

Abstract
Background
Common carp (Cyprinus carpio) is thought to have undergone one extra round of genome duplication compared to zebrafish. Transcriptome analysis has been used to study the existence and timing of genome duplication in species for which genome sequences are incomplete. Large-scale transcriptome data for the common carp genome should help reveal the timing of the additional duplication event.
Results
We have sequenced the transcriptome of common carp using 454 pyrosequencing. After assembling the 454 contigs and the published common carp sequences together, we obtained 49,669 contigs and identified genes using homology searches and an ab initio method. We identified 4,651 orthologous pairs between common carp and zebrafish and found 129,984 paralogous pairs within the common carp. An estimation of the synonymous substitution rate in the orthologous pairs indicated that common carp and zebrafish diverged 120 million years ago (MYA). We identified one round of genome duplication in common carp and estimated that it had occurred 5.6 to 11.3 MYA. In zebrafish, no genome duplication event after speciation was observed, suggesting that, compared to zebrafish, common carp had undergone an additional genome duplication event. We annotated the common carp contigs with Gene Ontology terms and KEGG pathways. Compared with zebrafish gene annotations, we found that a set of biological processes and pathways were enriched in common carp.
Conclusions
The assembled contigs helped us to estimate the time of the fourth-round of genome duplication in common carp. The resource that we have built as part of this study will help advance functional genomics and genome annotation studies in the future.
Keywords: Sequence assembly, Genome duplication, Common carp, Substitution rate, Speciation
Background
The Cyprininae family includes zebrafish (Danio rerio) and the several economically important cultivated carp, such as common carp (Cyprinus carpio), grass carp (Ctenopharyngodon idella), silver carp (Hypophthalmichthys molitrix) and bighead carp (Hypophthalmichthys nobilis). Teleosts are widely believed to have gone through an additional round of whole genome duplication referred to as the 3R hypothesis, as compared to mammals. This third round (3R) of whole genome duplication is specific to ray-finned fish and possibly occurred about 360 million years ago (MYA), preceding the divergence of the teleosts [1]. The 3R duplication may have led to the major diversification of the teleosts [2-4]. The chromosome number of common carp (n = 50) is twice that of most other Cyprinidae. Thus, it has been assumed that common carp have undergone a fourth round (4R) of genome duplication. Indeed, previous studies have found more copies of several genes and microsatellites in common carp than in most other Cyprinidae and various estimates of the time of the additional round of genome duplication have been made. An analysis of the c-myc genes in common carp estimated that the tetraploidization event occurred 58 MYA [5] while another study, based on other duplicated genes in common carp, reported a tetraploidization time of less than 16 MYA [6]. Using 59 microsatellites, David et al. [7] estimated that the 4R genome duplication had occurred about 12 MYA. However, these conflicting estimates of the time of genome duplication were all based on small data set. Thus, to obtain a more accurate estimate of the duplication time, it is necessary to use a larger data set.
For species for which the whole genome sequence is not yet available, transcriptome analysis is an alternative method that has been used to discover new genes and to investigate gene expression. A large set of common carp ESTs produced using Sanger sequencing has been developed and used to study traits in common carp [8-11]. More recently, second generation sequencing platforms have been applied to transcriptome sequencing [12-14], making the transcriptome more readily accessible. Transcriptome analysis is a power tool that has been used to study various genome feature, including genome duplication [15,16]. When the synonymous substitution rate (Ks) in two paralogous sequences is assumed to increase approximately linearly with age [17], paralogous pairs can be sorted along with their relative ages of duplication by estimating their Ks. A genome duplication event would result in a sharp increase in the number of paralogous genes, yielding a secondary peak in the Ks distribution of paralogous pairs. Therefore, a secondary peak in the paralogous Ks distribution indicates one genome duplication event. Large-scale transcriptome data for the common carp will help the study of the additional genome duplication and improve estimates of the timing of this event in this species.
Here, we have compared the contigs from common carp with zebrafish genes: 1) to assess the relative age of the separation between zebrafish and common carp; 2) to estimate the time of the additional genome duplication event in common carp; and 3) to determine biological processes and pathways enriched in common carp.
Results
Hybrid assembly of the common carp 454 contigs and public ESTs/mRNAs
The 454 pyrosequencing generated 242,261 reads, encompassing about 52.9 Mb of sequencing data. The average length of the 454 reads was 218 bp. After the initial adapter trimming and quality filtering, we assembled the remaining 241,170 cleaned reads using Celera assembler [18] and obtained 8,422 contigs and 60,910 singletons. We downloaded 34,067 common carp ESTs and 989 mRNAs from NCBI sequence database. After filtering out possible vector sequences, 33,259 cleaned ESTs and 987 cleaned mRNAs were assembled with 454 contigs/singletons into 51,065 contigs using CAP3 [19]. Further, to avoid redundant gene identification and annotation caused by alternative splicing, we performed an all-against-all BLASTN search on the CAP3 contigs. If the alignments of two sequences had 100% identity over 100 bp, they were considered to be from the same genes as the result of alternative splicing. We selected the longest contigs to represent these genes and finally obtained 49,669 unique contigs. The N50 length of this set of contigs was 654 bp.
Assessment of hybrid assembly
To assess the quality of our assembly, the CAP3 contigs were compared with 40 published paralogous mRNAs [20 pairs of paralogs, see Additional file 1: Table S1]. The 40 mRNAs were aligned with the CAP3 contigs using BLAT and the CAP3 contig that aligned best with each of the mRNAs was selected. Although the highest identity between these paralogous genes was 97%, all the mRNAs matched distinct CAP3 contigs with the full length sequences covered. This result indicated that our assembly was of high quality.
Without a genome sequence, it is difficult to determine splicing variants in a de novo transcriptome assembly [20]. Spliced variants in the dataset can lead to redundant gene prediction and artificial paralogs. Because splicing variants were from the same genes and share common exons, we filtered out possible alternative splicing variants using high sequence identity (100%) over long matched region (> 100 bp). We evaluated the reliability of this strategy using zebrafish transcripts from the Ensembl database [21] as a test dataset. In the Ensembl database, 11,227 zebrafish genes have 30,963 spliced variants and form 36,087 splicing pairs. We identified 31,957 pairs of spliced variants using our strategy; 28,434 of them were in the Ensembl splicing pairs. This result suggested that our strategy had the sensitivity of nearly 80% with a low error rate of only 11% (3,523 out of 31,957).
Gene prediction from common carp contigs
We used homolog searches and an ab initio prediction method to annotate the common carp contigs. By searching against three different protein databases using BLASTX with a cutoff e-value of 1e-5, we found 24,784 contigs that had BLASTX hits to at least one of these databases. Most contigs were homologous to sequences from the fish protein database (Table 1). As shown in Figure 1, the common carp contigs had the greatest number of hits against zebrafish sequences. After running the unmatched contigs against UTRdb [22], a database of 5' and 3' untranslated regions (UTRs) of eukaryotic mRNAs, we found another 3,658 contigs that were homologous to UTRs, indicating that these contigs might represent the UTRs of common carp protein-coding genes. An additional 14,524 contigs could be aligned to the NCBI nonredundant nucleotide database using BLASTN. Since these contigs had hits to known proteins or nucleotide sequences, they might be conserved genes.
Table 1.
Summary of common carp contig annotation
| Methods | Database | Number | |
|---|---|---|---|
| Homolog search | Protein-coding | Fish protein database* | 24,167 |
| UniProt database | 409 | ||
| NCBI nr protein database | 208 | ||
| UTRdb | 3,658 | ||
| NCBI nr nucleotide database | 14,524 | ||
| ab initio search | Protein-coding | CPC | 47 |
| Unknown | 6,656 |
* Fish protein database consists of protein sequences from Zebrafish, Fugu, Stickleback, Tetraodon and Medaka.
Figure 1.
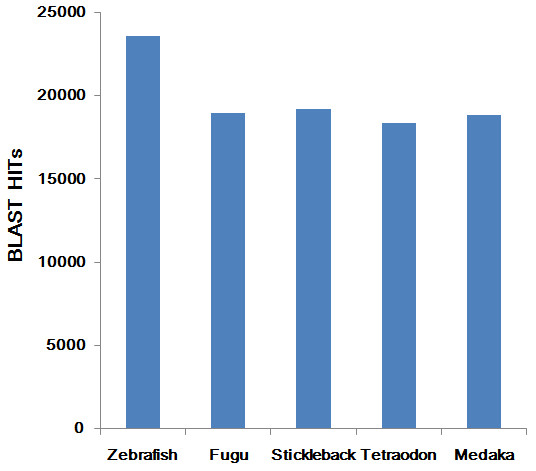
A bar plot showing the hits to protein sequences from five sequenced teleost species. Alignments of common carp contigs to protein sequences from Zebrafish, Fugu, Stickleback, Tetraodon and Medaka, respectively.
Finally, among the remaining unidentified contigs, an ab initio method using the CPC predicted that 47 of the contigs were potentially coding sequences. Because these contigs had no matches to known proteins, they might be common carp specific protein-coding genes. The remaining unknown 6,656 contigs had neither protein-coding potential nor known homologs, indicating that they were probably transcribed from common carp intergenic regions of the common carp genome. The complete annotation statistics are shown in Table 1.
To validate the reliability of the de novo assembly and the assembled contigs, gene expression was examined in RNAs from the pooled-tissues by PCR. Twenty contigs, including conserved genes, common carp specific protein-coding genes and unknown contigs [see Additional files 2: Methods S1 and 3: S2], were selected randomly. Primers were designed specifically for the selected contigs to avoid amplifying paralogs. The results showed that all the selected contigs could be amplified [see Additional file 4: Figure S1], indicating that these contigs were correctly assembled and truly expressed.
Genome speciation event deduced from orthologous pairs between common carp and zebrafish
We assumed that one round of genome duplication occurred in common carp after the speciation while zebrafish had no further genome duplication. A secondary peak in the orthologous Ks value distribution indicates a speciation events [16]. Therefore, we firstly estimated the genome speciation time based on the Ks distribution of orthologous pairs between the two species. We identified 4,651 orthologous pairs between common carp and zebrafish using the reciprocal best blast hit approach with a stringent e-value cutoff (1e-20) as described previously [16]. The Ks distribution of these 4,651 orthologous pairs showed a distinct secondary Ks peak at 0.42 (Figure 2). Considering a clock-like rate of synonymous substitution of 3.51 × 10-9 substitutions/synonymous site per year [7,17], the speciation between common carp and zebrafish was estimated to have occurred ~120 MYA. This estimated speciation time is earlier than the time predicted from previous reports based on individual genes [23].
Figure 2.
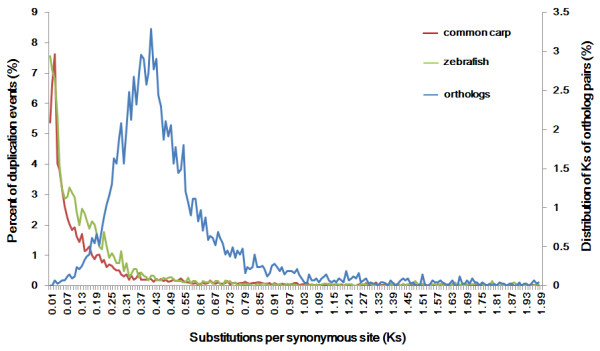
Ks value distribution to identify the genome duplication event and the speciation event. Data was grouped into bins of 0.01 Ks units for graphing. For common carp and zebrafish, the Ks distributions of duplication events were shown in red and green respectively. A secondary Ks peak within common carp indicated the genome duplication (red line). Given the rate of substitutions/synonymous site per year, the peak indicated the time of the 4R of genome duplication. Within zebrafish, no secondary peak in the Ks distribution of paralogous sequences was observed (green line). The Ks distribution of the orthologous pairs was plotted in blue line and showed a distinct secondary Ks peak, indicating the speciation time between these two species.
Genome duplication event estimated from Ks distribution of paralogous pairs
A secondary peak in the paralogous Ks distribution plot can provide signatures of genome duplication within a species [16]. To evaluate the quality of the paralogs identification by our strategy, we performed an all-against-all BLASTN using the same 20 paralogous pairs. A total of 18 pairs (90% of the real paralogous pairs) satisfied the criteria for paralogous identification [see Additional file 1: Table S1], indicating that the customized parameter was suitable for paralogs identification. We identified 129,984 and 46,385 paralogous pairs within common carp and zebrafish, respectively. These paralogous pairs were then organized into 4,689 and 869 gene families in carp and zebrafish respectively, by a single linkage clustering method. The numbers of sequences in the paralogous pairs and gene families are shown in Table 2 and the Ks distribution plots for the two species are shown in Figure 2. We observed an obvious secondary peak in the Ks distribution of paralogous sequences within common carp (with a mode at Ks = 0.02 to 0.04), indicating a genome duplication event. With the rate of 3.51 × 10-9 substitutions/synonymous site per year [7,17], this duplication was estimated to have occurred 5.6 to 11.3 MYA. This estimate is more recent than previous ones [5-7]. Together with the estimated speciation time, these data suggested that this duplication event occurred long after the speciation. Within zebrafish, no secondary peak in the Ks distribution of paralogous sequences was observed, indicating that after speciation no genome duplication event had occurred. Together, these data supported the hypothesis that common carp had undergone one extra round of genome duplication compared with zebrafish.
Table 2.
Number of sequences and paralogs within common carp and zebrafish
| Species | Sequences in final dataseta | Paralogous pairs | Paralogous sequencesb | Percentage of paralogsc | Gene familiesd | Duplication Event with median Ks < 2e |
|---|---|---|---|---|---|---|
| common carp | 49,669 | 129,984 | 19,159 | 38.6% | 4,689 | 8,190 |
| zebrafish | 25,348 | 46,385 | 3,774 | 14.9% | 869 | 2,721 |
a Number of the longest sequences.
b Number of paralogous sequences found in the final dataset using BLASTN search.
c Percentage of paralogous sequences found in the final dataset.
d Number of gene families constructed with paralogous sequences using single linkage clustering.
e Number of duplication events used in the distributions in Figure 2 and of which median Ks rates are < 2.
Comparison of biological process and pathway enriched in common carp and zebrafish
We determined that a whole genome duplication event occurred 5.6 to 11.3 MYA in the common carp. Since then, re-diploidization would have resulted in a pseudo-tetraploid state and differential evolution or loss of duplicated genes. A biological process and pathway comparison between common carp and zebrafish may provide hints of the possible consequences of whole genome duplication and following re-diploidization.
We assigned Gene Ontology (GO) terms to 47.2% of the common carp contigs (23,441 out of 49,669) using homologous assignments and Interproscan [24]. This percentage is similar to GO assignments in other fish [25,26]. We then used WEGO to find significantly enriched GO terms in common carp (p-value < 0.05) using zebrafish genes as the background. In the biological process category, a total of 15 GO terms (up to 2 level) were significantly overrepresented (p < 0.05) while 6 terms were either underrepresented or similar in the common carp relative to the zebrafish GO annotations (Figure 3).
Figure 3.
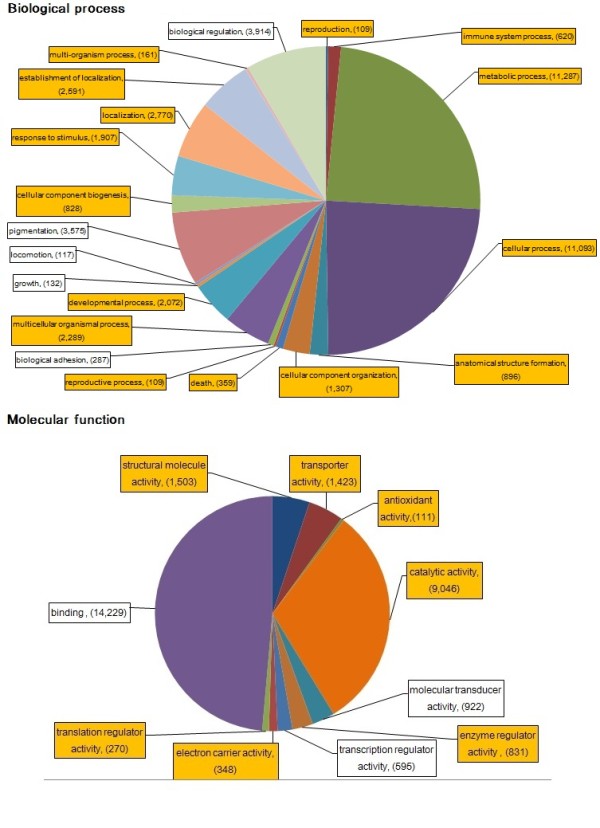
Distribution of common carp GO terms in biological process and molecular function categories. The relative proportion of GO terms is represented by more than 100 contigs for the biological process (A) and molecular function (B) categories in the GO vocabulary. The enriched GO terms in common carp (p < 0.05) were highlighted with orange.
To study the biological pathways involved, we mapped common carp contigs to pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) using KOBAS software and a total of 10,308 contigs were mapped to KEGG pathway. Zebrafish genes were also mapped to KEGG pathway and used as background to compare pathway differences between common carp and zebrafish. The statistically enriched pathways (corrected p-value < 0.05, Table 3) were consistent with the enriched GO biological process terms. For example, immune-related process, localization and response to stimulus were all indicated in both enriched GO processes and KEGG pathways. These data, therefore, provide an insight into the process and pathway enriched in common carp.
Table 3.
The enriched pathways in common carp identified by KOBAS (corrected p-value < 0.05)
| KEGG pathway | ID | Common carp contigs proportion | Zebrafish gene proportion | Biological process GO term (level 2)* |
|---|---|---|---|---|
| Protein digestion and absorption | ko04974 | 547/10308 | 126/7433 | multicellular organismal process |
| Glycolysis/Gluconeogenesis | ko00010 | 358/10308 | 76/7433 | metabolic process |
| Pancreatic secretion | ko04972 | 564/10308 | 172/7433 | Localization |
| Complement and coagulation cascades | ko04610 | 360/10308 | 105/7433 | immune system process |
| Starch and sucrose metabolism | ko00500 | 243/10308 | 56/7433 | metabolic process |
| Oxidative phosphorylation | ko00190 | 415/10308 | 139/7433 | metabolic process |
| Antigen processing and presentation | ko04612 | 278/10308 | 85/7433 | immune system process |
| Pyruvate metabolism | ko00620 | 175/10308 | 47/7433 | metabolic process |
| TCA cycle | ko00020 | 149/10308 | 36/7433 | metabolic process |
| Pentose phosphate pathway | ko00030 | 134/10308 | 31/7433 | cellular process |
| RNA transport | ko03013 | 352/10308 | 162/7433 | Localization |
| Mineral absorption | ko04978 | 134/10308 | 43/7433 | developmental process |
| PPAR signaling pathway | ko03320 | 232/10308 | 96/7433 | response to stimulus |
| Protein processing in endoplasmic reticulum | ko04141 | 400/10308 | 215/7433 | cellular process |
| Ribosome biogenesis in eukaryotes | ko03008 | 113/10308 | 42/7433 | cellular component biogenesis |
| RNA degradation | ko03018 | 153/10308 | 77/7433 | metabolic process |
* The enriched pathways correspond to biological process GO terms of level 2 in GO vocabulary. These biological processes were still enriched in common carp, indicating the consistence between GO term comparison and KEGG pathway analysis.
Discussion
Here, we performed transcriptome analysis to estimate the time of the fourth round (4R) of genome duplication in common carp. Earlier, several genes and microsatellites had been used to estimate this. In the present study, we generated an order-of-magnitude more contigs than previously, making the estimated time of the 4R duplication event more reliable.
Timothy et al. [27] reported that the pooled-tissue approach was highly effective in preparing libraries for efficient deep sequencing. RNA that has been pooled from multiple tissues can maximize the number of independent genes for which sequence data can be obtained [27]. Thus, we constructed a pooled-tissue cDNA library and sequenced the library. Although the number of sequencing reads that we obtained were lower than in other studies [28,29], the hybrid assembly built by including public EST/mRNAs improved the sequence coverage and allowed more common carp genes to be identified. The size of common carp genome has been estimated to be 1.7-2.0 Gb [30,31]. Assuming that 1% of the genome encodes mRNAs [26,32], the 49,669 contigs, accounting for 17 Mb, that we obtained provide an estimated coverage of the common carp transcriptome of at least 85%. This estimate is an indication that, in our study, the common carp transcriptome has been sampled broadly with good coverage. Previous studies have suggested that singletons may be biologically valuable [33]. Therefore, we included the singletons in all further analyses. The high coverage of the transcriptome helped in genome duplication detection and function comparison.
Although de novo transcriptome assembly can make the genes of non-model organisms with no available genome sequences more readily accessible, a number of challenges including gene duplication or paralogy and alternative splicing still exist. We used stringent parameters for the de novo assembly to help differentiate paralogous genes. We then assessed the correctness of the assembly by comparing it with published paralogous genes. These published genes all corresponded to distinct contigs, indicating that the parameters were robust enough to have avoided the creation of chimeric sequences from paralogous transcripts. Spliced variants present in de novo assembly dataset are another challenge because their presence can result in redundant gene predictions and paralogs identification. Although spliced variants are difficult to identify without the genome sequences, the strategy that we applied here would have filtered out a high proportion of the alternative variant with a low error rate [see Additional file 5: Figure S2].
To estimate the time of the 4R genome duplication event in the common carp, we estimated the Ks values for all orthologous and paralogous pairs. The clear signature of a duplication event within common carp that we obtained showed that the duplication was fairly recent. This is possibly one of the most recent genome duplications in vertebrates. As expected, we did not observe a paralogous peak in zebrafish. These data provided evidence for an additional round of genome duplication in common carp. However, with age increasing, an exponential decrease of density in the distribution of paralogous pairs as a result of the deletion of duplicated sequences would have occurred [34,35]. Because of this effect, common genome duplication events in common carp and zebrafish could not be found.
Zebrafish is the most closely related species to common carp; both belong to the Cyprinidae family. The common carp contigs that were generated in this study allowed us to conduct an initial comparative genome analysis between zebrafish and common carp. Because, one more round of genome duplication followed by re-diploidization occurred in the common carp, the comparison of GO biological process terms and KEGG pathways between common carp and zebrafish provided hints of the possible consequences of whole genome duplication and re-diploidization. Interestingly, compared to the zebrafish genes, the common carp contigs were enriched in immune-related terms and pathways, such as, complement and coagulation cascades, and antigen processing and presentation. We examined the functions of the published paralogous genes [see Additional file 1: Table S1] and found that 45% of them (9 pairs) were involved in immune-related pathways. The enriched immune-related contigs might be genes that assist carp in resisting pathogens or might be of help in adapting to different aquaculture growing environment.
Conclusions
The hybrid assembly of the 454 contigs and published ESTs/mRNAs in this study had significantly expanded the common carp EST resource and provided a valuable dataset for future gene discovery. Our comparative analysis between the common carp contigs and zebrafish genes estimated the time of the 4R of genome duplication event in common carp to be 5.6 to 11.3 MYA and revealed the enriched biological processes and pathways in common carp.
Methods
Hybrid assembly of the 454 contigs and public ESTs of common carp
All experimental procedures were conducted in conformity with institutional guidelines for the care and use of laboratory animals in Chinese Academy of Fishery Science, Beijing, China, and conformed to the National Institutes of Health Guide for Care and Use of Laboratory Animals. The animal work was approved by the academic committee in the Centre for Applied Aquatic Genomics (approval ID: 01/2011). Tissue samples were excised from brain, muscle and live of three mature German mirror carps. The tissues were cut into small pieces and immediately pooled into liquid nitrogen. Total RNA in all three tissues was purified using TRIZOL (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. Total RNA quality and quantity were checked by agarose gel electrophoresis containing formaldehyde and using a spectrophotometer. Poly(A) + RNA was purified from total cellular RNA using oligo dT primer. Full-length cDNA was synthesized from 2 μg of poly(A) + RNA using the Clontech SMART cDNA Library Construction Kit (Clontech, Mountain View, CA, USA) according to manufacturer's protocol. The cDNA was amplified using PCR Advantage II Polymerase in 16 thermo cycles with the following thermal profile: 7 s at 95°C, 20 s at 66°C, and 4 mins at 72°C. The amplified cDNA was subsequently purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA) to remove fragments of less than 300 bp.
Preparation of the 454 library was performed according to the supplier's instructions (454 Life Sciences, Branford, CT, USA). In summary, approximately 3 μg of amplified cDNA was nebulized and selected for lengths that ranged from 300 to 800 bp. The FLX specific adapters, adapter A (GCCTCCCTCGCGCCATCAG) and adapter B (GCCTTGCCAGCCCGCTCAG), were added to each fragmented cDNA, resulting in adapter A-DNA fragment-adapter B constructs. The DNA fragments were then denatured to generate single-stranded DNA which was then amplified by emulsion PCR for sequencing. The sequencing of the library was performed in one half-plate run on the 454 GS FLX machine. The entire set of reads used for the assembly was submitted to the NCBI Sequence Read Archive under the accession SRA009366 (Submission: SRA009366 by CAFS).
Before assembling the 454 sequencing reads, the adapter sequences were removed. If the ends of one read contained parts of either adapter A (GCCTCCCTCGCGCCATCAG) or adapter B (GCCTTGCCAGCCCGCTCAG), these nucleotides were removed. Next, the Solexa QA package [36] was used to filter out low-quality bases with the following parameters: -probcutoff 0.05 (the quality cutoff score below which the base-calling error was considered to be too high) and -454 (to trim 454 reads). Because homopolymers of poly(A/T) have low quality scores in 454 sequencing [37], this process filters out poly(A/T) sequences. The resulting high-quality (HQ) reads were then assembled using the Celera Assembler 6.1 [18] on a single multiprocessor computer. Most of the parameters were set to the default values; for example, overlapper = mer (a seed and extend overlap algorithm), unitigger = bog (a best overlap graph approach for building unitigs), and doOverlapTrimming = 1 (for overlap-based trimming). To avoid the putative mis-assembly of paralogous genes into chimera contigs, we manually collected 20 pairs of published common carp paralogous genes and analyzed their sequence identity using BLASTN [see Additional file 1: Table S1]. We found that the highest identity between these paralogous gene sequences was 97%. Therefore, the Assembler parameters that might influence sequence assembly were set as: utgErrorRate = 0.029 (the error rate above which the unitigger discards overlaps), ovlErrorRate = 0.029 (overlaps above this limit will not be detected), and cnsErrorRate = 0.029 (he error rate below which consensus finds alignments).
To increase transcriptome coverage, we downloaded 34,067 common carp ESTs from the UniGene database [38] and 989 common carp mRNAs from GenBank. Any vector contamination of the public ESTs/mRNAs was removed using the seqclean program [39]. The 454 contigs/singletons and cleaned ESTs/mRNAs were assembled into contigs using the CAP3 software with default parameters except that we used -p 98 because the highest identity that we found between the carp paralogous genes was 97%.
To avoid the identification of redundant genes as a result of alternative splicing, all-against-all BLASTN searches were performed using CAP3 contigs. If the alignment of two sequences had 100% identity over 100 bps, then they were considered as spliced variants and the longest contigs was selected to represent this gene.
Gene annotation
To identify the common carp protein-coding genes, we used BLASTX with an e-value of 1e-5 to run our assembled sequences against an in-house fish protein database of protein sequences from Zebrafish, Fugu, Stickleback, Tetraodon and Medaka that were downloaded from the Ensembl database [40]. The sequences that had no BLASTX hits to the sequences in the fish protein database were searched against the UniProt database [41] and the NCBI nonredundant protein database [42] using BLASTX with an e-value of 1e-5. Contigs that had no matches to either of these protein databases were aligned against UTRdb [22] because these sequences might represent the UTRs of protein-coding genes. Contigs that had hits to UTR sequences were considered to be protein-coding genes. These remaining unmatched contigs were searched against NCBI nonredundant nucleotide collection using BLASTN with an e-value of 0.05.
Because common carp might have some species-specific protein-coding genes, the contigs that had no matches to any of the known proteins or nucleotide sequences were run through the Coding Potential Calculator (CPC) [43] to predict their coding potential. If CPC predicted that the contig was a coding gene or if it predicted that the contig was non-coding but had an intact open reading frame over 100 amino acids long, then the contig was considered to be a protein-coding gene.
To compare the common carp contigs with the zebrafish genes, we downloaded 45,646 zebrafish protein-coding transcripts from the Ensembl database [40] and selected the longest transcript to represent each gene so as to avoid redundant GO comparison and ortholog identification.
Estimation of Ks in orthologs and paralogs
To identify putative orthologs between common carp and zebrafish, the sequences from common carp and zebrafish were aligned using the reciprocal BLAST (BLASTN) hit method of Blanc et al. [16] with an e-value of 1e-20. Two sequences were defined as orthologs if each of them was the best hit of the other and if the sequences were aligned over 300 bp.
The approach used to estimate the Ks of orthologous pairs was adapted from previous studies [15,16]. Briefly, we aligned the common carp contigs to its orthologous zebrafish protein sequence using BLASTX. The longest alignment was selected for analysis. The corresponding common carp sequences were extracted using their aligned coordinates and translated with the getorf program from the EMBOSS package [44]. The translated carp amino acid sequences were aligned against the orthologous zebrafish protein using Clustalw [45]. The corresponding codon alignments were produced using PAL2NAL [46]. Finally, Ks were estimated using a maximum likelihood method in the CODEML program (runmode-2) of the PAML package [47].
Paralogs within the common carp and zebrafish sequences were identified by all-against-all BLASTN searches. Because of length variations between the sequences, we modified the procedure of Ding et al. [48] and defined two sequences as paralogs if the aligned regions were over 70% of the shorter sequences. To estimate the Ks for common carp paralogs, we first aligned the sequence pairs using TBLASTX and then followed the same steps that were used in the Ks estimation pipeline in orthologous pairs. Because zebrafish genes have well-annotated protein sequences, the protein sequences of two paralogous genes were aligned with Clustalw [45] and the corresponding codon alignments were produced using PAL2NAL [46]. Ks was estimated based on the codon alignments.
Detection of genome speciation and duplication events
The detection of genome speciation and the duplication event was carried out according to the procedure described by Blanc et al. [16] which were based on the Ks distribution of orthologous and paralogous pairs, respectively. For detection, we used only those alignments that were longer than 30 amino acids and had Ks < 2 to minimize statistical artifacts caused by short alignments and the saturation of Ks [17].
The Ks frequencies of orthologous pairs were plotted and the Ks peak was used as a potential indicator of the genome speciation event. To detect genome duplication event, the common carp paralogous pairs were organized into gene families according to previous studies [15,16]. One contig was selected to represent one gene cluster, obviating possible redundant Ks from multiple entries of the same gene. A gene family of n members was assumed to be the results of n - 1 gene duplication events. A hierarchical clustering method described previously [16], was used to reconstruct the tentative phylogeny of each gene family. Briefly, in one gene family, all contigs were treated first as a separate cluster. Then, the two clusters (A and B) with the smallest Ks values were grouped into a new cluster containing all the contigs. The median Ks, obtained for all possible pairs between a contig in cluster A and a contig in cluster B, was used as the Ks for the new cluster. Every clustering was assumed to represent one gene duplication event. These steps were repeated until all the contigs were grouped into a single cluster. The obtained Ks values that were obtained in each clustering were plotted and the Ks peak was assumed to indicate the genome duplication event.
Biological process and pathway enriched in common carp compared with zebrafish
To study the biological processes enriched in common carp, we annotated each contig by assigning the GO terms associated with the top hit in the fish protein database to the common carp contig. If a contig could not be annotated with a homologous assignment, then Interproscan [24] was used with the default settings to annotate the contigs with GO terms. To study the pathways involved, we used the KOBAS software [49] to map common carp contigs to KEGG pathways based on sequence similarity.
We retrieved the GO terms for the zebrafish protein-coding genes from Ensembl database and kept non-redundant GO terms for every zebrafish gene because redundant GO terms might be assigned to the gene spliced variants. We used WEGO [50] to identify significantly enriched GO terms in the common carp contigs using zebrafish genes as the background. WEGO uses the Pearson Chi-Square test to indicate significant relationships between two input datasets. The biological process terms with p-value ≤ 0.05 were considered to be statistically enriched in common carp contigs. To study the enriched pathways by common carp, we assigned KEGG pathways to the zebrafish genes using KOBAS [49] and then used the software to compare the proportion of common carp contigs in each pathway against the proportion of zebrafish genes in the same pathway. KOBAS used fisher-exact test to identify significant pathways and then performed an FDR correction to reduce Type-1 errors. KEGG pathways with corrected p values ≤ 0.05 were considered to be statistically enriched in common carp.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JTW and JTL conducted the bioinformatic analysis and were involved in writing the manuscript. XFZ was responsible for samples preparation and 454 pyrosequencing. JTL and XWS supervised the entire study and provided assistance in manuscript preparation. All authors read and approved the final manuscript.
Supplementary Material
Table S1 Twenty pairs of published common carp paralogs.
Methods S1 Sample preparation and PCR validation of selected contigs.
Table S2 Primers designed specifically for the selected contigs that we assembled.
Figure S1 PCR products of the selected contigs.
Figure S2 Spliced variants detected under different alignment lengths using zebrafish variants as a test dataset.
Contributor Information
Jin-Tu Wang, Email: wangjintu_104@yahoo.com.cn.
Jiong-Tang Li, Email: lijt83@gmail.com.
Xiao-Feng Zhang, Email: zhangxf.yu@163.com.
Xiao-Wen Sun, Email: sunxw2002@163.com.
Acknowledgements
We thank Edanz group for manuscript editing. This study was supported by the grants from Research Foundation of Chinese Academy of Fishery Sciences (No. 2011 C016, No. 2012C014), China Ministry of Agriculture 948 Program (2011-G12), China Ministry of Science and Technology 863 Hi-Tech Research and Development Program (No. 2011AA100401) and National Department Public Benefit Research Foundation (No. 200903045). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Venkatesh B. Evolution and diversity of fish genomes. Current opinion in genetics & development. 2003;13(6):588–592. doi: 10.1016/j.gde.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL. Zebrafish hox clusters and vertebrate genome evolution. Science. 1998;282(5394):1711–1714. doi: 10.1126/science.282.5394.1711. [DOI] [PubMed] [Google Scholar]
- Meyer A, Mlaga-Trillo E. Vertebrate genomics: More fishy tales about Hox genes. Current biology. 1999;9(6):R210–R213. doi: 10.1016/S0960-9822(99)80131-6. [DOI] [PubMed] [Google Scholar]
- Taylor JS, Van de Peer Y, Braasch I, Meyer A. Comparative genomics provides evidence for an ancient genome duplication event in fish. Philosophical Transactions of the Royal Society of London Series B: Biological Sciences. 2001;356(1414):1661–1679. doi: 10.1098/rstb.2001.0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Okamoto N, Ikeda Y. Two c-myc genes from a tetraploid fish, the common carp (Cyprinus carpio) Gene. 1995;153(2):231–236. doi: 10.1016/0378-1119(94)00813-8. [DOI] [PubMed] [Google Scholar]
- Larhammar D, Risinger C. Molecular genetic aspects of tetraploidy in the common carp Cyprinus carpio. Mol Phylogenet Evol. 1994;3(1):59–68. doi: 10.1006/mpev.1994.1007. [DOI] [PubMed] [Google Scholar]
- David L, Blum S, Feldman MW, Lavi U, Hillel J. Recent duplication of the common carp (Cyprinus carpio L.) genome as revealed by analyses of microsatellite loci. Mol Biol Evol. 2003;20(9):1425–1434. doi: 10.1093/molbev/msg173. [DOI] [PubMed] [Google Scholar]
- Williams D, Li W, Hughes M, Gonzalez S, Vernon C, Vidal M, Jeney Z, Jeney G, Dixon P, McAndrew B. Genomic resources and microarrays for the common carp Cyprinus carpio L. Journal of Fish Biology. 2008;72(9):2095–2117. doi: 10.1111/j.1095-8649.2008.01875.x. [DOI] [Google Scholar]
- Kawano H, Kono T, Watanuki H, Savan R, Sakai M. Analysis of genes expressed in head kidney of common carp Cyprinus carpio L treated with cortisol. Comp Biochem Physiol B Biochem Mol Biol. 2003;136(4):875–886. doi: 10.1016/S1096-4959(03)00281-1. [DOI] [PubMed] [Google Scholar]
- Gracey AY, Fraser EJ, Li W, Fang Y, Taylor RR, Rogers J, Brass A, Cossins AR. Coping with cold: an integrative, multitissue analysis of the transcriptome of a poikilothermic vertebrate. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(48):16970–16975. doi: 10.1073/pnas.0403627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez SF, Chatziandreou N, Nielsen ME, Li W, Rogers J, Taylor R, Santos Y, Cossins A. Cutaneous immune responses in the common carp detected using transcript analysis. Mol Immunol. 2007;44(7):1664–1679. doi: 10.1016/j.molimm.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Parchman T, Geist K, Grahnen J, Benkman C, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010;11(1):180. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JE, Guelbeogo WM, Sanou A, Traor A, Vernick KD, Lazzaro BP. De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PLoS One. 2010;5(12):e14202. doi: 10.1371/journal.pone.0014202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen SD, Karlsen BO, Furmanek T, Andreassen M, Jorgensen TE, Bizuayehu TT, Breines R, Emblem A, Kettunen P, Luukko K. RNA deep sequencing of the Atlantic cod transcriptome. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics; 2010. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Rogers WL, Breton CM, Cai L, Malmberg RL. Transcriptome Analysis of Sarracenia, an Insectivorous Plant. DNA Research. 2011;18(4):253–261. doi: 10.1093/dnares/dsr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. The Plant Cell Online. 2004;16(7):1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH, Ere-Walker A. Molecular evolution. Sinauer Associates Sunderland (Massachusetts); 1997. p. 11. [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24(24):2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome research. 1999;9(9):868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Wang Z. Next-generation transcriptome assembly. Nature Reviews Genetics. 2011;12(10):671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S. Ensembl 2012. Nucleic acids research. 2012;40(D1):D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesole G, Liuni S, Grillo G, Ippedico M, Larizza A, Makalowski W, Saccone C. UTRdb: a specialized database of 5¡ä and 3¡ä untranslated regions of eukaryotic mRNAs. Nucleic acids research. 1999;27(1):188–191. doi: 10.1093/nar/27.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk CP, Hermsen TT, Westphal AH, Savelkoul HFJ, Stet RJM. A novel functional class I lineage in zebrafish (Danio rerio), carp (Cyprinus carpio), and large barbus (Barbus intermedius) showing an unusual conservation of the peptide binding domains. The Journal of Immunology. 2002;169(4):1936–1947. doi: 10.4049/jimmunol.169.4.1936. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan" Can integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Yinnan M, Feng D, Peng C, Jingqun A, Songnian H, Xinhua C. Transcriptome and expression profiling analysis revealed changes of multiple signaling pathways involved in immunity in the large yellow croaker during Aeromonas hydrophila infection. BMC Genomics. 2010;11(1):506. doi: 10.1186/1471-2164-11-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem M, Rexroad CE, Wang J, Thorgaard GH, Yao J. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics. 2010;11:564. doi: 10.1186/1471-2164-11-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TPL, Grosse WM, Freking BA, Roberts AJ, Stone RT, Casas E, Wray JE, White J, Cho J, Fahrenkrug SC. Sequence evaluation of four pooled-tissue normalized bovine cDNA libraries and construction of a gene index for cattle. Genome research. 2001;11(4):626–630. doi: 10.1101/gr.170101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt R, Pinheiro M, Egas C, Gomes P, Afonso M, Shank T, Santos R. High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics. 2010;11(1):559. doi: 10.1186/1471-2164-11-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der Joshua BM, Norman W, dePamphilis Claude WP. De novo characterization of the gametophyte transcriptome in bracken fern, Pteridium aquilinum. BMC Genomics. 2011;12(1):99. doi: 10.1186/1471-2164-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima Y, Yamamoto K. Cellular DNA contents of fishes determined by flow cytometry. La Kromosomo II. 1990;57:1871–1888. [Google Scholar]
- Tiersch TR, Chandler RW, Wachtel SS, Elias S. Reference standards for flow cytometry and application in comparative studies of nuclear DNA content. Cytometry. 1989;10(6):706–710. doi: 10.1002/cyto.990100606. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Meyer E, Aglyamova G, Wang S, Buchanan-Carter J, Abrego D, Colbourne J, Willis B, Matz M. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 2009;10(1):219. doi: 10.1186/1471-2164-10-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary demography of duplicate genes. Journal of Structural and Functional Genomics. 2003;3(1):35–44. doi: 10.1023/A:1022696612931. [DOI] [PubMed] [Google Scholar]
- Patrick B. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Blaxter M. Comparing de novo assemblers for 454 transcriptome data. BMC Genomics. 2010;11(1):571. doi: 10.1186/1471-2164-11-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA. Database resources of the National Center for Biotechnology. Nucleic acids research. 2003;31(1):28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- seqclean. http://compbio.dfci.harvard.edu/tgi/software/
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T. The Ensembl genome database project. Nucleic acids research. 2002;30(1):38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M. The universal protein resource (UniProt) Nucleic acids research. 2005;33(suppl 1):D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S. Database resources of the national center for biotechnology information. Nucleic acids research. 2011;39(suppl 1):D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic acids research. 2007;35(suppl 2):W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends in Genetics. 2000;16(6):276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic acids research. 2006;34(suppl 2):W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Ding G, Sun Y, Li H, Wang Z, Fan H, Wang C, Yang D, Li Y. EPGD: a comprehensive web resource for integrating and displaying eukaryotic paralog/paralogon information. Nucleic acids research. 2008;36(suppl 1):D255–D262. doi: 10.1093/nar/gkm924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic acids research. 2011;39(2):316–322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L. WEGO: a web tool for plotting GO annotations. Nucleic acids research. 2006;34(suppl 2):W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Twenty pairs of published common carp paralogs.
Methods S1 Sample preparation and PCR validation of selected contigs.
Table S2 Primers designed specifically for the selected contigs that we assembled.
Figure S1 PCR products of the selected contigs.
Figure S2 Spliced variants detected under different alignment lengths using zebrafish variants as a test dataset.