

Abstract
Evolutionary biologists have largely left the search for solutions to the drug resistance crisis to biomedical scientists, physicians, veterinarians and public health specialists. We believe this is because the vast majority of professional evolutionary biologists consider the evolutionary science of drug resistance to be conceptually uninteresting. Using malaria as case study, we argue that it is not. We review examples of evolutionary thinking that challenge various fallacies dominating antimalarial therapy, and discuss open problems that need evolutionary insight. These problems are unlikely to be resolved by biomedical scientists ungrounded in evolutionary biology. Involvement by evolutionary biologists in the science of drug resistance requires no intellectual compromises: the problems are as conceptually challenging as they are important.
Keywords: antibiotic resistance, drug resistance, malaria, Plasmodium
Introduction
Drug resistance causes immense human suffering globally and is one of the best documented examples of evolution in real time. No self-respecting introductory evolution text fails to mention this, and several professional evolution societies give this as a major practical argument for the teaching of evolution and continued investment in evolutionary science (Meagher and Futuyma 2001). Yet the drug resistance field is – with a few outstanding exceptions – dominated by people with no training in evolutionary biology. Indeed, the microbiologists, clinicians and public health practitioners who publish on drug resistance do not even much use the word ‘evolve’– they more naturally use ‘emerge’, ‘spread’ or ‘arise’ (Antonovics et al. 2007). The vast majority of publications on the evolution of antibiotic resistance are in the medical field and not in academic evolutionary biology or genetics journals (Antonovics et al. 2007).
Why do so few professional evolutionary biologists work on drug resistance evolution, particularly given the commercial and grant money involved? This is certainly a specific instance of the remarkable antipathy of most evolutionary biologists to utilitarian science (the journal Evolutionary Applications appeared 17 years after the first issue of Ecological Applications), an antipathy historians of science have yet to explain. But in the case of drug resistance, the overwhelming volume of data does make assimilating the relevant natural history a challenge, especially as the data are elucidated by physicians, veterinarians, microbiologists and public health specialists, so that a foreign, often pathogen-specific jargon and intellectual culture dominates. Moreover, drug resistance, like many biomedical problems, is perhaps not so interesting to those attracted to evolutionary biology by a passion for ‘natural’ natural history.
But we think it goes deeper than this. Based on an entirely ad hoc, nonrandom sample of our colleagues (largely the evolutionary biologists we meet at conferences), we believe the main reason evolutionary biologists avoid drug resistance is that evolutionary biologists consider drug resistance to be conceptually uninteresting. And at one level it is. As Antonovics et al. (2007) point out, ‘the evolution of antibiotic resistance, while critically important from a medical view point, is no longer in and of itself a novel finding in evolutionary biology’. This is true of course, but the evolutionary processes which determine patterns of drug resistance are a different issue. Our straw poll reveals that most professional evolutionary biologists consider these processes conceptually simple (mutation, selection, fixation), and any solutions largely obvious (combination therapy, reduced drug use). The general feeling seems to be that drug resistance provides excellent examples with which to begin evolution classes and introductory textbooks, and an excellent vehicle to get across basic population genetics. But it is not believed to be an intellectually challenging pursuit around which to structure an interesting evolutionary research program.
Here we attempt to counter this view. We believe there is a strong case for advanced classes in drug resistance evolution, and also that there are numerous conceptually challenging problems in drug resistance evolution to which evolutionary biologists can make unique contributions. The solution of these problems would both be intellectually rewarding and could reduce human suffering. We make this case using malaria, with which we are most familiar, but we believe similar arguments can be made for almost any infectious disease.
We illustrate our case by reviewing a series of fallacies which were, or still are, held by the malaria community (albeit here translated into evolutionary language), and we finish with a number of very open evolutionary research questions. Throughout, we have picked examples which we think are both interesting and challenging, and which demonstrate the practical contribution evolutionary biology is making, or could make, to help alleviate the medical problems caused by drug resistance. By way of an aside, we emphasize that none of this is intended as an argument against fundamental evolutionary research (clearly everything we discuss here builds on that foundation), or a critique of those biologists – evolutionary or otherwise – currently engaging with drug resistance. Our point is that the proportion of evolutionary biologists working on drug resistance is tiny compared with the importance and size of the problem – and the conceptual interest of the issues involved.
The malaria drug resistance problem
Malaria parasites have evolved resistance to all classes of antimalarials that have gone into widespread use, except for the recently deployed artemisinin derivatives (Roll Back Malaria 2008). Resistance was first reported from the field between 1 and 15 years after introduction, depending on the drug (Fig. 1; Peters 1987, Hyde 2005) with drugs failing (i.e. being withdrawn from use by national authorities) years or even decades after that. For instance, chloroquine was widely deployed after the Second World War, with resistance first seen in the field in 1957 in Thailand (Talisuna et al. 2004). Molecular evolution studies show that chloroquine resistance arose only a handful of times, from which it spread world wide (Fig. 2A). It never arose in Africa. Chloroquine was first withdrawn as a first line drug from Thailand in 1973 (Talisuna et al. 2004) and is now recommended only for central America, where parasites are still susceptible (WHO 2008). High level sulphadoxine–pyrimethamine (SP) resistance was observed within the same year as it was introduced in Thailand in 1967 (Talisuna et al. 2004), but replaced chloroquine as first line treatment in most African countries in the early 1990s. Resistance against SP is now widespread. Similar to chloroquine resistance, the major cause of SP resistance in Africa is thought to be a consequence of a selective sweep from a single introduction from southeast Asia (Fig. 2B) (Roper et al. 2004). There may also have been an African origin in Kenya (McCollum et al. 2006) which seems not to have spread far. Current hopes rest on artemisinin and its derivatives, which have become the key element in current malaria control plans (Roll Back Malaria 2008). Artemisinins are used in co-formulations with other antimalarial agents (artemisinin combination therapy, ACT) in an attempt to minimize the chances of resistance arising. So far this seems to be working, although there are recent reports of parasites with reduced sensitivity to some ACTs (White 2008) and artemisinin resistance can be readily generated in the laboratory (Afonso et al. 2006; Puri and Chandra 2006).
Figure 1.

History of the introduction of antimalarial drugs and the first detection of resistance in the field. The following abbreviations are used: CQ, chloroquine; PG, proguanil; Pyr, pyrimethamine; SP, sulphadoxine–pyrimethamine; Mef, mefloquine; Hal, halofantrine; ACTs, artemisinin combination therapies; Ato, atovaquone; Ato-PG, atovaquone–proguanil combination (malarone); LD, LapDap (chlorproguanil–dapsone). R as suffix denotes resistance. Figure redrawn from Hyde (2005).
Figure 2.
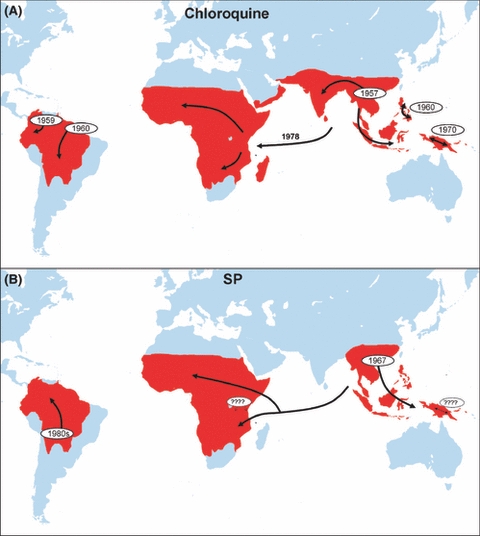
The history of chloroquine and high level pyrimethamine–sulphadoxine (SP) resistance inferred from molecular evolution studies. Chloroquine resistance has spread globally from selective sweeps from five independent origins, none of them in Africa where the health burden of drug resistance is greatest. Resistance to SP is tracked by analyses of the dhfr gene which primarily confers resistance to the pyrimethamine component. The timing of two of the independent origins is unclear. SP resistance may have several local origins in Kenya (denoted by ‘????’), but the majority of SP-resistant infections are a consequence of a selective sweep from a single origin in South East Asia. Figure 2A is redrawn from Wellems (2004), Fig. 2B is a summary of data from Cortese et al. (2002), Nair et al. (2003), Roper et al. (2003, 2004), McCollum et al. (2006, 2007, 2008), Maiga et al. (2007), Mita et al. (2007), Hayton and Su (2008), Saito-Nakano et al. (2008).
The evolution of drug resistance by malaria parasites is now accepted as inevitable by the World Health Organization (WHO 2006), and a key component of the recently released global malaria action plan (GMAP) is an explicit plan for a drug delivery pipeline – intended as open-ended so long as malaria parasites still exist (Roll Back Malaria 2008). This is the so called ‘drug treadmill’, the rolling-out of new drugs which will inevitably fail in the face of parasite evolution. The GMAP estimates of the cost of this pipeline are as follows. The basic research budget is estimated as $US34 million per year, and the development cost of bringing a new compound to market at $US250 million over 10 years. The budget for reformulation of compounds already in use (new combinations for instance) is put at $US25 million over 2–6 years. Given the rate at which existing drugs are rendered useless by evolution (in many cases, faster than the speed with which new compounds can go through regulatory processes), and the few useful compounds currently available, GMAP estimates that two new active ingredients for preventive therapy and six new ingredients for therapeutic use will need to be discovered and brought to market in the coming decade. Over the same period, 14 reformulations of existing and new compounds will need to be developed. Thus, the costs of the pipeline for R&D alone (i.e. excluding the production and deployment costs) will be in excess of $US2.5 billion for the coming decade to get things up to speed. Once the currently inadequate drug arsenal is rebuilt, $US1.5 billion will be required every decade that follows. These are incredible amounts of money for a disease affecting some of the poorest people on the planet.
The challenge for evolutionary biologists is to devise ways to slow the drug treadmill. This includes the demolition of any evolutionarily-naïve medical orthodoxies which drive the treadmill faster. The speed of the treadmill is set primarily by the rate at which mutations conferring resistance escape the clutches of stochastic loss and establish in a population, and then the rate at which they subsequently spread. The WHO considers a drug ineffective once 10% of the parasites in a population have become resistant (WHO 2006, p. 15). Reviews of malaria drug resistance from a population genetics perspective are provided by Hastings and D'Alessandro (2000), Hastings (2001), Koella and Antia (2003), Barnes and White (2005) and Mackinnon (2005), and from the more dominant drug discovery, biochemistry or pharmacokinetic perspective by Hastings et al. (2002), White (2004), Hyde (2005), Barnes et al. (2008), Hayton and Su (2008) and Stepniewska and White (2008). The current WHO guidelines for drug use at national and individual patient levels are published by the WHO (2006).
Fallacy 1: Drugs active against transmission stages slow the spread of resistance
It is apparently conventional wisdom among malariologists that the spread of resistance can be slowed by drugs which target the parasite stages responsible for infecting mosquitoes (sexual stages called gametocytes). For instance Mendez et al. (2002, p. 237) state that ‘Antimalarial drugs and drug combinations designed to eliminate both asexual and sexual parasites may deserve priority […] because they will reduce the spread of drug resistance in its earliest stages’. Similarly, the WHO (2006, p. 141) says that ‘Reducing transmission is fundamental to the curtailment of drug resistance’, and Barnes and White (2005, p. 230) state that ‘...reducing the carriage of gametocytes […] is necessary to limit the transmission of malaria parasites and the spread of antimalarial resistance’. The intuition behind this orthodoxy is that attacking transmission stages reduces the chances of transmitting a resistant mutant.
But as Hastings (2006a) has pointed out, this argument makes little evolutionary sense. It is true that gametocytocidal drugs will reduce transmission, but they will do so most strongly for sensitive parasites. The relative fitness of resistant and sensitive strains determines the rate of spread of resistance, and this will be increased by drugs targeting transmission stages. Imagine an individual infected with susceptible parasites and a few resistant mutants. If drug treatment kills all susceptible gametocytes, only the few resistant gametocytes will remain. Now imagine treatment with a drug which kills only the replicative (asexual) stages. Susceptible gametocytes remain viable in the blood for weeks, and so will co-occur with the resistant gametocytes. The relative fitness of the resistant mutants is lower in the second scenario compared to the first one. All else being equal, gametocytocidal drugs will enhance the rate at which resistant parasites rise in frequency in a population.
This can turn into a significant effect because small relative fitness differences compound through time. Using a population genetics model, Hastings (2006a) has shown that where drug use is common in a population, the enhanced fitness advantage conferred by gametocytocidal drugs on resistant strains can reduce the useful therapeutic lifespan of a drug by about a year (15%) compared to nongametocytocidal therapy. There may still be sound reasons for using drugs which target transmission stages (e.g. reductions in infectiousness, reducing case incidence, or an incidental side-effect of high lethality against blood stages), but resistance management is not one of them. Indeed, these other reasons need to be weighed against the enhanced resistance evolution that such drugs will prompt.
Fallacy 2: Drugs with long half lives are preferable
Drugs that are slowly eliminated from the body after treatment have several clinical advantages. Clearly, they provide longer term protection against re-infection. For SP, this can be up to 2 months, which in a high transmission region can help prevent novel infections interfering with recovery or generating new symptoms. Slowly eliminated drugs also require fewer administrations to achieve clearance, reducing problems of patient compliance. However, as Watkins and Mosobo (1993) pointed out, drugs with long clearance times also impose stronger selection for drug resistance. This is because, for similar treatment rates, parasites are substantially more likely to encounter drugs with long half lives. If a course of artesunate persists for 5 days, the drug pressure exerted by SP is 10 times greater (Hastings et al. 2002). Drug half life is typically left out of models of drug resistance, yet it may be one of the most powerful determinants of the useful lifespan of a drug (Hastings et al. 2002). From the resistance management perspective, drugs which are rapidly eliminated from the body are preferable.
An important corollary of this argument is that half lives of drugs used for combination therapy should be similar (Hastings et al. 2002; Hastings and Watkins 2006). The more dissimilar the elimination rates, the greater the chances that resistance to one of the component compounds can become established in a population, thus effectively reducing combination therapy to monotherapy. To achieve clearance with artemisinins alone takes a 7-day treatment regime. Because adherence to a 7-day course is typically poor, the current WHO policy is to combine it with a slowly eliminated antimalarial drug (WHO 2006; White 2008). Recent reports of the failure of these combinations seem to be due to the failure of the partner compound (e.g. Wongsrichanalai and Meshnick 2008). This is likely to continue whenever the partner is a slowly cleared compound. If so, rapid reformulation of ACTs is going to be an open-ended necessity, or there will need to be an abandonment of the aim of complete clearance following ACT.
Fallacy 3: De novo resistance mutations are the main enemy
Current malaria treatment guidelines for uncomplicated malaria are radical parasitologic cure (WHO 2006). This is achieved by the administration of sufficiently high and repetitive drug dosages to ensure a kill of every parasite in an infection, and recommended patient treatment regimes are explicitly designed to do this. A major motivator behind this is that ‘[r]esistance can be prevented, or its onset slowed considerably, by […] ensuring very high cure rates through full adherence to correct dose regimens’ (WHO 2006, p. 12). The underlying reasoning is that complete parasitologic cure (i) reduces parasite biomass and thus the chances of resistance mutations occurring (e.g. White 2004; WHO 2006, p. 165), and (ii) minimizes the number of parasites exposed to sub-curative drug dosages which favor ‘tolerant’ parasites (e.g. Hastings and Watkins 2006). Tolerant parasites are mutants which are not fully resistant but are able to survive subcurative doses and so are a mutational step towards full resistance (Hastings and Watkins 2006).
However, there are very few data demonstrating that resistance arising de novo within a patient is a clinically relevant source of drug failure in malaria patients. Indeed, as we summarized above, the evolutionary history of resistance to two of the major antimalarials, chloroquine and SP, argues that it is effectively zero. Resistance to both drugs arose just a handful of times and spread worldwide (Fig. 2). Indeed, chloroquine resistance seems never to have arisen de novo in Africa: it was imported from Asia. So far as is known, every patient in Africa with chloroquine-resistant parasites got them from other people, never from mutational processes within their own infections. Most high-level SP resistance in Africa was similarly due to a single selective sweep of resistance introduced from SE Asia (Fig. 2).
Given this, the widespread conventional wisdom that patients should take a full course of chloroquine to slow resistance evolution makes little sense. Indeed, chloroquine resistance clearly failed to arise in Africa despite widespread underdosing as a consequence of the economically driven noncompliance and low quality drugs (Djimde et al. 1998; Goodman et al. 2007; Bate et al. 2008). Moreover, the recommended patient treatment regimes of overwhelming drug treatment, way beyond what is needed on clinical grounds, imposes the strongest possible selection in favor of resistance, possibly for little clinical gain. Indeed, there is an inconsistency at the heart of the current WHO (2006) guidelines. Correctly, there is a strong argument for reducing unnecessary use of antimalarials at a population level, so as to minimize selection for resistance. In contrast, the recommendation at the single patient level is overwhelming drug use even when there is no clinical need. This maximizes selection for resistance.
De novo resistance may not be irrelevant for all antimalarials, and where single point mutations confer high level resistance as, for example against atovaquone (White 2004), de novo mutations may be a serious issue clinically. But for at least high level chloroquine and SP resistance, for which there is the best data on the evolutionary history, resistance arose so rarely that the de novo origin of resistance can be ignored as a clinical concern. The explanation for the rare origins is almost certainly because complete resistance with high viability involves multiple mutations (Hastings and Watkins 2006), and so requires a highly unlikely series of mutational events to occur simultaneously. Current combination therapy recommendations are – rightly – designed to make artemisinin resistance similarly unlikely (WHO 2006). When resistance against artemisinins does arise, as it inevitably will, WHO will need to consider patient treatment regimes that will minimize the spread of resistance – not to continue to manage individual malaria patients against the extraordinarily unlikely possibility that every patient will be the source of a second origin. There is no strong argument for treating malaria as if it were a highly mutable pathogen like HIV, and nor is it a bacterium which can easily acquire resistance by lateral gene transfer.
Fallacy 4: Genetic trade-offs alone determine the magnitude of the costs of resistance
Much circumstantial evidence suggests that resistant malaria parasites have a lower fitness than sensitive parasites in the absence of chemotherapy (Walliker et al. 2005; Felger and Beck 2008). Suggestive evidence for a cost of resistance comes from progressive increases in drug sensitivity in populations where drug use has been discontinued. This has been seen in Malawi (Kublin et al. 2003; Mita et al. 2003; Laufer et al. 2006), Tanzania (Temu et al. 2006), South-Africa (Raman et al. 2008), Thailand (Thaithong et al. 1988), and China (Liu et al. 1995), although there are also areas where a decrease of resistance has not been observed (e.g. McCollum et al. 2007; Yang et al. 2007). Seasonal variation in the frequency of resistant alleles in eastern Sudan and The Gambia is also consistent with costs of resistance. When there is low to no transmission during the dry season, and hence few new malaria cases and essentially no drug use, resistance alleles drop in frequency among the chronically infected patients who source the next outbreak. During the wet season, when high transmission ensures many new disease cases and hence high drug usage, resistance alleles rise in frequency (Abdel-Muhsin et al. 2004; Ord et al. 2007).
As in other pathogens, costs of resistance in malaria presumably arise from the metabolic costs of efflux or detoxification, or reduced biochemical efficiency associated with target site mutations (Hastings and Donnelly 2005); in other words, genetic trade-offs. Most models of malaria drug resistance evolution recognize these costs of resistance, but, if included at all, they are typically taken as a fixed and relatively modest parameter (e.g. a selective disadvantage s, so that the fitness of resistant mutant is 1 − s, where s in the order of 0.1 or less). Although not much discussed, we believe there is a widely held assumption that these costs can be mitigated by compensatory mutations, as they can be in bacteria and HIV (Levin et al. 2000), so that s can drop further through time. Such selection processes might explain some of the sequential mutational steps associated with chloroquine and SP resistance (Hastings and Donnelly 2005; Hastings and Watkins 2006).
Yet the natural history of malaria makes it highly unlikely that the costs of resistance can be captured by a fixed parameter like ‘s’, and moreover suggests that the costs can often be much larger under some ecologic circumstances. This is because the costs of resistance are a function of the interactions between coinfecting strains within the host. Indeed, this in-host ecology maybe the primary determinant of the magnitude of the costs of resistance. The natural history is as follows.
Human malaria infections frequently consist of more than one Plasmodium genotype (Arnot 1998; Babiker et al. 1999; Smith et al. 1999; Bruce et al. 2000; Jafari et al. 2004), so that coexistence of sensitive and resistant parasites is common – and indeed may even be the rule, especially when resistance is beginning to spread through a population. Mixed infections arise from inoculations of genetically diverse parasites by a single mosquito, or contemporaneous bites by multiple mosquitoes infected with different parasites.
A substantial body of correlational epidemiologic evidence is consistent with crowding effects within infections, where population densities of individual genotypes are suppressed when other genotypes are present (Daubersies et al. 1996; Mercereau-Puijalon 1996; Smith et al. 1999; Bruce et al. 2000; Hastings 2003; Talisuna et al. 2006). Direct experimental evidence of crowding cannot be ethically obtained from human infections, but in the rodent malaria model Plasmodium chabaudi in laboratory mice, we and others have experimentally demonstrated that strong crowding effects occur. Replicative and transmission stage densities of individual clones within an infection are severely suppressed when coinfecting strains are present (e.g. Jarra and Brown 1985; Taylor et al. 1997; de Roode et al. 2004, 2005; Bell et al. 2006; Wargo et al. 2007). Competitive suppression within hosts also substantially reduces transmission of individual clones to mosquitoes (de Roode et al. 2005). Therefore, the removal of sensitive strains by chemotherapy leads to competitive release of resistant strains (de Roode et al. 2004; Wargo et al. 2007).
We have found in our experiments with rodent malarias that differences in clone performance are greatly magnified by this crowding effect. An example is given in Fig. 3. Pyrimethamine-resistant and sensitive clones are shown. Alone, the resistance clone produces fewer transmission stages. However, when the two clones coinfect the same host, the difference is amplified by clonal competition. We are currently doing experiments to see whether this competitive disadvantage increases as more sensitive coinfecting strains are added. In high transmission regions, infections can consist of five or more clones; if crowding increasing with the number of clones, the fitness disadvantage of resistance could substantially increase with the force of infection.
Figure 3.
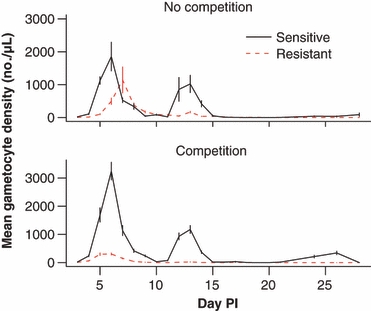
An example of how competition between parasites within infections magnifies differences in performance between sensitive and resistant parasite lines. Top panel – the performance of the two clones when in separate infections. Bottom panel – the performance of the two clones together in the same infection. The lower performance of the resistant clone is further lowered by competition. Plotted points are the mean (±SEM) density of transmission stages of Plasmodium chabaudi in peripheral blood through time from three to six laboratory mice per group (S. Huijben, A. R. Wargo, B. H. K. Chan, D. Drew, A. F. Read, unpublished data). Parasite densities were quantified by real time quantitative RT-PCR (Drew and Reece 2007).
Thus, the within-host ecology is likely to be a primary determinant of the strength of selection of any resistant mutant in the absence of chemotherapy: the ecologic circumstances can magnify fitness differences way beyond those due simply to ecology-independent genetic trade-offs (Hastings and D'Alessandro 2000; Mackinnon 2005; Hastings 2006b). Except perhaps where single clone infections dominate (as can be the case in low transmission regions; Arnot 1998), infection context is almost everything. Within-host genetic diversity is in turn determined by the epidemiology (force of infection), since this is what determines frequency of mixed infections in a population. We suggest that standard population genetics approaches to model drug resistance are likely to be of very limited value in malaria. Explicit evolutionary epidemiologic models (Restif 2009) are needed instead. They are in their infancy in this context (e.g. Hastings 2006b), but there is no escaping this complexity: the epidemiology determines the strength of selection and hence the evolution, and the evolution in turn determines the epidemiologic dynamics.
A highly contentious and unorthodox possibility is suggested by the above considerations (Wargo et al. 2007). Crowding by drug sensitive parasites will suppress transmission stage densities of resistant parasites in untreated hosts. This suggests it might be possible to harness these within-host dynamics for human benefit: the use of patient treatment regimes which do not remove all the sensitive parasites may restrict the transmission of resistance. Some evidence that this might be feasible comes from one of our experiments with rodent malaria (Wargo et al. 2007). We found that treating mice with half the normal dose of antimalarials alleviated the symptoms as effectively as a full dose, but a degree of in-host competition was retained, with the consequence that the transmission potential of the resistant clone was significantly less than in mice given standard doses. Considerably more work needs to be done to evaluate the merits of abandoning the parasitologic cure orthodoxy which currently form the basis of WHO (2006) patient treatment guidelines, but we note that overwhelming chemotherapy is also the way to most effectively up-select resistant mutants in laboratory settings (e.g. Peters 1987), and that host immunity can very effectively clear parasites, especially following drug treatment (Cravo et al. 2001). The theoretical and experimental analysis of the possibility of optimizing patient treatment regimes with respect to both patient health and resistance management is long overdue. For instance, would the best regime actually be what is currently considered heretical: take drug treatment until the patient feels better, then further treatment if there are any symptom-associated relapses?
Fallacy 5: Fixation of resistance is inevitable if drug pressure is maintained
A very interesting implication of in-host competition is that the costs of resistance must be frequency-dependent. When a resistant mutant first becomes established in a population, it will typically share its host with competitively more able sensitive strains. As resistance becomes more frequent, resistant strains will increasingly share their hosts with other resistant strains. Because competition magnifies differences in competitive ability as described above, this means that the costs of resistance will be highest early in the spread process, and will decline as resistant strains are increasingly likely to be competing with strains with similar competitive abilities.
Moreover, the benefits of resistance will be similarly frequency dependent. For malaria, the benefits of resistance accrue from two sources: (i) improved survival in a drug-treated host, and (ii) removal of competitors (Hastings and D'Alessandro 2000; de Roode et al. 2004). This competitive release, whereby the resistant clone is able to expand into ‘niche space’ emptied by chemotherapy has the potential to greatly magnify the survival benefits of resistance – and indeed, it can shorten the therapeutically useful lifespan of a drug many-fold below that expected if resistance evolution were powered only by the survival advantage (Hastings and D'Alessandro 2000). Direct evidence of competitive release cannot be ethically obtained for humans, but in rodent malaria infections it is seen following both prophylatic and therapeutic chemotherapy (Fig. 4; de Roode et al. 2004; Wargo et al. 2007). As this potent selective advantage arises only when a resistant clone is in a coinfection with sensitive clones, it will become progressively weaker as resistance spreads in a population.
Figure 4.

Competitive release of resistant parasites following the removal of sensitive competitors by chemotherapy. The total number of resistant parasites present in infections where sensitive parasites have been removed by drug treatment or allowed to remain (no drugs) are shown. Plotted points are the mean (±SEM) cumulative total number of Plasmodium chabaudi parasites present in peripheral blood of mice, based on three to five infections per group. Therapeutic treatment is applied when the hosts first start to show symptoms of malaria (weight loss, anemia); prophylactic treatment is applied at the time host are infected. Data from Wargo et al. (2007), and de Roode et al. (2004) respectively.
Thus, both the costs and benefits of resistance depend on the frequency of resistance in a population. Hastings (2006b) has pointed out that this means that resistance evolution is likely to have dynamics which are considerably more complex than the standard S-shaped curve of rising allele frequencies through time seen in introductory population genetics textbooks. He suggests that this might explain why, for several different countries and drugs, resistance has apparently stabilized at frequencies well short of fixation (e.g. over 8 years in eastern Sudan, chloroquine resistance fluctuated seasonally around an apparently stable equilibrium frequency of 40%; Babiker et al. 2005).
More generally, given the enormous regional and seasonal variation in the force of infection, which is the major determinant of the genetic diversity of malaria infections (Arnot 1998), it may be that there are profound regional differences in the patient treatment regimes and drug deployment strategies which are optimal for resistance management. Existing global recommendations (WHO 2006) may be too simplistic.
Open questions
In addition to the unresolved issues which arise in the context of the preceding fallacies, there are a very large number of other open issues which seem to us unlikely to be resolved without the input of professional evolutionary biologists. Consider, for example, the following:
Why did resistance to chloroquine and SP become established so rarely when resistance spread so globally? In particular, why is so much drug resistance arising in Southeast Asia? There are at least five hypotheses (Klein et al. 2008), most of which focus on the observation that drug resistance seems to have arisen in areas of low or unstable transmission (White and Pongtavornpinyo 2003).
Will vector control enhance the spread of drug resistance? Historically, resistance seems to have emerged predominantly in low-transmission areas and spread more effectively in low transmission areas (White 2004; Klein et al. 2008). The large-scale deployment of bednets envisaged by GMAP is aimed at reducing malaria transmission (Roll Back Malaria 2008). Will this lead to more rapid resistance evolution, and necessitate a faster drug pipeline? The influence of transmission rate on resistance evolution has been hotly debated. Several arguments have been put forward that high transmission intensity promotes the spread of drug-resistance. Clonal diversity in infections is higher, exacerbating benefits of resistance, as discussed above. Higher transmission also means that, for a given level of drug use, more parasites will be exposed to drug selection (Mackinnon and Hastings 1998). On the other hand, genetically diverse infections will generate more outcrossed progeny infections, and will thereby lead to the destruction of multi-locus resistance genotypes (Talisuna et al. 2004; Mackinnon 2005). Moreover, areas with low transmission intensity typically harbor fewer immune individuals, who have (i) a higher parasite biomass and so more mutations (White and Pongtavornpinyo 2003), (ii) more symptomatic infections, and hence stronger drug selection (Talisuna et al. 2004; Mackinnon 2005), and (iii) a reduced capacity to clear drug-resistant parasites (Cravo et al. 2001). How these and other conflicting forces play out has yet to be established.
Is the WHO-recommended radical parasite cure really optimal for either patient treatment or resistance management? We have already questioned above whether radical parasite cure really is the best way of both treating patients and managing resistance evolution. Analyses of the question could also consider the following. In high transmission regions, where people receive more than one infective bite per day (Arnot 1998; Beier et al. 1999; Hay et al. 2000), does radical cure of a symptomatic infection have a sufficiently large clinical beneficial effect to offset the greatly enhanced exposure of parasites to drugs? Does complete parasite clearance make it easier for new parasites to invade?
Will chemotherapy select for more virulent or less virulent parasites? Chemotherapy could enhance the circulation of more virulent strains by keeping alive patients who would otherwise have died from virulent infections (Gandon et al. 2001; Porco et al. 2005). It could also be that drug tolerance varies with virulence, for instance if more rapidly replicating parasites are more vulnerable to drugs (higher metabolic sensitivity) or less vulnerable (faster population recovery once drug pressure has stopped). For one clonal lineage of a rodent malaria, less virulent parasites were more strongly suppressed by subcurative chemotherapy than more virulent parasites, suggesting that virulence evolution could indeed proceed in parallel with classical resistance evolution (Schneider et al. 2008).
Will the HIV epidemic increase the rate of antimalarial resistance evolution? There are about 18% more malaria parasites in sub-Saharan Africa as a result of the HIV-associated immunosuppression (Van Geertruyden et al. 2008). Does this increase in parasite number increase the chance of resistance mutations becoming established? If HIV-infection increases the severity of malaria or impairs immune clearance, will drug use become more common, strengthening the selection for resistance?
Coda
We hope that this review of some recent work and ideas in malaria drug resistance has made our general point that, from the perspective of evolutionary science, there is nothing fundamentally uninteresting or easy about drug resistance – and that solutions to the issues could have profound impacts on human health and wellbeing. Evolutionary biologists could conceivably contribute as much as drug discovery specialists (and much more cheaply). It is very hard to imagine that the world will indefinitely fund a malaria drug discovery pipeline at $US1.5 billion per decade, or indeed that there is an unlimited supply of drug classes to be discovered. Using the compounds we already have in the pipeline more effectively is a very high priority. Evolutionary geneticists have and continue to play a crucial role in reconstructing the history of drug resistance (e.g. Fig. 2). The challenge is to add to this an understanding of the processes that shaped this history, and use that understanding to change the future.
For those evolutionary biologists interested in general principles, the issues we have raised here in the context of malaria are relevant across a diverse range of pathogens, from RNA viruses to worms. Our bias is that, at least when it comes to policy and patient treatment, there has been too much focus on simple generalities and not enough focus on the important consequences of disease-specific natural history and indeed location-specific epidemiology. It may also be that considering the drug resistance problem alongside other problems of resistance management, such as mosquito resistance to insecticides, pest resistance to GM crops, and weed resistance to herbicides, would provide novel insights for human health, especially since for some of these, the evolutionary analysis is more advanced (e.g. Labbé et al. 2007), and evolutionary biologists have had a profound impact on policy and implementation on the ground (e.g. Bates et al. 2005).
A major focus of evolutionary biology has been the adaptation of traits where group and individual interests conflict. This way of thinking will undoubtedly prove to be a fertile area in drug resistance too, not least as a guide to the identification of drug targets (Andre and Godelle 2005). But there is also an urgent need to identify resistance management strategies which are good for the group (the currently uninfected, and the patients of the future) without being detrimental to individual patients seeking primary health care right now. In the limit, there is a trade-off between patient treatment and resistance management (the latter being optimized when very few patients are treated with a drug). But such trade-offs are extreme cases. Even where it is necessary to treat effectively large numbers of patients, there are many ways patients can be treated, and among those that similarly restore patient health will be some which are better at resistance management than others. As we have pointed out above, clinical cure is the object of patient treatment, and this need not require parasitologic cure. From a public health perspective, what is the best way to treat patients, impact transmission, and slow the spread of resistance?
More generally, there is a real need to engage with those who deliver and receive health care, and the economists and social scientists who study the process. What sort of resistance management strategies can patients, physicians and public health planners cope with, particularly if they involve an understanding of evolution?
Acknowledgments
Members of the Read group have shaped our thinking on this topic, but particularly Marg Mackinnon, Petra Schneider, Jaap de Roode and Andrew Wargo. SH is funded by the Darwin Trust of the University of Edinburgh.
Literature cited
- Abdel-Muhsin AMA, Mackinnon MJ, Ali E, Nassir EKA, Suleiman S, Ahmed S, Walliker D, et al. Evolution of drug-resistance genes in Plasmodium falciparum in an area of seasonal malaria transmission in Eastern Sudan. Journal of Infectious Diseases. 2004;189:1239–1244. doi: 10.1086/382509. [DOI] [PubMed] [Google Scholar]
- Afonso A, Hunt P, Cheesman S, Alves AC, Cunha CV, Do Rosario V, Cravo P. Malaria parasites can develop stable resistance to artemisinin but lack mutations in candidate genes atp6 (encoding the sarcoplasmic and endoplasmic reticulum Ca2+ ATPase), tctpmdr1, and cg10. Antimicrobial Agents and Chemotherapy. 2006;50:480–489. doi: 10.1128/AAC.50.2.480-489.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre JB, Godelle B. Multicellular organization in bacteria as a target for drug therapy. Ecology Letters. 2005;8:800–810. [Google Scholar]
- Antonovics J, Abbate JL, Baker CH, Daley D, Hood ME, Jenkins CE, Johnson LJ, et al. Evolution by any other name: antibiotic resistance and avoidance of the E-word. PLoS Biology. 2007;5:137–140. doi: 10.1371/journal.pbio.0050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnot DE. Clone multiplicity of Plasmodium falciparum infections in individuals exposed to variable levels of disease transmission. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1998;92:580–585. doi: 10.1016/s0035-9203(98)90773-8. [DOI] [PubMed] [Google Scholar]
- Babiker HA, Ranford-Cartwright LC, Walliker D. 3. Genetic structure and dynamics of Plasmodium falciparum infections in the Kilombero region of Tanzania. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:11. doi: 10.1016/s0035-9203(99)90321-8. [DOI] [PubMed] [Google Scholar]
- Babiker HA, Satti G, Ferguson H, Bayoumi R, Walliker D. Drug resistant Plasmodium falciparum in an area of seasonal transmission. Acta Tropica. 2005;94:260–268. doi: 10.1016/j.actatropica.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Barnes KI, White NJ. Population biology and antimalarial resistance: the transmission of antimalarial drug resistance in Plasmodium falciparum. Acta Tropica. 2005;94:230–240. doi: 10.1016/j.actatropica.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Barnes KI, Watkins WM, White NJ. Antimalarial dosing regimens and drug resistance. Trends in Parasitology. 2008;24:127–134. doi: 10.1016/j.pt.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Bate R, Coticelli P, Tren R, Attaran A. Antimalarial drug quality in the most severyly malarious parts of Africa – a six country study. PLoS ONE. 2008;3:e2132. doi: 10.1371/journal.pone.0002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates SL, Zhao JZ, Roush RT, Shelton AM. Insect resistance management in GM crops: past, present and future. Nature Biotechnology. 2005;23:57–62. doi: 10.1038/nbt1056. [DOI] [PubMed] [Google Scholar]
- Beier JC, Killeen GF, Githure JI. Short report: entomologic inoculation rates and Plasmodium falciparum malaria prevalence in Africa. American Journal of Tropical Medicine and Hygiene. 1999;61:109–113. doi: 10.4269/ajtmh.1999.61.109. [DOI] [PubMed] [Google Scholar]
- Bell AS, De Roode JC, Sim D, Read AF. Within-host competition in genetically diverse malaria infections: parasite virulence and competitive success. Evolution. 2006;60:1358–1371. [PubMed] [Google Scholar]
- Bruce MC, Donnelly CA, Alpers MP, Galinski MR, Barnwell JW, Walliker D, Day KP. Cross-species interactions between malaria parasites in humans. Science. 2000;287:845–848. doi: 10.1126/science.287.5454.845. [DOI] [PubMed] [Google Scholar]
- Cortese JF, Caraballo A, Contreras CE, Plowe CV. Origin and dissemination of Plasmodium falciparum drug-resistance mutations in South America. Journal of Infectious Diseases. 2002;186:999–1006. doi: 10.1086/342946. [DOI] [PubMed] [Google Scholar]
- Cravo P, Culleton R, Hunt P, Walliker D, Mackinnon MJ. Antimalarial drugs clear resistant parasites from partially immune hosts. Antimicrobial Agents and Chemotherapy. 2001;45:2897–2901. doi: 10.1128/AAC.45.10.2897-2901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubersies P, SallenaveSales S, Magne S, Trape JF, Contamin H, Fandeur T, Rogier C, et al. Rapid turnover of Plasmodium falciparum populations in asymptomatic individuals living in a high transmission area. American Journal of Tropical Medicine and Hygiene. 1996;54:18–26. doi: 10.4269/ajtmh.1996.54.18. [DOI] [PubMed] [Google Scholar]
- Djimde AA, Plowe CV, Diop S, Dicko A, Wellems TE, Doumbo O. Use of animalarial drugs in Mali: policy versus reality. American Journal of Tropical Medicine and Hygiene. 1998;59:376–379. doi: 10.4269/ajtmh.1998.59.376. [DOI] [PubMed] [Google Scholar]
- Drew DR, Reece SE. Development of reverse-transcription PCR techniques to analyse the density and sex ratio of gametocytes in genetically diverse Plasmodium chabaudi infections. Molecular and Biochemical Parasitology. 2007;156:199–209. doi: 10.1016/j.molbiopara.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felger I, Beck HP. Fitness costs of resistance to antimalarial drugs. Trends in Parasitology. 2008;24:331–333. doi: 10.1016/j.pt.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Gandon S, Mackinnon MJ, Nee S, Read AF. Imperfect vaccines and the evolution of pathogen virulence. Nature. 2001;414:751–756. doi: 10.1038/414751a. [DOI] [PubMed] [Google Scholar]
- Goodman C, Brieger W, Unwin A, Mills A, Meek S, Greer G. Medicine sellers and malaria treatment in sub-Saharan Africa: what do they do and how can their practice be improved? American Journal of Tropical Medicine and Hygiene. 2007;77:203–218. [PMC free article] [PubMed] [Google Scholar]
- Van Geertruyden JP, Menten J, Colebunders R, Korenromp E, D'Alessandro U. The impact of HIV-1 on the malaria parasite biomass in adults in sub-Saharan Africa contributes to the emergence of antimalarial drug resistance. Malaria Journal. 2008;7:13. doi: 10.1186/1475-2875-7-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings IM. Modelling parasite drug resistance: lessons for management and control strategies. Tropical Medicine & International Health. 2001;6:883–890. doi: 10.1046/j.1365-3156.2001.00800.x. [DOI] [PubMed] [Google Scholar]
- Hastings IM. Malaria control and the evolution of drug resistance: an intriguing link. Trends in Parasitology. 2003;19:70–73. doi: 10.1016/s1471-4922(02)00017-x. [DOI] [PubMed] [Google Scholar]
- Hastings IM. Gametocytocidal activity in antimalarial drugs speeds the spread of drug resistance. Tropical Medicine & International Health. 2006a;11:1206–1217. doi: 10.1111/j.1365-3156.2006.01668.x. [DOI] [PubMed] [Google Scholar]
- Hastings IM. Complex dynamics and stability of resistance to antimalarial drugs. Parasitology. 2006b;132:615–624. doi: 10.1017/S0031182005009790. [DOI] [PubMed] [Google Scholar]
- Hastings IM, D'Alessandro U. Modelling a predictable disaster: the rise and spread of drug-resistant malaria. Parasitology Today. 2000;16:340–347. doi: 10.1016/s0169-4758(00)01707-5. [DOI] [PubMed] [Google Scholar]
- Hastings IM, Donnelly MJ. The impact of antimalarial drug resistance mutations on parasite fitness, and its implications for the evolution of resistance. Drug Resistance Updates. 2005;8:43–50. doi: 10.1016/j.drup.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Hastings IM, Watkins WM. Tolerance is the key to understanding antimalarial drug resistance. Trends in Parasitology. 2006;22:71–77. doi: 10.1016/j.pt.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Hastings IM, Watkins WM, White NJ. The evolution of drug-resistant malaria: the role of drug elimination half-life. Philosophical Transactions of the Royal Society of London Series B-Biological Sciences. 2002;357:505–519. doi: 10.1098/rstb.2001.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay SI, Rogers DJ, Toomer JF, Snow RW. Annual Plasmodium falciparum entomological inoculation rates (EIR) across Africa: literature survey, internet access and review. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2000;94:113–127. doi: 10.1016/s0035-9203(00)90246-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayton K, Su XZ. Drug resistance and genetic mapping in Plasmodium falciparum. Current Genetics. 2008;54:223–239. doi: 10.1007/s00294-008-0214-x. [DOI] [PubMed] [Google Scholar]
- Hyde JE. Drug-resistant malaria. Trends in Parasitology. 2005;21:494–498. doi: 10.1016/j.pt.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari S, Le Bras J, Bouchaud O, Durand R. Plasmodium falciparum clonal population dynamics during malaria treatment. Journal of Infectious Diseases. 2004;189:195–203. doi: 10.1086/380910. [DOI] [PubMed] [Google Scholar]
- Jarra W, Brown KN. Protective immunity to malaria: studies with cloned lines of Plasmodium chabaudi and P. berghei in CBA/Ca mice. I. The effectiveness and inter- and intra-species specificity of immunity induced by infection. Parasite Immunology. 1985;7:595–606. doi: 10.1111/j.1365-3024.1985.tb00103.x. [DOI] [PubMed] [Google Scholar]
- Klein EY, Smith DL, Boni MF, Laxminarayan R. Clinically immune hosts as a refuge for drug-sensitive malaria parasites. Malaria Journal. 2008;7:67. doi: 10.1186/1475-2875-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koella JC, Antia R. Epidemiological models for the spread of anti-malarial resistance. Malaria Journal. 2003;2:3. doi: 10.1186/1475-2875-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kublin JG, Cortese JF, Njuniu EM, Mukadam RAG, Wirima JJ, Kazembe PN, Djimde AA, et al. Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. Journal of Infectious Diseases. 2003;187:1870–1875. doi: 10.1086/375419. [DOI] [PubMed] [Google Scholar]
- Labbe P, Berticat C, Berthomieu A, Unal S, Bernard C, Weill M, Lenormand T. Forty years of erratic insecticide resistance evolution in the mosquito Culex pipiens. PLoS Genetics. 2007;3:2190–2199. doi: 10.1371/journal.pgen.0030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, Taylor TE, et al. Return of chloroquine antimalarial efficacy in Malawi. New England Journal of Medicine. 2006;355:1959–1966. doi: 10.1056/NEJMoa062032. [DOI] [PubMed] [Google Scholar]
- Levin BR, Perrot V, Walker N. Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics. 2000;154:985–997. doi: 10.1093/genetics/154.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DQ, Liu RJ, Ren DX, Gao DQ, Zhang CY, Qiu CP, Cai XZ, et al. Changes in the resistance of Plasmodium falciparum to chloroquine in Hainan, China. Bulletin of the World Health Organization. 1995;73:483–486. [PMC free article] [PubMed] [Google Scholar]
- Mackinnon MJ. Drug resistance models for malaria. Acta Tropica. 2005;94:207–217. doi: 10.1016/j.actatropica.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Hastings IM. The evolution of multiple drug resistance in malaria parasites. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1998;92:188–195. doi: 10.1016/s0035-9203(98)90745-3. [DOI] [PubMed] [Google Scholar]
- Maiga O, Djimde AA, Hubert V, Renard E, Aubouy A, Kironde F, Nsimba B, et al. A shared Asian origin of the triple-mutant dhfr allele in Plasmodium falciparum from sites across Africa. Journal of Infectious diseases. 2007;196:165–172. doi: 10.1086/518512. [DOI] [PubMed] [Google Scholar]
- McCollum AM, Poe AC, Hamel M, Huber C, Zhou ZY, Shi YP, Ouma P, et al. Antifolate resistance in Plasmodium falciparum: multiple origins and identification of novel dhfr alleles. The Journal of Infectious Diseases. 2006;194:189–197. doi: 10.1086/504687. [DOI] [PubMed] [Google Scholar]
- McCollum AM, Mueller K, Villegas L, Udhayakumar V, Escalante AA. Common origin and fixation of Plasmodium falciparum dhfr and dhps mutations associated with sulfadoxine-pyrimethamine resistance in a low-transmission area in South America. Antimicrobial Agents and Chemotherapy. 2007;51:2085–2091. doi: 10.1128/AAC.01228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum AM, Basco LK, Tahar R, Udhayakumar V, Escalante AA. Hitchhiking and selective sweeps of Plasmodium falciparum sulfadoxine and pyrimethamine resistance alleles in a population from central Africa. Antimicrobial Agents and Chemotherapy. 2008;52:4089–4097. doi: 10.1128/AAC.00623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher TR, Futuyma D. Executive document: evolution, science, and society – Foreword: evolution in the century of biology. American Naturalist. 2001;158:1–46. doi: 10.1086/323109. [DOI] [PubMed] [Google Scholar]
- Mendez F, Munoz A, Carrasquilla G, Jurado D, Arevalo-Herrera M, Cortese JF, Plowe CV. Determinants of treatment response to sulfadoxine-pyrimethamine and subsequent transmission potential in falciparum malaria. American Journal of Epidemiology. 2002;156:230–238. doi: 10.1093/aje/kwf030. [DOI] [PubMed] [Google Scholar]
- Mercereau-Puijalon O. Revisiting host/parasite interactions: molecular analysis of parasites collected during longitudinal and cross-sectional surveys in humans. Parasite Immunology. 1996;18:173–180. doi: 10.1046/j.1365-3024.1996.d01-79.x. [DOI] [PubMed] [Google Scholar]
- Mita T, Kaneko A, Lum JK, Bwijo B, Takechi N, Zungu IL, Tsukahara T, et al. Recovery of chloroquine sensitivity and low prevalence of the Plasmodium falciparum chloroquine resistance transporter gene mutation K76T following the discontinuance of chloroquine use in Malawi. American Journal of Tropical Medicine and Hygiene. 2003;68:413–415. [PubMed] [Google Scholar]
- Mita T, Tanabe K, Takahashi N, Tsukahara T, Eto H, Dysoley L, Ohmae H, et al. Independent evolution of pyrimethamine resistance in Plasmodium falciparum isolates in melanesia. Antimicrobial Agents and Chemotherapy. 2007;51:1071–1077. doi: 10.1128/AAC.01186-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann JP, et al. A selective sweep driven by pyrimethamine treatment in southeast Asian malaria parasites. Molecular Biology and Evolution. 2003;20:1526–1536. doi: 10.1093/molbev/msg162. [DOI] [PubMed] [Google Scholar]
- Ord R, Alexander N, Dunyo S, Hallett R, Jawara M, Targett G, Drakeley CJ, et al. Seasonal carriage of pfcrt and pfmdr1 alleles in Gambian Plasmodium falciparum imply reduced fitness of chloroquine-resistant parasites. Journal of Infectious Diseases. 2007;196:1613–1619. doi: 10.1086/522154. [DOI] [PubMed] [Google Scholar]
- Peters W. Chemotherapy and Drug Resistance in Malaria. Vol. 1. London: Academic Press; 1987. [Google Scholar]
- Porco TC, Lloyd-Smith JO, Gross KL, Galvani AP. The effect of treatment on pathogen virulence. Journal of Theoretical Biology. 2005;233:91–102. doi: 10.1016/j.jtbi.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri SK, Chandra R. Plasmodium vinckei: selection of a strain exhibiting stable resistance to arteether. Experimental Parasitology. 2006;114:129–132. doi: 10.1016/j.exppara.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Raman J, Sharp B, Kleinschmidt I, Roper C, Streat E, Kelly V, Barnes KI. Differential effect of regional drug pressure on dihydrofolate reductase and dihydropteroate synthetase mutations in southern Mozambique. American Journal of Tropical Medicine and Hygiene. 2008;78:256–261. [PMC free article] [PubMed] [Google Scholar]
- Restif O. Evolutionary epidemiology 20 years on: challenges and prospects. Infection, Genetics and Evolution. 2009;9:108–123. doi: 10.1016/j.meegid.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Roll Back Malaria. The Global Malaria Action Plan. 2008. http://www.rbm.who.int/gmap/gmap.pdf (accessed on 2 January 2009) [Google Scholar]
- De Roode JC, Culleton R, Bell AS, Read AF. Competitive release of drug resistance following drug treatment of mixed Plasmodium chabaudi infections. Malaria Journal. 2004;3:33. doi: 10.1186/1475-2875-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roode JC, Pansini R, Cheesman SJ, Helinski MEH, Huijben S, Wargo AR, Bell AS, et al. Virulence and competitive ability in genetically diverse malaria infections. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:7624–7628. doi: 10.1073/pnas.0500078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper C, Pearce R, Bredenkamp B, Gumede J, Drakeley C, Mosha F, Chandramohan D, et al. Antifolate antimalarial resistance in southeast Africa: a population-based analysis. Lancet. 2003;361:1174–1181. doi: 10.1016/S0140-6736(03)12951-0. [DOI] [PubMed] [Google Scholar]
- Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- Saito-Nakano Y, Tanabe K, Kamei K, Iwagami M, Komaki-Yasuda K, Kawazu S, Kano S, et al. Genetic evidence for Plasmodium falciparum resistance to chloroquine and pyrimethamine in Indochina and the Western Pacific between 1984 and 1998. American Journal of Tropical Medicine and Hygiene. 2008;79:613–619. [PubMed] [Google Scholar]
- Schneider P, Chan BHK, Reece SE, Read AF. Does the drug sensitivity of malaria parasites depend on their virulence? Malaria Journal. 2008;7:257. doi: 10.1186/1475-2875-7-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Felger I, Tanner M, Beck H-P. The epidemiology of multiple Plasmodium falciparum infections. 11. Premunition in Plasmodium falciparum infection: insights from the epidemiology of multiple infections. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:S59–S64. doi: 10.1016/s0035-9203(99)90329-2. [DOI] [PubMed] [Google Scholar]
- Stepniewska K, White NJ. Pharmacokinetic determinants of the window of selection for antimalarial drug resistance. Antimicrobial Agents and Chemotherapy. 2008;52:1589–1596. doi: 10.1128/AAC.00903-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talisuna AO, Bloland P, D'Alessandro U. History, dynamics, and public health importance of malaria parasite resistance. Clinical Microbiology Reviews. 2004;17:235–254. doi: 10.1128/CMR.17.1.235-254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talisuna AO, Erhart A, Samarasinghe S, Van Overmeir C, Speybroeck N, D'Alessandro U. Malaria transmission intensity and the rate of spread of chloroquine resistant Plasmodium falciparum: why have theoretical models generated conflicting results? Infection, Genetics and Evolution. 2006;6:241–248. doi: 10.1016/j.meegid.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Taylor LH, Walliker D, Read AF. Mixed-genotype infections of malaria parasites: within-host dynamics and transmission success of competing clones. Proceedings of the Royal Society of London Series B. 1997;264:927–935. doi: 10.1098/rspb.1997.0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temu EA, Kimani I, Tuno N, Kawada H, Minjas JN, Takagi M. Monitoring chloroquine resistance using Plasmodium falciparum parasites isolated from wild mosquitoes in Tanzania. American Journal of Tropical Medicine and Hygiene. 2006;75:1182–1187. [PubMed] [Google Scholar]
- Thaithong S, Suebsaeng L, Rooney W, Beale GH. Evidence of increased chloroquine sensitivity in Thai isolates of Plasmodium falciparum. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1988;82:37–38. doi: 10.1016/0035-9203(88)90255-6. [DOI] [PubMed] [Google Scholar]
- Walliker D, Hunt P, Babiker H. Fitness of drug-resistant malaria parasites. Acta Tropica. 2005;94:251–259. doi: 10.1016/j.actatropica.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Wargo AR, Huijben S, De Roode JC, Shepherd J, Read AF. Competitive release and facilitation of drug-resistant parasites after therapeutic chemotherapy in a rodent malaria model. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19914–19919. doi: 10.1073/pnas.0707766104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WM, Mosobo M. Treatment of Plasmodium falciparum malaria with pyrimethamine-sulfadoxine – selective pressure for resistance is a function of long elimination half-life. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1993;87:75–78. doi: 10.1016/0035-9203(93)90431-o. [DOI] [PubMed] [Google Scholar]
- Wellems TE. Transporter of a malaria catastrophe. Nature Medicine. 2004;10:1169–1171. doi: 10.1038/nm1104-1169. [DOI] [PubMed] [Google Scholar]
- White NJ. Antimalarial drug resistance. Journal of Clinical Investigation. 2004;113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White NJ. Qinghaosu (Artemisinin): the price of success. Science. 2008;320:330–334. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- White NJ, Pongtavornpinyo W. The de novo selection of drug-resistant malaria parasites. Proceedings of the Royal Society of London Series B-Biological Sciences. 2003;270:545–554. doi: 10.1098/rspb.2002.2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. WHO Guidelines for the Treatment of Malaria. Geneva, Switzerland: WHO; 2006. http://www.who.int/malaria/docs/TreatmentGuidelines2006.pdf (accessed on 2 January 2009) [Google Scholar]
- WHO. World Malaria Report 2008. Geneva, Switzerland: WHO; 2008. http://malaria.who.int/wmr2008/malaria2008.pdf (accessed on 2 January 2009) [Google Scholar]
- Wongsrichanalai C, Meshnick SR. Declining artesunate-mefloquine efficacy against falciparum malaria on the Cambodia-Thailand border. Emerging Infectious Diseases. 2008;14:716–719. doi: 10.3201/eid1405.071601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZQ, Zhang ZX, Sun XD, Wan WL, Cui L, Zhang X, Zhong DB, et al. Molecular analysis of chloroquine resistance in Plasmodium falciparum in Yunnan Province, China. Tropical Medicine & International Health. 2007;12:1051–1060. doi: 10.1111/j.1365-3156.2007.01882.x. [DOI] [PubMed] [Google Scholar]