 2005; Lampidis et al., 1994; Vega et al., 1998). Members of this family appear to require the binding of a small molecule cofactor in order to induce an allosteric change that leads to protein activation (Harman, 2001). While the identity of the putative PrfA cofactor remains unknown, a number of mutations have been identified within PrfA that appear to result in protein activation in the absence of cofactor binding (prfA* mutations) (Miner et al., 2008a, b; Ripio et al., 1997; Shetron-Rama et al., 2003; Vega et al., 2004; Wong & Freitag, 2004). These mutations map to different regions of prfA, but have generally clustered either near the putative PrfA cofactor binding site or near residues spanning amino acids 140–155 in the PrfA coding sequence. The most well characterized prfA* mutation, prfA G145S, induces the repositioning of the PrfA helix–turn–helix DNA-binding domain and results in a substantial increase in PrfA DNA-binding affinity (Eiting et al., 2005; Ripio et al., 1997; Vega et al., 1998). Substitutions at L140 also dramatically increase PrfA DNA binding (Miner et al., 2008b; Wong & Freitag, 2004); however, the mechanisms leading to PrfA activation for other mutations (Y63C, E77K and G155S) are less clear (Miner et al., 2008b; Mueller & Freitag, 2005). What has become apparent is that PrfA activation results in a variety of L. monocytogenes physiological changes that increase bacterial fitness within an infected host while decreasing fitness in broth culture (Bruno & Freitag, 2010; Wong & Freitag, 2004). Strains containing prfA* mutations are hyperinvasive for tissue culture cells and hypervirulent in murine models of infection (Bruno & Freitag, 2010; Freitag & Portnoy, 1994; Mueller & Freitag, 2005); however, the mutants are defective in flagella-mediated swimming motility and are more susceptible to both acid and salt stress (Bruno & Freitag, 2010; Port & Freitag, 2007; Shetron-Rama et al., 2003; Wong & Freitag, 2004). Environment-responsive regulation of PrfA activation thus appears essential for maximizing L. monocytogenes fitness both inside and outside of host cells.
While PrfA shares extensive structural homology with its well-studied family member Escherichia coli Crp, it differs from Crp in having an extended C-terminal domain consisting of three alpha helices (Eiting et al., 2005; Lampidis et al., 1994). This extended C-terminal domain has been postulated to stabilize PrfA DNA binding, and purified PrfA protein does indeed bind DNA with higher affinity than purified Crp lacking its cAMP cofactor (Eiting et al., 2005; Herler et al., 2001; Lampidis et al., 1994; Scortti et al., 2007; Vega et al., 1998). A mutation within the C terminus of PrfA (K220T) has been reported to inactivate PrfA (Roche et al., 2003; Velge et al., 2007), as have deletions at the C terminus (Herler et al., 2001). Here we present the characterization of a novel prfA P219S mutation within the extended C-terminal region of PrfA that serves to constitutively activate the protein, thus confirming a role for this region in the regulation of PrfA activity.
Methods
Bacterial strains, plasmids and growth conditions.
L. monocytogenes and E. coli strains used in this study are listed in Table 1. E. coli XL1-Blue (Agilent Technologies), One Shot TOP10 (Invitrogen), NEB 5αF′Iq (New England Biolabs) and SM10 were used as host strains for maintenance and propagation of recombinant plasmids. L. monocytogenes and E. coli strains were grown at 37 °C in brain heart infusion (BHI) media (Difco) and Luria broth (LB) (Invitrogen). Bacteria containing the L. monocytogenes integration plasmid pPL2 (Lauer et al., 2002) were maintained on media containing chloramphenicol (25 µg ml−1 for E. coli and 7.5 µg ml−1 for L. monocytogenes). Bacteria containing the 6× histidine-tagged vector pQE30 (Qiagen) were maintained in E. coli with 100 µg ampicillin ml−1. Streptomycin (200 µg ml−1) was used in selection of L. monocytogenes following bacterial conjugation and for isolation of bacteria from infected mice.
Table 1. Bacterial strains and plasmids used in this study.
| Strain or plasmid | Description | Source or reference |
| Strains | ||
| NF-L1124 | L. monocytogenes 10403S actA–gus–neo–plcB | Miner et al. (2008a) |
| NF-L1003 | L. monocytogenes ΔprfA in 10403S actA–gus–plcB | Wong & Freitag (2004) |
| NF-L1041 | L. monocytogenes NF-L1003 with pPL2–prfA | Wong & Freitag (2004) |
| NF-L1226 | L. monocytogenes NF-L1003 with pPL2–prfA G145S | Port & Freitag (2007) |
| NF-L1452 | L. monocytogenes NF-L1003 with pPL2–prfA P219S | This work |
| XL1-Blue | E. coli cloning strain | Agilent Technologies |
| NEB 5αF′Iq | E. coli protein expression strain | New England Biolabs |
| Plasmids | ||
| pQE30 | N-terminal His-tagged expression vector | Qiagen |
| pNF1019 | pPL2 site-specific integration vector with full-length prfA and all promoters | Wong & Freitag (2004) |
| pNF1223 | pNF-1019 prfA G145S | Port & Freitag (2007) |
| pNF1163 | pNF-1019 prfA P219S | This work |
| pNF1797 | pQE30 prfA P219S | This work |
| pNF2076 | pQE30 prfA wild-type | This work |
| pNF3028 | pQE30 prfA G145S | This work |
Ethylene methylsulphonate (EMS) mutagenesis and β-glucuronidase assays.
L. monocytogenes strain NF-L1124 containing an actA–gus–neo reporter fusion was chemically mutagenized using EMS (Sigma-Aldrich) as described previously (Shetron-Rama et al., 2003). Bacteria expressing high levels of actA were selected on indicator plates containing 50 µg 5-bromo-4-chloro-3-indolyl β-d-glucuronide (XG) ml−1 and 10 µg neomycin ml−1. Mutants that formed blue colonies on the XG indicator plates and were neomycin-resistant (in contrast with the wild-type which formed white colonies and were neomycin-sensitive) were selected for further analysis. Quantitative liquid culture enzymic assays for β-glucuronidase (GUS) activity were performed to confirm the increase in actA promoter expression levels. For GUS assays, overnight cultures of L. monocytogenes were diluted 1 : 50 in fresh BHI and grown with shaking at 37 °C. At various time points, the OD600 was determined for each culture and 1 ml of each sample was centrifuged to recover bacteria. Bacterial pellets were resuspended in 1 ml ABT buffer [1 M potassium phosphate (pH 7.0), 0.1 M NaCl, 1 % Triton] and GUS activity was measured as described by Youngman (1987) with the substitution of 4-methylumbelliferyl β-d-glucuronide (Sigma-Aldrich) in place of 4-methylumbelliferyl β-d-galactoside. The prfA promoter and coding regions of the strains that showed an increase in actA–gus–neo activity over wild-type levels were amplified by PCR and their DNA sequences were obtained.
Plasmid and bacterial mutant construction.
The prfA P219S mutation was introduced into plasmid pNF1019, a pPL2 site-specific phage integration plasmid containing a wild-type copy of prfA with all promoters required for expression (Wong & Freitag, 2004), using the Quick Change site-directed mutagenesis kit (Stratagene) with primer pairs 5′-CTCAAAAGATATGCCTCTAAATTAGATGAATGGTTTTATTTAGCATGTCC-3′ and 5′-GGACATGCTAAATAAAACCATTCATCTAATTTAGAGGCATATCTTTTGAG-3′. Letters in bold type indicate the mutagenesis of proline (CCT) to serine (TCT) on both forward and reverse strands. The resulting plasmid, pNF1163, was conjugated into strain NF-L1003, which contains an in-frame deletion within prfA as well as the actA–gus reporter gene fusion, resulting in strain NF-L1452. For generation of purified PrfA proteins, the coding sequence of prfA was amplified by PCR from L. monocytogenes strains 10403S (wild-type), prfA P219S (NF-L1452) and prfA G145S (NF-L1226) using primer pairs 5′-AAAGGTACCAACGCTCAAGCAGAAG-3′ and 5′-GGCTGCAGTTTAATTTAATTTTCCCCAAG-3′ and cloned into a pQE30 Expression vector (Qiagen), which contains an N-terminal 6×histidine tag and an IPTG-inducible promoter. Letters in bold type indicate the second codon and stop codon of the prfA coding sequence and italicized letters indicate the KpnI and PstI restriction endonuclease sites used for cloning the PCR fragment into pQE30. The resulting construct was initially propagated in E. coli TOP10 cells, isolated and then transformed into NEB 5αF′Iq. An overnight culture containing the expression construct was diluted 1 : 50 in fresh LB broth and the culture was incubated at 37 °C (with shaking) until an OD600 of 0.5 was reached. To induce expression of the PrfA protein, 1 mM IPTG (Inalco) was added to the culture and induction was allowed to proceed for 3–4 h. The bacterial cells were recovered by centrifugation followed by sonication with four repeated 10 s bursts and 1 min cooling on ice. The soluble fraction containing the N-His-PrfA protein was collected and purified using the His-Pur Purification kit (Thermo Scientific). Protein concentration was determined using a BCA Protein Assay kit (Thermo Scientific).
Assessment of growth in broth culture.
Overnight cultures of strains grown in BHI were resuspended in fresh BHI media at a dilution of 1 : 20 and growth at 37 °C with shaking was assessed by measuring the OD600 at the indicated time points.
Measurement of haemolytic activity.
Stationary-phase bacterial cultures were diluted 1 : 10 into LB medium and grown at 37 °C for 5 h with shaking. OD600 was determined, cultures were normalized to OD600 0.5 and 1 ml of each culture was centrifuged at 13 000 g for 5 min. The supernatant was collected and was assayed for haemolytic activity with sheep erythrocytes washed with PBS (Gibco), as described previously (Camilli et al., 1989). Haemolytic activity was determined as the reciprocal of the supernatant dilution at which 50 % lysis of erythrocytes was observed.
Assessment of phospholipase activity.
plcB-dependent phospholipase production was assayed on egg yolk agar plates (Alonzo et al., 2009; Mueller & Freitag, 2005). Antibiotic-free chicken egg yolk was added in a 1 : 1 (v/v) ratio to PBS and vortexed to form a suspension. A 5 ml aliquot of egg yolk suspension was added to 100 ml molten LB medium plus 0.2 % activated charcoal (Sigma-Aldrich) and 25 mM glucose-6-phosphate (Sigma-Aldrich) (Bitar et al., 2008); 10 ml of this mixture was poured into Petri dishes. Bacterial strains were gently streaked onto the surface of the plate and incubated at 37 °C for 48 h. Phospholipase activity was visualized as a zone of opacity surrounding bacterial streaks.
Western blot analysis of PrfA protein.
PrfA was detected within cytoplasmic fractions isolated from bacterial whole-cell extracts. A 25 ml culture of each L. monocytogenes strain was grown to mid-exponential phase in BHI at 37 °C with shaking. Cells were normalized to OD600 0.8 and centrifuged and the bacterial pellets were resuspended in 1 ml PBS. Mutanolysin (100 U; Sigma-Aldrich) was added and the suspension was incubated at 37 °C for 2 h. A 10 µl volume of 100× Protease Inhibitor Cocktail (Calbiochem) was added to the mutanolysin-treated cells, followed by sonication with three 40 s pulses with 2 min cooling on ice in between each pulse. Cellular debris was centrifuged at 24 000 g for 20 min at 4 °C and the supernatant containing the cytoplasmic components was collected and stored at −20 °C until further use. For detection of PrfA, 10 µl of the isolated cytoplasmic fraction was mixed with 10 µl 2× Laemmli sample buffer (Bio-Rad), boiled for 5 min then separated by SDS-PAGE. Protein samples were transferred onto PVDF membranes. PrfA was detected using a 1 : 500 dilution of a monoclonal antibody directed against PrfA in 1× PBST (PBS plus 0.05 % Tween-20) followed by incubation with a 1 : 2500 dilution of a polyclonal goat–anti-mouse secondary antibody conjugated to alkaline phosphatase (SouthernBiotech). Bands were visualized colorimetrically with the addition of 10 ml BCIP/NBT Plus solution (SouthernBiotech). Densitometry was determined using ImageJ software (http://rsbweb.nih.gov/ij/download.html).
Limited proteolysis.
Limited proteolytic digestion of His-purified proteins with subtilisin (Sigma-Aldrich) was done as previously described by Miner et al. (2008b) with slight modifications. In brief, 2 µg purified protein was incubated with 500 ng subtilisin for 10 and 30 min at room temperature followed by the addition of 1 mM PMSF to terminate the reaction. Samples were boiled for 5 min in SDS-loading buffer and fragments were separated by SDS electrophoresis on a NuPAGE 4–12 % Bistris gel (Invitrogen). Bands were visualized by staining with Bio-Safe Coomassie G-250 (Bio-Rad).
Circular dichroism (CD).
His-purified protein was purified and isolated as described above (Plasmid and bacterial mutant construction). Following purification, eluted protein samples were dialysed in 10 mM NaH2PO4 (pH 8) overnight at 4 °C and the concentration of the dialysed protein that remained in solution was determined using a BCA protein assay kit (Thermo Scientific). Samples (400 µl) containing 250 µg purified protein ml−1 were then analysed at room temperature on a Jasco J-710 CD spectrometer (UIC Center for Structural Biology, Chicago, Illinois) in a 2 mm rectangular quartz cuvette. CD spectra parameters were set to read at wavelengths between 180 and 310 nm, continuous scanning mode, 2 nm data pitch, a scanning speed of 100 nm s−1, response of 1 s, band width of 1.0 nm and an accumulation of 3. Data analysis was done using the SpectraManager software program.
Protein chemical cross-linking.
Chemical cross-linking was done as previously described by Miner et al. (2008b) with minor modifications. In brief, after separation of samples by SDS-PAGE, proteins were transferred to PVDF membranes. Proteins were detected using a 1 : 500 dilution of a monoclonal antibody directed against PrfA in 1× PBST followed by incubation with a 1 : 2500 dilution of a polyclonal goat–anti-mouse secondary antibody conjugated to alkaline phosphatase (SouthernBiotech). Bands were visualized colorimetrically with the addition of a BCIP/NBT Plus solution (Southern Biotech).
Electrophorectic mobility shift assays (EMSAs).
Primer pairs used to amplify the hly promoter DNA fragment (~100 bp) were 5′-TCCTATCTTAAAGTGACTTTATGTT-3′ and 5′-GCTTCTAAAGATGAAACGCAATATTA-3′. The 3′ end primer was purchased with a biotin label (Sigma-Aldrich). EMSAs were done as described previously (Miner et al., 2008b) with slight modifications. In brief, after DNA-binding reactions and electrophoresis, protein/DNA samples were transferred onto nylon membranes followed by detection using the Pierce chemiluminescent nucleic acid detection module (Thermo Scientific).
Intracellular growth.
Bacterial intracellular growth assays in Potorous tridactylis kidney epithelial cells (PtK2) were performed as described previously (Marquis et al., 1995; Mueller & Freitag, 2005; Wong & Freitag, 2004; Xayarath et al., 2009). In brief, monolayers of cells were grown on glass coverslips to confluence and infected with bacterial strains with an m.o.i. of 100 : 1. One hour after infection of PtK2 cells, monolayers were washed three times in PBS and 5 µg gentamicin ml−1 was added to kill extracellular bacteria. At the indicated time points, coverslips were removed and tissue culture cells were lysed in 5 ml sterile H2O to release intracellular bacteria for enumeration of intracellular growth, or the coverslips were processed for microscopy.
Plaque assays.
Plaque assays were conducted as described previously (Sun et al., 1990). Briefly, murine L2 fibroblasts were grown to confluence in six-well microtitre plates and infected with 20 µl of a normalized 1 : 20 dilution of overnight culture grown at 37 °C in BHI with shaking (m.o.i. 10 : 1). One hour post-infection, L2-infected monolayers were washed and 5 µg gentamicin ml−1 was added to kill extracellular bacteria. Three days post-infection, Neutral Red (Sigma-Aldrich) was added and plaques were visualized and measured using a micrometer (Finescale).
Mouse infections.
All animal procedures were approved by the Institutional Animal Care and Use Committees (IACUC) and performed in the Biological Resources Laboratory at the University of Illinois at Chicago. Overnight bacterial cultures were diluted 1 : 20 into fresh media and grown to OD600 ~0.6. A 1 ml volume of culture (corresponding to 6×108 c.f.u. ml−1) was washed, diluted and resuspended in PBS to a final concentration of 1×105 c.f.u. ml−1. Female 8–10-week-old ND4 Swiss Webster mice (Harlan Laboratories) received injections of 200 µl PBS containing 2×104 c.f.u. L. monocytogenes via the tail vein. Mice were killed and livers and spleens were harvested 72 h post-infection. Organs were homogenized with a Tissue Master 125 homogenizer (Omni International) and dilutions were plated onto BHI streptomycin (200 µg ml−1) plates.
Swimming motility assays.
Swimming motility was evaluated on semisolid [0.3 % (w/v) agar] BHI medium. The plates were inoculated with 5 µl mid-exponential phase (OD600 ~0.6) bacterial cultures grown in BHI at 37 °C. Inoculated plates were then incubated at 37 °C for 48 h. The diameter of the swimming colony in millimetres was measured at various time points, and swimming motility was expressed in millimetres travelled from the edge of the original drop diameter of inoculation. Each strain was assayed in triplicate.
Results
Identification of a novel prfA* mutation within the extended C-terminal region of PrfA
To identify unique and previously uncharacterized mutations resulting in the constitutive activation of PrfA, a L. monocytogenes strain containing an actA–gus–neo–plcB transcriptional reporter gene fusion was mutagenized with EMS, a chemical mutagen that most commonly induces G/C to A/T transitions. L. monocytogenes mutants exhibiting activation of PrfA activity were selected based on high-level actA promoter expression as indicated by increased levels of neomycin resistance and by enhanced blue colony colour on indicator plates containing the GUS substrate XG. Out of approximately 1×1010 total bacteria present in six independently EMS-treated pools, 282 mutants were isolated as dark blue neomycin-resistant colonies. The prfA promoter and coding regions of 60 of these isolates were sequenced. A total of 42 mutants contained mutations within prfA, with the largest group (46 %) containing the previously described prfA* G145S mutation. Other previously described prfA* mutations were also identified (prfA L140F, G155S and E77K). Only one of the 42 sequenced mutants possessed a novel prfA mutation not previously associated with PrfA* activity; this mutant contained a C to T transition resulting in a serine substitution for a proline at position 219 (P219S). This mutant was selected for further analysis.
Based on the PrfA crystal structure (Eiting et al., 2005), the prfA P219S mutation is located in αH, one of the three C-terminal alpha helices present in PrfA but absent from Crp (Fig. 1a). These three alpha helices have been proposed to stabilize the C-terminal PrfA DNA-binding domain and to participate in homodimer interactions, such that residues within the three helices of one monomer form hydrogen bonds to β loops of the second monomer (Eiting et al., 2005; Herler et al., 2001). The proline residue at position 219 is the first amino acid of the αH helix, contributing to the transition from αG to αH (Fig. 1a).
Fig. 1.

Location of the PrfA P219S mutation. (a) Ribbon modelling of the PrfA dimer using DeepView-Swiss Pdb viewer v4.0 (http://spdbv.vital-it.ch/). The monomers that make up the dimer are coloured either light or dark grey. The DNA-binding helix–turn–helix helices are shown in blue and the three helices unique to PrfA that are located at the distal end of the C terminus are coloured pink (αG and αI) and magenta (αH). The prfA P219S mutation is located in αH and the proline residue at this specific location is shown in orange. (b) Ribbon modelling as done in (a) highlighting the tunnel suggested by Eiting et al. (2005) to be the putative ligand-binding pocket as indicated by the grey arrow. Colours for domains and secondary structures are the same as in (a), with the addition of αD shown in light blue. The specific residue Y63 (in β5) is shown in green and G145 (in αD) is in red. The G145 side chain is buried within helix D and is difficult to visualize but has been further highlighted by the black arrow pointing to its location.
The prfA P219S mutation results in constitutive activation of PrfA
The prfA P219S mutation was introduced into a L. monocytogenes ΔprfA actA–gus–plcB strain as a single copy on plasmid pPL2 to confirm that the mutation was sufficient to confer PrfA activation. In addition to prfA P219S, wild-type prfA and prfA G145S were also introduced into ΔprfA actA–gus–plcB strains as single copies using the plasmid integration vector pPL2 which contains all three promoters required for full prfA expression and complementation (Wong & Freitag, 2004). All comparisons of PrfA activity were thus carried out in isogenic ΔprfA actA–gus–plcB strains containing either wild-type or mutant alleles of prfA. The introduction of pPL2–prfA P219S resulted in a blue colony colour on plates containing the XG indicator for GUS activity, similar to that observed for strains containing pPL2-prfA G145S (data not shown). Strains containing the prfA P219S allele exhibited no obvious growth defect when grown in broth culture (Fig. 2a), but did display high levels of GUS activity in broth culture, similar to the levels observed for the highly activated prfA* G145S mutant (Fig. 2b). In addition, overnight cultures of the prfA P219S strain grown in BHI at 37 °C without shaking settled to the bottom of culture tubes, exhibiting a phenotype similar to that reported for other high-activity prfA* mutant cultures (Wong & Freitag, 2004) (data not shown).
Fig. 2.

The prfA P219S mutation dramatically increases PrfA-dependent virulence gene expression in broth culture. (a) Growth curve of the prfA P219S mutant (▵) compared with wild-type (•), prfA G145S (▪) and a ΔprfA strain (▪). Overnight cultures of strains grown in BHI were diluted 1 : 20 in fresh BHI and grown at 37 °C with shaking and the OD600 was measured at the indicated time points. The prfA P219S mutant grew similarly to all strains grown in rich media. (b) PrfA-dependent activation of actA expression as indicated by GUS activity. L. monocytogenes strains containing actA–gus transcriptional reporter fusions in the presence of prfA G145S (▪), prfA P219S (▵) or wild-type prfA (•) were grown in BHI at 37 °C with shaking. GUS activity indicative of actA expression was assessed at the indicated time points. Each data point is the mean±95 % confidence interval GUS activity measured in duplicate and the data shown are representative of at least two independent experiments. (c) Detection of PrfA-dependent LLO-associated haemolytic activity. Secreted LLO activity was assessed by measuring lysis of sheep erythrocytes from serial dilutions of bacterial culture supernatants. Haemolytic activity is represented as the reciprocal of the supernatant dilution at which 50 % lysis of erythrocytes was observed. Data shown represent the mean±sem activity measured in triplicate for two independent experiments. (d) Detection of PrfA-dependent PC-PLC activity. PC-PLC-associated phospholipase activity was assessed on egg yolk agar plates containing 0.2 % activated charcoal and 25 mM glucose 6-phosphate following incubation at 37 °C for 24 h. The white precipitate surrounding the bacterial streak is indicative of PC-PLC activity. Data are representative of at least three independent experiments.
The expression of listeriolysin O (LLO) and the broad-range phospholipase PC-PLC is increased following PrfA activation (Alonzo et al., 2009; Portnoy et al., 1988; Shetron-Rama et al., 2003; Wong & Freitag, 2004). Consistent with the increase observed for actA promoter activity, the prfA P219S mutant exhibited increased LLO and PC-PLC activity with levels that were similar to those observed for the prfA* G145S mutant (Fig. 2c, d). In addition, Western blot analysis using antibody directed against PrfA indicated that PrfA protein levels were approximately twofold higher in strains containing the prfA P219S and prfA* G145S mutations in comparison with strains containing wild-type prfA (Fig. 3a), a result consistent with increased prfA expression resulting from PrfA-dependent activation of the upstream plcA–prfA promoter (Camilli et al., 1993; Freitag & Portnoy, 1994). These data indicate that the prfA P219S mutation results in the constitutive activation of PrfA to a level that resembles that of the highly activated prfA* G145S mutant strains.
Fig. 3.

The prfA P219S mutation influences PrfA protein conformation and dimer formation. (a) Western blot analysis of PrfA protein. Top panel: 20 µl isolated bacterial cytoplasmic fractions, normalized based on cell density, were loaded onto a 12 % Bis–Tris polyacrylamide gel and proteins were separated by gel electrophoresis. Polypeptides were visualized by staining with Coomassie blue. Bottom panel: PrfA protein was detected in the cytoplasmic fractions of the various L. monocytogenes strains using a monoclonal antibody directed against PrfA. Approximately twofold more PrfA protein was detected in both the prfA P219S and G145S mutant strains compared with the wild-type strain. Numbers below the Western panel indicate relative amounts of protein as determined by densitometry using ImageJ software (http://rsbweb.nih.gov/ij/download.html) in comparison to the levels of wild-type PrfA detected. (b) Comparison of PrfA protein conformation by limited proteolytic digestion. Samples (2 µg) of purified PrfA G145S, PrfA P219S or of the wild-type protein were digested with 500 ng subtilisin for 10 and 30 min at room temperature. Protein fragments were separated by SDS-PAGE and visualized by Coomassie staining. Untreated protein samples (uncut) and heat-denatured samples treated with subtilisin (Sub cont) were included as controls to demonstrate that denatured samples were equally susceptible to enzymic digestion. Arrows indicate the position of subtilisin and asterisks denote fragments that were not observed for the wild-type protein. PrfA P219S was more susceptible to proteolytic digestion than the wild-type protein but was also distinguishable from PrfA G145S. Gels are representative of three independent experiments. (c) CD analysis of the purified PrfA proteins. CD far-UV absorption signals between wavelengths of 180 and 310 nm were measured for 250 µg purified protein ml−1 in 400 µl 10 mM NaH2PO4 at room temperature. The graph is representative of two independent experiments run in triplicate. (d) PrfA homodimer formation as assessed by chemical cross-linking of purified PrfA. Samples (500 ng) of purified PrfA proteins were chemically cross-linked with 10 µM sulpho-ethylene glycol bis[succinimidylsuccinate] (S-EGS) for 1 h at room temperature followed by SDS-PAGE and Western blotting for detection of PrfA dimers. The PrfA P219S mutant formed homodimers with an efficiency that was less than that observed for wild-type PrfA but greater than that observed for the PrfA* G145S mutant. The ratio of PrfA dimer to monomer formation was assessed by densitometry using ImageJ software (http://rsb.info.nih.gov/ij/download). Data are representative of at least three independent experiments.
The prfA P219S mutation alters PrfA conformation
Previously described prfA* mutations have been reported to alter PrfA conformation in ways that can be detected by limited proteolytic digestion (Miner et al., 2008b). Limited proteolysis enables a rapid assessment of protein conformational changes that can distinguish between active and inactive forms of Crp (Crp* mutants or Crp with or without cAMP) as well as PrfA and PrfA* mutants (Harman et al., 1986; Miner et al., 2008b; Tan et al., 1991). Polypeptide fragments resulting from limited protease digestion were separated by SDS-PAGE and visualized by Coomassie staining (Fig. 3b). Heat-denatured PrfA samples were also subjected to subtilisin digestion to confirm that all protein samples were equally susceptible to proteolytic cleavage when denatured (Fig. 3b, position of subtilisin indicated by arrow). Limited proteolytic digestion of wild-type PrfA, PrfA P219S and PrfA* G145S proteins indicated that both PrfA G145S and PrfA P219S were more susceptible to proteolytic digestion in comparison to the wild-type protein (Fig. 3b). The PrfA P219S protein was also distinguishable from PrfA G145S, indicating that the P219S mutation resulted in PrfA conformational changes that are distinct from those imposed by G145S.
To further assess conformational changes conferred by the P219S mutation, CD was used to compare protein secondary structure. Interestingly, both the PrfA P219S and PrfA G145S proteins exhibited similar CD profiles that were distinct from the spectrum observed for wild-type PrfA (Fig. 3c). PrfA P219S and PrfA G145S exhibited a more negative signal in the 208–222 nm range when compared with wild-type PrfA, suggestive of a higher alpha helical content. Thus, while PrfA P219S can be structurally distinguished from both wild-type PrfA and PrfA G145S via limited protease digestion, CD analysis suggests that overall PrfA P219S may adopt a confirmation more closely related to that of the constitutively activated PrfA G145S mutant than that of wild-type PrfA.
The prfA P219S mutation modestly reduces PrfA dimer formation
PrfA crystal structure analysis indicates that PrfA forms homodimers, and homodimers have also been observed via chemical cross-linking (Eiting et al., 2005; Velge et al., 2007). Mutationally activated forms of PrfA form homodimers; however, this homodimer formation is reduced in comparison with the wild-type protein (Miner et al., 2008b). To assess the ability of purified PrfA P219S to form homodimers in vitro, purified protein was incubated with two distinct chemical cross-linking agents [sulpho-ethylene glycol bis[succinimidylsuccinate] and Bis suberate (BS3)] and analysed by SDS-PAGE gel electrophoresis. Wild-type PrfA protein was observed to readily form dimers under these conditions, whereas the PrfA* G145S mutant exhibited reduced dimer formation as expected (Fig. 3d, the similar BS3 data are not shown). Interestingly, despite its resemblance thus far in other assays to highly activated forms of PrfA, the PrfA P219S mutant formed dimers with an efficiency that was somewhat less than that of wild-type PrfA but greater than that of PrfA G145S.
PrfA P219S modestly enhances PrfA DNA-binding affinity
Highly activated PrfA G145S and PrfA L140F mutant proteins bind target DNA with an affinity that is significantly greater than that exhibited by the wild-type protein (Eiting et al., 2005; Miner et al., 2008b; Vega et al., 1998). Given that the extended C-terminal region of PrfA has been proposed to enhance the DNA-binding stability of PrfA, we sought to compare DNA binding between the PrfA P219S mutant with wild-type and highly activated PrfA G145S. EMSAs confirmed that purified PrfA G145S bound a DNA fragment containing the hly promoter with significantly higher affinity than wild-type protein (Fig. 4). Unexpectedly, although the PrfA P219S mutant stimulated high levels of PrfA-dependent hly-encoded LLO activity, the mutant protein displayed only a modest enhancement in DNA-binding activity in comparison with wild-type PrfA (Fig. 4). The PrfA P219S mutation therefore affects PrfA function in a way that is distinct from that of the PrfA G145S mutation.
Fig. 4.
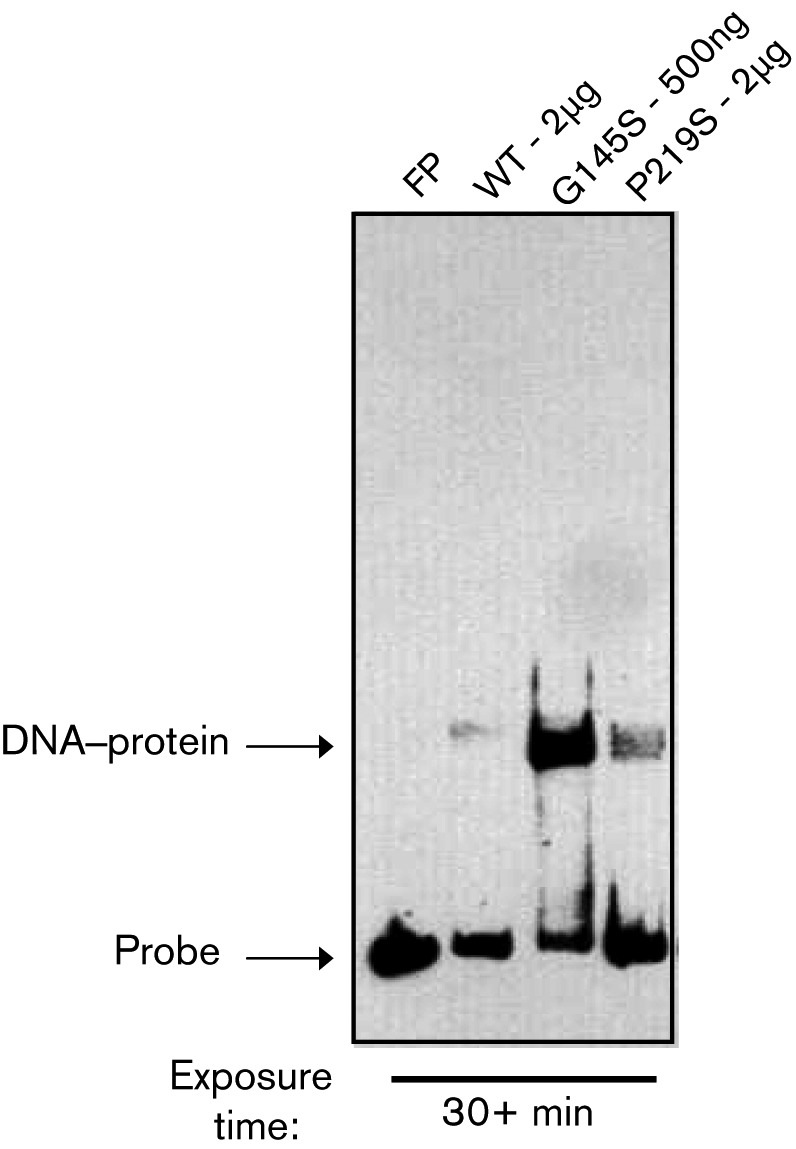
PrfA P219S does not exhibit high-affinity in vitro DNA binding. The binding of purified PrfA wild-type and mutant protein to a biotin-labelled hly promoter DNA fragment was assessed by EMSA. FP, Free probe, no protein added. Data are representative of at least three independent experiments.
L. monocytogenes prfA P219S strains are hyperinvasive for tissue culture cells
PrfA activation leads to the increased expression of a number of gene products that contribute to bacterial invasion, including InlA, InlB, ActA and LLO (Freitag et al., 2009; Ireton & Cossart, 1997; Ramaswamy et al., 2007; Scortti et al., 2007; Vázquez-Boland et al., 2001). Consistent with the ability of the prfA P219S mutation to induce PrfA activation, L. monocytogenes prfA P219S mutants were hyperinvasive for both epithelial and fibroblast cell lines (Fig. 5). Cells infected with the prfA P219S mutants had significantly increased numbers of intracellular bacteria (up to 13-fold higher) at multiple time points post-infection of PtK2 epithelial cells, while the rate of intracellular bacterial replication was similar to that observed for wild-type bacteria (approximately 57 min doubling time for prfA P219S versus 61 min doubling time for wild-type) (Fig. 5a). Given the similar rate of intracellular bacterial replication, the increased numbers of prfA P219S bacteria at 3 h after infection (2 h after gentamicin treatment) are consistent with enhanced bacterial invasion of PtK2 epithelial cells. The prfA P219S mutant also appeared hyperinvasive for L2 fibroblast cells as indicated by the increased number of plaques formed by the mutant following the infection of monolayers in comparison with those infected with wild-type L. monocytogenes (Fig. 5b). Approximately 10-fold greater numbers of plaques were formed by the prfA P219S strain versus wild-type bacteria at a similar m.o.i. (Fig. 5b). The prfA P219S mutation therefore serves to enhance bacterial invasion of host cells in tissue culture.
Fig. 5.

Strains containing prfA P219S are hyperinvasive for tissue culture cells. (a) Bacterial invasion and intracellular growth in monolayers of PtK2 epithelial cells. PtK2 epithelial cells grown on glass coverslips were infected with the indicated strains at an m.o.i. of 100 : 1. Monolayers were washed and gentamicin was added 1 h post-infection (p.i.); coverslips were removed at the indicated time points for lysis of host cells and enumeration of intracellular bacteria. The number of bacteria recovered from the prfA P219S mutant was significantly different from the number recovered from the wild-type strain at 3, 5 and 7 h p.i., while the differences between the prfA G145S mutant and wild-type were significant at 3 and 7 h p.i. **P≤0.005, ***P≤0.0005 using an unpaired two-tailed Student’s t-test (GraphPad Prism v.5.0A). In addition, the P219S mutant was significantly different from the G145S mutant at 3 and 5 h p.i. (**P≤0.005). Data shown represent the mean±sem of three independent experiments done in triplicate. (b) Bacterial infection of L2 fibroblast cells. The ability of the prfA P219S mutant and wild-type L. monocytogenes to invade, multiply and spread cell-to-cell within fibroblast tissue culture cell monolayers was determined by assessing plaque formation following infection with an m.o.i. of 10 : 1 or 1 : 1. At 1 h p.i. the cells were washed and gentamicin was added. Plaques were visualized 3 days p.i. Data shown are representative of three independent experiments done in duplicate.
The prfA P219S mutant strain is hypervirulent in mice
Highly activated prfA* mutants have been shown to be hypervirulent in murine models of infection (Bruno & Freitag, 2010; Wong et al., 2004). When mice were infected with 1×104 c.f.u. of either wild-type or the prfA P219S mutant via tail vein injection, approximately 5- and 10-fold more bacteria were recovered from the livers and spleens, respectively, of mice infected with the mutant strain (Fig. 6). Taken together, these results clearly indicate that the prfA P219S mutation results in high-level activation of PrfA as well as increased bacterial virulence.
Fig. 6.

The L. monocytogenes prfA P219S mutant is hypervirulent in vivo. Swiss Webster mice were intravenously infected with 2×104 c.f.u. through the tail vein. The livers and spleens were harvested 48 h post-infection, homogenized and enumerated for bacterial burdens. Each data point represents one mouse and the solid lines denote the median for each data group. Data were obtained from two independent experiments. Asterisks indicate statistical significance; **P≤0.005 using an unpaired two-tailed Student’s t-test (GraphPad Prism v.5.0A).
Unlike L. monocytogenes prfA G145S mutants, prfA P219S mutants are not defective for swimming motility
Outside of host cells, it has been previously demonstrated that prfA* mutants exhibit defects in swimming motility that can be readily detected in soft agar media (Shetron-Rama et al., 2003; Wong & Freitag, 2004). Swimming motility defects would be anticipated to compromise bacterial fitness in environments outside of host cells, as well as potentially reducing bacterial invasion of the intestinal epithelium (O’Neil & Marquis, 2006). It has been reported that gene products that contribute to flagella biosynthesis and chemotaxis, such as FlaA and MotA, are downregulated following PrfA activation, suggesting an inverse relationship between motility and prfA activity (Port & Freitag, 2007; Toledo-Arana et al., 2009). To examine the motility of prfA P219S strains, L. monocytogenes swimming motility was assessed on semisolid BHI media at 37 °C over the course of 2 days. Defects in the initiation of bacterial swimming motility were readily detectable for prfA G145S but not for prfA P219S or wild-type strains during the first 24 h of growth (Fig. 7a). After approximately 24 h, prfA G145S strains exhibited rates of swimming motility that were similar to those observed for prfA P219S, wild-type and ΔprfA strains (Fig. 7b). These results indicate that PrfA activation does not prohibit the assembly of a functional flagellum nor interfere with bacterial chemotaxis. Interestingly, for strains containing some prfA* mutations (such as G145S), PrfA activation delays the initiation of swimming motility, whereas other prfA* strains (P219S) show no indication of motility defects.
Fig. 7.

prfA G145S but not P219S strains are delayed for initiation of swimming motility. Measurement of bacterial spread in soft agar. The diameter of bacterial spread (in mm) from the original inoculation site was measured at various time points up to 48 h for plates incubated at 37 °C. A ΔflaA mutant, which is defective for motility, was used as a negative control. The prfA G145S mutant strain is slow to initiate swimming motility, but then moves at a rate that is similar to that of wild-type, prfA P219S and prfA deletion strains. •, wild-type; ▴, P219S; ◊, ΔprfA; ▪, G145S; ▽, ΔflaA.
Discussion
PrfA activation appears to represent a critical switch that enables L. monocytogenes to transition from an environmental bacterium to a pathogen that can access and exploit the mammalian cytosol as a bacterial replication niche (Freitag et al., 2009; Toledo-Arana et al., 2009). While the signal that triggers PrfA activation remains unknown, the isolation of prfA* mutant strains has provided insight into the effects of PrfA activation on bacterial fitness and physiology (Bruno & Freitag, 2010; Mueller & Freitag, 2005; Shetron-Rama et al., 2003; Wong & Freitag, 2004). Phenotypic characterization of prfA* mutants has established the critical importance of environmental regulation of PrfA to optimize bacterial survival in distinct habitats both outside and inside mammalian cells. Here we describe the isolation of a novel prfA* mutation located within the distal C-terminal region of PrfA. This region has not been previously associated with PrfA activation and it is absent from the structurally related family member Crp (Eiting et al., 2005; Lampidis et al., 1994). While loss-of-function mutations have previously been associated with the PrfA C terminus (Herler et al., 2001; Roche et al., 2003; Velge et al., 2007), our results are the first, to our knowledge, to show that mutations within this region are capable of conferring high-level constitutive activation to this key virulence regulator.
The prfA P219S mutation is located within the αH helix at the C-terminal end of PrfA. Based on the PrfA crystal structure, the three C-terminal alpha helices (αG, αH and αI) form an extended region that has been postulated to contribute to stabilization of PrfA DNA binding through both intra- and intermolecular homodimer interactions (Eiting et al., 2005; Herler et al., 2001). Within an individual PrfA monomer, hydrogen bonds are formed between helices αH and αI with helices αC and αD; PrfA mutants lacking the last 17 C-terminal residues (αH and αI) lose all ability to bind DNA (Eiting et al., 2005; Herler et al., 2001). In addition, the intermolecular contacts made between the helices of αG, αH and αI of one PrfA monomer and β6 and β7 in the N terminus of the second monomer reportedly stabilize the monomer–monomer interface (Eiting et al., 2005). Residues participating in these contacts include K220 and Q223 in αH and T82 in β6–β7. The K220 residue has previously been implicated as playing an important role in PrfA activity in that a K220T mutant exhibited reduced PrfA DNA binding without affecting dimer formation (Roche et al., 2003; Velge et al., 2007). It was thus somewhat surprising that mutants containing the adjacent P219S substitution exhibited only modest alterations in both PrfA DNA binding and PrfA dimer formation, but nevertheless exhibited high level activation of PrfA and a corresponding dramatic increase in PrfA-dependent gene expression.
How does the prfA P219S substitution induce PrfA activation? In the absence of structural data for this mutant protein, the precise mechanism remains unclear. PrfA activation in the absence of a significant increase in PrfA in vitro DNA-binding affinity has been reported for one additional prfA* mutant, prfA Y63C (Miner et al., 2008b). The prfA Y63C mutation is located within a structural pocket that has been suggested to serve as the PrfA cofactor-binding site (Fig. 1b) and thus has been speculated to influence cofactor binding (Miner et al., 2008b). The prfA P219S mutation is not located near the putative cofactor-binding region and is not therefore anticipated to enhance or influence cofactor binding unless it does so via a distal conformational effect. Such a distal effect is possible given that the P219 residue is located at the end of PrfA αH, near the transition of helix G to helix H. αH is in close proximity to αD, which contains G145 (Fig. 1b), for which the substitution of serine results in a repositioning of the PrfA helix–turn–helix DNA-binding domain and an increase in PrfA DNA-binding affinity (Eiting et al., 2005). While it is possible that the P219S substitution alters the conformation of αH so as to mimic an activated state, the change imposed must be distinct from that conferred by G145S as there is no evident increase in PrfA P219S DNA-binding affinity (Fig. 4). Alternatively, it has been postulated that a component of the phosphoenol pyruvate phosphotransferase system may bind and sequester PrfA, thereby preventing PrfA binding at target promoters (Joseph & Goebel, 2007; Marr et al., 2006; Mertins et al., 2007). If this is the case, it is possible that the prfA P219S mutation disrupts this inhibitory interaction and prevents sequestration of PrfA, leaving it free to bind and activate its target promoters. Although the precise in vivo mechanism underlying activation of PrfA remains speculative, the isolation and characterization of prfA* mutant proteins has made it readily apparent that there are multiple types of PrfA activation via amino acid substitution.
Acknowledgements
We thank Erika Guerrero for assistance with PrfA CD studies and members of the Freitag lab and the UIC (Gram) Positive Thinking group for helpful discussions. This work was supported by Public Health Service grant AI41816 (N. E. F.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding source.
Abbreviations:
- GUS
β-glucuronidase
- CD
circular dichroism
- EMS
ethylene methylsulphonate
- EMSA
electrophoretic mobility shift assay
- LLO
listeriolysin O
- XG
5-bromo-4-chloro-3-indolyl β-d-glucuronide
References
- Alonzo F., III, Port G. C., Cao M., Freitag N. E. (2009). The posttranslocation chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. Infect Immun 77, 2612–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitar A. P., Cao M., Marquis H. (2008). The metalloprotease of Listeria monocytogenes is activated by intramolecular autocatalysis. J Bacteriol 190, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno J. C., Jr, Freitag N. E. (2010). Constitutive activation of PrfA tilts the balance of Listeria monocytogenes fitness towards life within the host versus environmental survival. PLoS ONE 5, e15138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A., Paynton C. R., Portnoy D. A. (1989). Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc Natl Acad Sci U S A 86, 5522–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A., Tilney L. G., Portnoy D. A. (1993). Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol 8, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2004). Preliminary FoodNet data on the incidence of infection with pathogens transmitted commonly through food – selected sites, United States, 2003. MMWR Morb Mortal Wkly Rep 53, 338–343. [PubMed] [Google Scholar]
- Cossart P. (2007). Listeriology (1926–2007): the rise of a model pathogen. Microbes Infect 9, 1143–1146. [DOI] [PubMed] [Google Scholar]
- Cossart P., Toledo-Arana A. (2008). Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect 10, 1041–1050. [DOI] [PubMed] [Google Scholar]
- Czuprynski C. J. (2005). Listeria monocytogenes: silage, sandwiches and science. Anim Health Res Rev 6, 211–217. [DOI] [PubMed] [Google Scholar]
- Delgado A. R. (2008). Listeriosis in pregnancy. J Midwifery Womens Health 53, 255–259. [DOI] [PubMed] [Google Scholar]
- Drevets D. A., Bronze M. S. (2008). Listeria monocytogenes: epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol Med Microbiol 53, 151–165. [DOI] [PubMed] [Google Scholar]
- Dussurget O. (2008). New insights into determinants of Listeria monocytogenes virulence. Int Rev Cell Mol Biol 270, 1–38. [DOI] [PubMed] [Google Scholar]
- Eiting M., Hagelüken G., Schubert W. D., Heinz D. W. (2005). The mutation G145S in PrfA, a key virulence regulator of Listeria monocytogenes, increases DNA-binding affinity by stabilizing the HTH motif. Mol Microbiol 56, 433–446. [DOI] [PubMed] [Google Scholar]
- Freitag N. E. (2006). From hot dogs to host cells: how the bacterial pathogen Listeria monocytogenes regulates virulence gene expression. Future Microbiol 1, 89–101. [DOI] [PubMed] [Google Scholar]
- Freitag N. E., Portnoy D. A. (1994). Dual promoters of the Listeria monocytogenes prfA transcriptional activator appear essential in vitro but are redundant in vivo. Mol Microbiol 12, 845–853. [DOI] [PubMed] [Google Scholar]
- Freitag N. E., Port G. C., Miner M. D. (2009). Listeria monocytogenes – from saprophyte to intracellular pathogen. Nat Rev Microbiol 7, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi M., Chikindas M. L. (2007). Listeria: a foodborne pathogen that knows how to survive. Int J Food Microbiol 113, 1–15. [DOI] [PubMed] [Google Scholar]
- Gray M. J., Freitag N. E., Boor K. J. (2006). How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect Immun 74, 2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman J. G. (2001). Allosteric regulation of the cAMP receptor protein. Biochim Biophys Acta 1547, 1–17. [DOI] [PubMed] [Google Scholar]
- Harman J. G., McKenney K., Peterkofsky A. (1986). Structure–function analysis of three cAMP-independent forms of the cAMP receptor protein. J Biol Chem 261, 16332–16339. [PubMed] [Google Scholar]
- Herler M., Bubert A., Goetz M., Vega Y., Vazquez-Boland J. A., Goebel W. (2001). Positive selection of mutations leading to loss or reduction of transcriptional activity of PrfA, the central regulator of Listeria monocytogenes virulence. J Bacteriol 183, 5562–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton K., Cossart P. (1997). Host–pathogen interactions during entry and actin-based movement of Listeria monocytogenes. Annu Rev Genet 31, 113–138. [DOI] [PubMed] [Google Scholar]
- Joseph B., Goebel W. (2007). Life of Listeria monocytogenes in the host cells’ cytosol. Microbes Infect 9, 1188–1195. [DOI] [PubMed] [Google Scholar]
- Kreft J., Vázquez-Boland J. A., Altrock S., Dominguez-Bernal G., Goebel W. (2002). Pathogenicity islands and other virulence elements in Listeria. Curr Top Microbiol Immunol 264, 109–125. [PubMed] [Google Scholar]
- Lampidis R., Gross R., Sokolovic Z., Goebel W., Kreft J. (1994). The virulence regulator protein of Listeria ivanovii is highly homologous to PrfA from Listeria monocytogenes and both belong to the Crp–Fnr family of transcription regulators. Mol Microbiol 13, 141–151. [DOI] [PubMed] [Google Scholar]
- Lauer P., Chow M. Y., Loessner M. J., Portnoy D. A., Calendar R. (2002). Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol 184, 4177–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M. (2007). Human listeriosis and animal models. Microbes Infect 9, 1216–1225. [DOI] [PubMed] [Google Scholar]
- Lynch M., Painter J., Woodruff R., Braden C., Centers for Disease Control and Prevention (2006). Surveillance for foodborne-disease outbreaks – United States, 1998–2002. MMWR Surveill Summ 55, 1–42. [PubMed] [Google Scholar]
- Marquis H., Doshi V., Portnoy D. A. (1995). The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect Immun 63, 4531–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr A. K., Joseph B., Mertins S., Ecke R., Müller-Altrock S., Goebel W. (2006). Overexpression of PrfA leads to growth inhibition of Listeria monocytogenes in glucose-containing culture media by interfering with glucose uptake. J Bacteriol 188, 3887–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead P. S., Dunne E. F., Graves L., Wiedmann M., Patrick M., Hunter S., Salehi E., Mostashari F., Craig A., et al. (2006). Nationwide outbreak of listeriosis due to contaminated meat. Epidemiol Infect 134, 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins S., Joseph B., Goetz M., Ecke R., Seidel G., Sprehe M., Hillen W., Goebel W., Müller-Altrock S. (2007). Interference of components of the phosphoenolpyruvate phosphotransferase system with the central virulence gene regulator PrfA of Listeria monocytogenes. J Bacteriol 189, 473–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner M. D., Port G. C., Bouwer H. G., Chang J. C., Freitag N. E. (2008a). A novel prfA mutation that promotes Listeria monocytogenes cytosol entry but reduces bacterial spread and cytotoxicity. Microb Pathog 45, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner M. D., Port G. C., Freitag N. E. (2008b). Functional impact of mutational activation on the Listeria monocytogenes central virulence regulator PrfA. Microbiology 154, 3579–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller K. J., Freitag N. E. (2005). Pleiotropic enhancement of bacterial pathogenesis resulting from the constitutive activation of the Listeria monocytogenes regulatory factor PrfA. Infect Immun 73, 1917–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil H. S., Marquis H. (2006). Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect Immun 74, 6675–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port G. C., Freitag N. E. (2007). Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect Immun 75, 5886–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy D. A., Jacks P. S., Hinrichs D. J. (1988). Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med 167, 1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V., Cresence V. M., Rejitha J. S., Lekshmi M. U., Dharsana K. S., Prasad S. P., Vijila H. M. (2007). Listeria – review of epidemiology and pathogenesis. J Microbiol Immunol Infect 40, 4–13. [PubMed] [Google Scholar]
- Ripio M. T., Domínguez-Bernal G., Lara M., Suárez M., Vazquez-Boland J. A. (1997). A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J Bacteriol 179, 1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S. M., Gracieux P., Albert I., Gouali M., Jacquet C., Martin P. M., Velge P. (2003). Experimental validation of low virulence in field strains of Listeria monocytogenes. Infect Immun 71, 3429–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortti M., Monzó H. J., Lacharme-Lora L., Lewis D. A., Vázquez-Boland J. A. (2007). The PrfA virulence regulon. Microbes Infect 9, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Shetron-Rama L. M., Marquis H., Bouwer H. G., Freitag N. E. (2002). Intracellular induction of Listeria monocytogenes actA expression. Infect Immun 70, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetron-Rama L. M., Mueller K., Bravo J. M., Bouwer H. G., Way S. S., Freitag N. E. (2003). Isolation of Listeria monocytogenes mutants with high-level in vitro expression of host cytosol-induced gene products. Mol Microbiol 48, 1537–1551. [DOI] [PubMed] [Google Scholar]
- Smith M. A., Takeuchi K., Anderson G., Ware G. O., McClure H. M., Raybourne R. B., Mytle N., Doyle M. P. (2008). Dose–response model for Listeria monocytogenes-induced stillbirths in nonhuman primates. Infect Immun 76, 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B., Kemp M., Ethelberg S., Schiellerup P., Bruun B. G., Gerner-Smidt P., Christensen J. J. (2009). Listeria monocytogenes: maternal-foetal infections in Denmark 1994–2005. Scand J Infect Dis 41, 21–25. [DOI] [PubMed] [Google Scholar]
- Sun A. N., Camilli A., Portnoy D. A. (1990). Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun 58, 3770–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan B., Gerner-Smidt P. (2007). The epidemiology of human listeriosis. Microbes Infect 9, 1236–1243. [DOI] [PubMed] [Google Scholar]
- Swaminathan B., Gerner-Smidt P., Whichard J. M. (2006). Foodborne disease trends and reports. Foodborne Pathog Dis 3, 316–318. [DOI] [PubMed] [Google Scholar]
- Tan G. S., Kelly P., Kim J., Wartell R. M. (1991). Comparison of cAMP receptor protein (CRP) and a cAMP-independent form of CRP by Raman spectroscopy and DNA binding. Biochemistry 30, 5076–5080. [DOI] [PubMed] [Google Scholar]
- Toledo-Arana A., Dussurget O., Nikitas G., Sesto N., Guet-Revillet H., Balestrino D., Loh E., Gripenland J., Tiensuu T., et al. (2009). The Listeria transcriptional landscape from saprophytism to virulence. Nature 459, 950–956. [DOI] [PubMed] [Google Scholar]
- Vázquez-Boland J. A., Kuhn M., Berche P., Chakraborty T., Domínguez-Bernal G., Goebel W., González-Zorn B., Wehland J., Kreft J. (2001). Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14, 584–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega Y., Dickneite C., Ripio M. T., Böckmann R., González-Zorn B., Novella S., Domínguez-Bernal G., Goebel W., Vázquez-Boland J. A. (1998). Functional similarities between the Listeria monocytogenes virulence regulator PrfA and cyclic AMP receptor protein: the PrfA* (Gly145Ser) mutation increases binding affinity for target DNA. J Bacteriol 180, 6655–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega Y., Rauch M., Banfield M. J., Ermolaeva S., Scortti M., Goebel W., Vázquez-Boland J. A. (2004). New Listeria monocytogenes prfA* mutants, transcriptional properties of PrfA* proteins and structure–function of the virulence regulator PrfA. Mol Microbiol 52, 1553–1565. [DOI] [PubMed] [Google Scholar]
- Velge P., Herler M., Johansson J., Roche S. M., Témoin S., Fedorov A. A., Gracieux P., Almo S. C., Goebel W., Cossart P. (2007). A naturally occurring mutation K220T in the pleiotropic activator PrfA of Listeria monocytogenes results in a loss of virulence due to decreasing DNA-binding affinity. Microbiology 153, 995–1005. [DOI] [PubMed] [Google Scholar]
- Wong K. K., Freitag N. E. (2004). A novel mutation within the central Listeria monocytogenes regulator PrfA that results in constitutive expression of virulence gene products. J Bacteriol 186, 6265–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K. K., Bouwer H. G., Freitag N. E. (2004). Evidence implicating the 5′ untranslated region of Listeria monocytogenes actA in the regulation of bacterial actin-based motility. Cell Microbiol 6, 155–166. [DOI] [PubMed] [Google Scholar]
- Xayarath B., Marquis H., Port G. C., Freitag N. E. (2009). Listeria monocytogenes CtaP is a multifunctional cysteine transport-associated protein required for bacterial pathogenesis. Mol Microbiol 74, 956–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman P. (1987). Plasmid vectors recovering and exploiting Tn917 transposons in Bacillus and other Gram-positive bacteria. In Plasmids: a Practical Approach, 1st edn, pp 79–103. Edited by Hardy K. G. Oxford: IRL Press [Google Scholar]