

Abstract
Heart disease is common in both humans and chimpanzees, manifesting typically as sudden cardiac arrest or progressive heart failure. Surprisingly, although chimpanzees are our closest evolutionary relatives, the major cause of heart disease is different in the two species. Histopathology data of affected chimpanzee hearts from two primate centers, and analysis of literature indicate that sudden death in chimpanzees (and in gorillas and orangutans) is commonly associated with diffuse interstitial myocardial fibrosis of unknown cause. In contrast, most human heart disease results from coronary artery atherosclerosis, which occludes myocardial blood supply, causing ischemic damage. The typical myocardial infarction of humans due to coronary artery thrombosis is rare in these apes, despite their human-like coronary-risk-prone blood lipid profiles. Instead, chimpanzee ‘heart attacks’ are likely due to arrythmias triggered by myocardial fibrosis. Why do humans not often suffer from the fibrotic heart disease so common in our closest evolutionary cousins? Conversely, why do chimpanzees not have the kind of heart disease so common in humans? The answers could be of value to medical care, as well as to understanding human evolution. A preliminary attempt is made to explore possibilities at the histological level, with a focus on glycosylation changes.
Keywords: atherosclerosis, evolution, great ape, heart attacks, heart disease, heart failure, hominids, myocardial fibrosis
Introduction
The commonest cause of human deaths in most developed and developing countries is atherosclerosis, a chronic progressive disease that affects large arteries, including the coronary blood vessels (Rosamond et al. 2007). Atherosclerosis is a form of chronic inflammation that arises because hyperlipidemia and modified lipoproteins cause accumulation of cholesterol-laden macrophages and inflammatory cells below the endothelial lining of the large arteries (Glass and Witztum 2001; Binder et al. 2002; Hansson 2005; Steinberg 2006). Other processes accelerating atherosclerosis include naturally occurring anti-endothelial antibodies (Binder et al. 2005), mechanical stress in blood vessels such as high blood pressure and turbulent flow (Traub and Berk 1998; Escobar 2002); autoimmune diseases (Sherer and Shoenfeld 2006; Soltesz et al. 2007); accumulation of advanced glycation end-products arising from high blood glucose in diabetics (Goldin et al. 2006), the ‘metabolic syndrome’ associated with high triglycerides and suppressed high density lipoprotein (HDL) levels (Eckel et al. 2005; Meerarani et al. 2006); genetic variations affecting the low density lipoprotein (LDL) receptor (Horton et al. 2007); and, chronic inflammation associated with gingivitis, arthritis, obesity, or cigarette smoking (Haynes and Stanford 2003; Libby and Ridker 2004; Gonzalez-Gay et al. 2006; Fantuzzi and Mazzone 2007; Yanbaeva et al. 2007). Thus, well-known risk factors for atherosclerosis include hypercholesterolemia, diabetes, obesity, hypertension, cigarette smoking, family history of heart disease and a sedentary lifestyle.
While atherosclerosis can cause vascular disease in many anatomical locations, the most prominently affected organ in humans is the heart. Coronary atherosclerosis frequently manifests as a ‘heart attack’, in which the patient becomes suddenly ill with chest pain, sweating and shortness of breath, resulting from obstruction of a major coronary artery that provides blood supply to the heart muscle (White and Chew 2008). Such acute coronary occlusion often results in ischemic death of the cardiac muscle regions that are perfused by the artery in question, and can even result in sudden death (Davies and Thomas 1984; Farb et al. 1995; Virmani et al. 2001). If the patient survives, there is a slow repair and remodeling process, wherein the ischemic muscle is mostly replaced by scar tissue, as cardiomyocytes cannot regenerate significantly. Those who survive an initial coronary occlusion often have recurrent coronary events and/or develop progressive heart failure due to loss of cardiac muscle function and the continued progression of the underlying disease process. In other humans who do not suffer such an acute cardiac event, the primary manifestation of atherosclerotic coronary artery disease can be progressive heart failure, due to the gradual loss of blood supply and ischemic damage to the myocardium (Mosterd and Hoes 2007; Boden 2008).
Chimpanzees are our closest evolutionary relatives, with nearly 99% identity of many of our homologous protein sequences (The Chimpanzee Sequencing, Analysis Consortium, 2005; Varki and Nelson 2007; Varki et al. 2008). Thus, at first glance it is not surprising that published data about chimpanzees cared for in captivity lists heart failure and/or sudden death due to ‘heart attacks’ as being among the commonest causes of death (Schmidt 1978; Munson and Montali 1990; Hubbard et al. 1991). However, closer examination of the clinical and pathological findings indicates that the mechanisms causing heart disease in humans and chimpanzees are quite different. This remarkable and surprising difference is the subject of this article, in which we present some new data, and also synthesize existing reports on the subject. We also address the limited data available on the other Hominids (‘Great Apes’) that have been studied (Gorillas and Orangutans), showing that they are more like chimpanzees than humans in this respect. Overall, we conclude that rather than representing a similarity, heart disease is an instance where there are unexplained human-specific differences from the other Hominids. Finally, we provide preliminary evidence for differences in extracellular matrix and glycosylation patterns of human and great ape myocardium, which could potentially be relevant to understanding this difference.
Materials and methods
Animals
The chimpanzees (Pan troglodytes) maintained at both the Yerkes National Primate Research Center (YNPRC) and the Primate Foundation of Arizona (PFA) were socially housed in indoor/outdoor enclosures of various sizes and configurations. They were fed low fat (<5%) commercial primate diets from Purina Mills (Monkey Diet Jumbo 5037 at YNPRC; Fiber Plus 5049 and High Protein 5045 Monkey Diets at PFA), with daily fruit and vegetable supplements.
Clinical data collection and analysis
Prior literature on the subject of heart disease in chimpanzees and other great apes was surveyed (Gray et al. 1981; Cousins 1983; Hansen et al. 1984; McNamara et al. 1987; Munson and Montali 1990; Hubbard et al. 1991; Schulman et al. 1995; Lowenstine 2003; Doane et al. 2006; Lammey et al. 2008, 2008; Seiler et al. 2008). The medical records of all adult chimpanzees (>10 years of age), cared for over many decades at the Yerkes National Primate Center in Atlanta (1966–2008) and at the PFA (1987–2007) were reviewed to determine the frequency of major illnesses and causes of death. Records were included in the final summary only if a terminal necropsy had been done to confirm the likely cause of death.
Histological staining of myocardial tissue sections
Paraffin sections of myocardium were deparaffinized, rehydrated, and stained to analyze morphology, using the routine hematoxylin and eosin staining. To ensure comparability, sections from human and great ape hearts were paired and stained on the same slide, whenever possible. Duplicate slides were stained using the Masson's trichrome stain to detect collagen, following the manufacturer's instructions. Other sets of slides were blocked for endogenous biotin and overlaid with predetermined dilutions of three different biotinylated lectins in Tris-buffered saline with 0.5% Tween (TBST), containing 1 mm calcium chloride, 1 mm magnesium chloride, and 1 mm manganese chloride, or with secondary reagent alone. After incubation in a humid chamber for 30 min, unbound lectins were washed off using the same buffer. Sections were then overlaid with alkaline phosphatase labeled streptavidin (Jackson ImmunoResearch, West Grove, PA) for 30 min in a humid chamber. After more washes, bound lectins were detected using freshly made alkaline phosphatase substrate Vector Blue (Vector labs, Burlingame, CA, USA), and nuclei were counterstained with nuclear fast red (Vector Labs).
Results and discussion
Heart disease is the commonest cause of death in recent series of captive chimpanzees
Prior to the last two decades, infection was the commonest cause of death in chimpanzees (see for example the Yerkes experience from 1966 to 1991 shown in Table 1). However, with improvements in infection control by vaccination and antibiotics, heart disease has now become the commonest cause of death at all centers surveyed (see Tables 1 and 2). Further analysis was focused on cases in which sudden deaths and/or heart failure occurred without any other obvious antecedent or precipitating illness.
Table 1.
Primary causes of death in adult* Yerkes chimpanzees
| 1966–1991 | 1992–2008 | |
|---|---|---|
| General category | n (%) | n (%) |
| Meningitis | 4 (11) | – |
| Heart disease | 4 (11) | 21 (36) |
| Enterocolitis | 6 (16) | – |
| Pneumonia | 2 (5) | – |
| Renal disease | 2 (5) | 9 (16) |
| Trauma | 1 (3) | 7 (12) |
| Miscellaneous | 19 (50) | 21 (36) |
| 38 | 58 |
>10 years of age.
Table 2.
Major causes of death in adult* chimpanzees in recent times
| PFA† 1992–2006 | SNPRC‡ 1982–2006 | APF§ 2001–2006 | YNPRC¶ 1992–2008 | |
|---|---|---|---|---|
| Heart disease | 1 (14%) | 30 (35%) | 13 (36%) | 21 (36%) |
| Pneumonia | 1 | ns* | – | – |
| Renal disease | 2 | ns | 9 | 9 |
| Trauma | – | ns | 3 | 7 |
| Miscellaneous | 4 | 57 | 11 | 21 |
| Total | 7 | 87 | 36 | 58 |
Early reports on heart disease in chimpanzees and other great apes
Although many series describing chimpanzee deaths in captivity list heart disease as a major cause of death, several of these reports also commented that gross coronary atherosclerosis was not seen at necropsy (Munson and Montali 1990; Hubbard et al. 1991; Lowenstine 2003). These series as well as various case reports also mention fibrosis of the myocardium in gorillas, orangutans, and chimpanzees (Gray et al. 1981; Cousins 1983; Hansen et al. 1984; McNamara et al. 1987; Munson and Montali 1990; Schulman et al. 1995) (see Table 3 for a summary of some examples). Meeting abstracts, nonpeer-reviewed literature, and news reports also contain several examples of unexplained sudden deaths of captive great apes, both in primate centers and zoos.
Table 3.
Examples of prior publications describing unexplained myocardial fibrosis in chimpanzees and other great apes
| Reference | Cases | Total | Notes |
|---|---|---|---|
| Hansen et al. (1984) | 1 | 1 | Case study of a 26-year-old male chimpanzee with myocardial fibrosis. |
| Munson and Montali (1990) | 1 | 1 | Review of medical and pathology records of great apes at the National Zoological Park. One chimpanzee of unspecified age was examined, and reported to have cardiac fibrosis and amyloidosis. Two orangutans also reported to have cardiac fibrosis. |
| Hubbard et al. (1991) | 6 | 33 | Review of chimpanzee medical records between 1981 and 1989 revealed 6 deaths due to myocardial disease. All had similar lesions including lymphocytic myocarditis, myocardial fibrosis, mineralization and necrosis. |
| Schulman et al. (1995) | 11 | 19 | A retrospective study describing fibrosing cardiomyopathy in 11 male captive lowland gorillas - out of a cohort of 16 male and 3 female individuals ranging from 9–38 years of age who died of various causes, including 7 sudden deaths. |
| Doane et al. (2006) | 29 | 36 | Sudden cardiac death was leading cause of death. All such animals had interstitial myocardial fibrosis. |
| Lammey et al. (2008) | 12 | 13 | 12 of 13 animals that died of sudden cardiac death had myocardial fibrosis. This is a subset of the animals examined in Lammey at al, (Lammey et al. 2008), below. |
| Lammey et al. (2008) | 29 | 36 | Sudden cardiac death (SCD) in 13 (11 male and 2 female) chimpanzees was leading cause of death. Cardiac arrhythmias (ventricular ectopy) seen in 15 (12 male and 3 female). Histologic examination showed IMF in 29 (81%). Systemic hypertension noted only in 3 cases. No other causes of IMF noted. |
| Seiler et al. (2008) | 50 | 87 | Prevalence of heart disease in chimpanzees was 68% and prevalence of IMF was 51%. Prevalence at death was higher in males (60%) than in females (29%). Heart disease was primary cause of death in 35%. No evidence that diet, environment, viral agents, experimental use or disease exposure contributed to IMF in chimpanzees. |
High frequency of interstitial myocardial fibrosis and cardiomyopathy at necropsy in chimpanzees
A 2005 meeting in La Jolla (‘Understanding Great Apes in the Genomic Era’) included a review of the experience of the Yerkes Primate Center, concluding that extensive myocardial fibrosis was in fact a very common cause of sudden death and heart failure in chimpanzees, particularly among males. Presentation of these findings generated considerable interest and discussion, leading to confirmation at other institutions, including papers showing a high frequency of cardiac anomalies in chimpanzees such as arrhythmias (Doane et al. 2006) and myocardial fibrosis (Lammey et al. 2008, 2008; Seiler et al. 2008). In these reports as well, a higher incidence of both pathologies was observed in males than in females. While these recent reports (see Table 3) did comment on human heart disease, they did not draw the striking contrast with regard to the different underlying pathological processes, i.e., coronary atherosclerosis as the main mechanism in humans, and myocardial fibrosis in chimpanzees. While different terms have been used to describe the latter pathology, we hereafter refer to it as interstitial myocardial fibrosis (IMF).
Here we have returned to the originally reported Yerkes series and combined it with experience from the PFA. A total of 16 deaths out of 52 in animals 10 years or older were attributed to heart disease in animals that died at the Yerkes Primate Research Center between 1992 and 2005 and were carefully examined with regard to the histological sections of affected hearts. This included nine animals (eight male; one female) in which sudden unexpected death occurred and three animals in which acute onset of severe illness (two male; one female) was noted. Indeed, all these deaths were associated with this unusual form of pathological IMF. While normal myocardium from humans and from chimpanzees are histologically quite similar (Fig. 1 uppermost panels), no human counterpart was observed of the mild, moderate or extensive IMF seen in chimpanzees (see Fig. 1 for an example of extensive fibrosis). The chimpanzee heart fibrosis was observed to be distributed haphazardly throughout the cardiac muscle, with no predilection for areas immediately surrounding blood vessels, as can be seen in some human hearts (Fig. 1). The fibrosis could be more obviously seen in Masson's trichrome stains, which reveal collagen fibers (see Fig. 1). Mild to moderate myocardial fibrosis was also seen in a number of chimpanzees that died of other causes during the period from 1992 to 2005 at the Yerkes Center. In total, 14 males and four females presented at necropsy with this type of diffuse IMF during this period.
Figure 1.
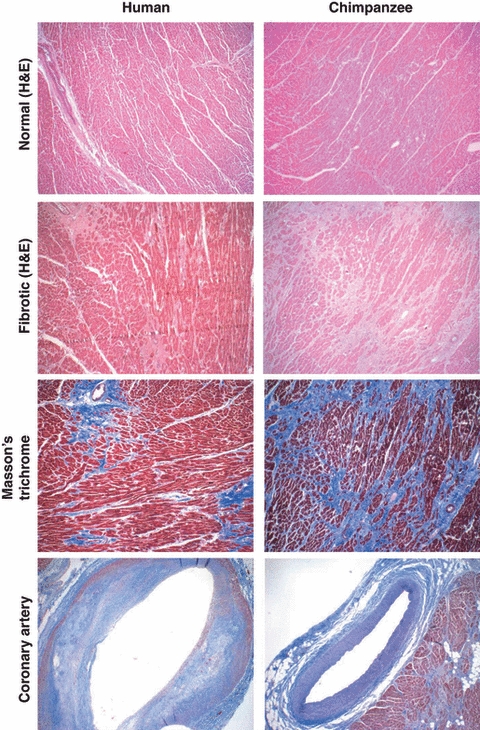
Examples of histological comparisons of human and chimpanzee hearts and coronary blood vessels. Top panels: Hematoxylin and eosin stains of normal myocardium sections from humans and chimpanzees look very similar. Upper middle panels: Example of fibrosis immediately surrounding blood vessels, as seen in some human hearts, and example of extensive interstitial myocardial fibrosis in chimpanzee heart. Lower middle panels: Collagen fibrosis is more clearly seen in Masson's trichrome stain. Bottom panels: Atherosclerotic coronary artery typically seen in humans (note the subendothelial plaques), compared with a typical uninvolved coronary artery in a chimpanzee, as seen in Masson's Trichrome stain.
Notably, although mild to moderate atherosclerosis was observed in the aorta and other major blood vessels in some of the chimpanzee necropsies, no major blockages of the coronary arteries were observed in any case (atherosclerosis of the Circle of Willis in the brain blood supply was sometimes seen, associated with strokes). Figure 1 shows a typical patent coronary artery in a chimpanzee, in contrast to the severely affected coronary arteries typically seen in humans. There were also no reports at necropsy of localized loss of myocardial perfusion in chimpanzees, which would manifest as either segmental regional pallor or localized myocardial necrosis.
Of course, while interesting and corroborated by similar surveys in geographically distinct primate centers throughout the United States, these findings are not without caveats. The relatively small sample size, the retrospective nature of necropsy data, and the inability to conduct controlled studies of the effects of experimental perturbations on IMF progression in great apes are notable examples. Nonetheless, the differences between the human and great ape cardiac pathologies are quite striking.
Rarity of chimpanzee-like IMF pathology in humans
The cardiac pathology described above for chimpanzees is in striking contrast to that seen in humans following a coronary occlusive event, where the pathological features of the myocardium are quite distinct, and major atherothrombotic occlusions of the coronary arteries are common. In fact, according to the literature, evidence of coronary thrombosis or plaque disruption can be found in up to ∼70% of postmortem cases of sudden death (Davies and Thomas 1984; Farb et al. 1995; Virmani et al. 2001). In contrast, while limited or focal cardiac fibrosis can be seen in humans (especially in patients with chronic hypertension or diabetes) (Diez 2007; Caglayan et al. 2008), there is no well-recognized common human pathological counterpart of the diffuse IMF seen in the great ape population.
Association of myocardial fibrosis with sudden death or congestive heart failure in chimpanzees
In the case of humans with sudden death following a coronary occlusion, the pathological mechanisms involved are obvious, i.e., sudden hypoxemia of the myocardium, with resulting loss of functions and/or generation of abnormal cardiac rhythms (arrhythmias) (White and Chew 2008). While there is a strong association of myocardial fibrosis with sudden deaths in chimpanzees (many with no obvious antecedent illness), the pathophysiological mechanisms leading to death are less clear. Based on a recent report of a high frequency of electrocardiographic abnormalities in chimpanzees (Doane et al. 2006), malignant arrythmias are the most likely precipitating event. In contrast, in a series of 270 humans presenting with sudden cardiac death, only six showed evidence of IMF-like features (John et al. 2004). Thus, sudden deaths of cardiac etiology appear to arise predominantly from different pathological processes between humans and chimpanzees, despite the loose application of the term ‘heart attack’ to both types of events.
Progressive congestive heart failure with or without any acute terminal cardiac event is another common cause of death in chimpanzees (six in this series at Yerkes) and in humans. Again, while the commonest cause of such progressive congestive heart failure in humans is ischemic heart disease due to atherosclerotic coronary disease (Rosamond et al. 2007), the associated pathology in chimpanzees is instead the aforementioned IMF and cardiomyopathy. We of course recognize that no rigorous study has or can be performed in which the environmental and nutritional variables of the captive chimpanzee are imposed on humans. Thus, it is difficult to argue conclusively that humans would never get significant IMF under the identical conditions. We also recognize that the captive lifestyle includes a diet low in cholesterol and a major portion of calories accounted for by fruits and vegetables, factors that could reduce risk the atherosclerosis. However, as discussed below, great apes actually appear to have a proatherogenic lipid profile.
Similar IMF pathology also occurs in the other great ape deaths with cardiac disease
In addition to the literature on IMF in chimpanzees (Hansen et al. 1984; Munson and Montali 1990; Hubbard et al. 1991; Lowenstine 2003; Lammey et al. 2008, 2008; Seiler et al. 2008), there are reports of sudden deaths or heart failure in the other closely related hominids (gorillas and orangutans) attributed to cardiac events, again associated with similar descriptions of myocardial fibrosis (Gray et al. 1981; Cousins 1983; Hansen et al. 1984; McNamara et al. 1987; Munson and Montali 1990; Schulman et al. 1995; Lowenstine 2003). Such reports tend to emphasize the similarity of chimpanzees and other great apes to humans, by noting that cardiovascular events are a major cause of death in all of these hominids. However, most also acknowledge the lack of severe atherosclerosis in most of these apes and the rarity of coronary occlusive disease. Indeed, a search of the extant literature revealed only rare instances of acute coronary thrombosis in chimpanzees and gorillas (Manning 1942; Vastesaeger et al. 1975; Blaton and Peeters 1976; Gray et al. 1981), typically under dietary or blood lipid circumstances that would have been expected to maximize the risk of severe atherosclerosis in humans.
Great apes often have proatherogenic blood lipid profiles
It is well-recognized that spontaneous atherosclerosis does not occur in rodents, which are the evolutionary clade closest to primates. Even in genetically manipulated models with very high cholesterol levels, it is difficult to show the severe atherosclerotic features that are common in humans (e.g., occlusive thrombosis). Thus, although genetically modified apoe−/− mice can suffer from myocardial infarcts, the frequency is rare (Calara et al. 2001). Watanabe heritable hyperlipidemic rabbits can also develop spontaneous myocardial infarcts, but these animals are genetically modified, and still do not often show coronary plaque rupture and thrombosis, as seen in humans (Shiomi et al. 2003). This difficulty in inducing advanced lesions in rodent and rabbit models is in stark contrast to the prevalence of severe atherosclerosis in humans, which occurs even in some individuals with mildly elevated LDL levels and no other obvious risk factors (Vasan et al. 2005). Indeed, despite statin treatment of their hypercholesterolemia 70% of such patients still have adverse cardiovascular events (Steinberg 2008).
The clinical manifestations of atherosclerosis also tend to be much commoner in humans than in nonhuman primates. While captive great apes have a relatively healthy diet from the perspective of atherosclerotic risk (low in cholesterol and saturated fat), this is not reflected in their serum lipid profiles. At the Yerkes Primate Center, a mean total cholesterol level of 211.13 ± 38.85 mg/dL was found in a population of 106 male captive chimpanzees over their 50-plus-year lifespan (Herndon and Tigges 2001). Similar ranges were noted at PFA (Howell et al. 2003), and at another center (Hainsey et al. 1993). Figure 2 shows a age-based longitudinal comparison between cholesterol levels in normal humans participating in the Framingham Heart Study (Abbott et al. 1983) and captive chimpanzees at the Yerkes Primate Center (Herndon and Tigges 2001). Plotting total serum cholesterol (mg/dL) as a function of age (decade of life) reveals two striking observations. First, we can see that the mean total serum cholesterol in this captive chimpanzee population is well above the mean in the human population, at every decade of life, in both males and females (Fig. 2). Second and most intriguingly, we see that unlike the case in humans, captive chimpanzees have high serum cholesterol levels even in the early decades of life (Fig. 2). Thus, the chimpanzee vasculature is exposed to high cholesterol levels throughout adolescence and adulthood. Indeed the levels in very young chimpanzees fall into a high enough range wherein pharmacological statin therapy would now be seriously considered in children (Daniels and Greer 2008).
Figure 2.
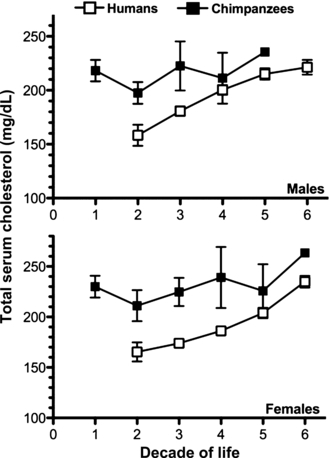
Longitudinal age-based comparison of total serum cholesterol in human and captive chimpanzee populations. Chimpanzee and human cholesterol levels were taken from two independent, original articles and plotted here, on the same axis, for comparison. Total serum cholesterol data from both publications was grouped by sex and age in decades. Human data were acquired from 6757 participants in the Framingham Heart Study, aged 15–79 years (Abbott et al. 1983). Chimpanzee data were acquired from 252 chimpanzees, aged 0–60, living in captivity at the Yerkes Primate Center (Herndon and Tigges 2001). Data are plotted as mean ± 95% CI. Error bars are not shown for the oldest chimps (5th decade in males, n = 1; 6th decade in females, n = 3) due to the very small sample size. Note that the total serum cholesterol (Y axis) starts at 100 mg/dL, to make the differences clearer.
Furthermore, evidence also indicates that chimpanzee serum cholesterol profiles have similar LDL and HDL components as humans. For instance, LDL cholesterol levels of captive chimpanzees (171 ± 16 mg/dL, n = 4) were found to be well above matched human levels (108 ± 2 mg/dL, n = 5) and already in a range comparable to the majority of at-risk humans (Vastesaeger et al. 1975; Blaton and Peeters 1976). Chimpanzee LDL cholesterol can be further increased experimentally by atherogenic diets (Vastesaeger et al. 1975; Blaton and Peeters 1976; Srinivasan et al. 1976). This augmentation in plasma cholesterol can be attributed to an increase in LDL levels with a concomitant suppression of HDL levels (Srinivasan et al. 1976), similar to that seen in hypercholesterolemic humans. Thus, many chimpanzees have serum cholesterol profiles that would be considered pro-atherogenic and requiring treatment in humans.
Additionally, chimpanzees and all other great apes are homozygous for the ancestral APOE4 allele (Hanlon and Rubinsztein 1995; Hacia et al. 1999), a well-known risk factor for severe atherosclerosis in humans (Eichner et al. 1993; Ilveskoski et al. 1999; Altenburg et al. 2007). Furthermore, chimpanzees have high levels of lipoprotein-associated phospholipase A(2) or LP(a) (Doucet et al. 1994; Huby et al. 2001), another independent cardiovascular risk marker (Corson et al. 2008). Despite hypercholesterolemia, high LP(a) levels and having the high-risk APOE4 allele, no deaths by myocardial infarction have been cataloged in the autopsy records of chimpanzees at major US primate centers. Thus, unlike the case with humans (Steinberg 2006), serum lipid profiles in great apes do not seem to correlate with risk, nor provide an explanation for the rarity of coronary artery disease. Of course, detailed studies of other aspects of great ape lipid profiles (e.g., HDL levels, HDL/LDL ratios, apo B/apo A-I ratios) (Fernandez and Webb 2008) need to be studied, to ascertain if there are other explanations.
Two mysteries to be solved
When taken together, the data suggest that there are two independent mysteries to be solved. First, why do humans not suffer from a high frequency of diffuse IMF similar to that seen in evolutionarily related great apes? Is this due to specific evolutionary genetic changes in the human lineage that protects us from this disease process? Conversely, could it be related to some as yet unknown exposure of apes to some pathogenic agent (toxin or virus) in captivity? In this regard, it would be very interesting to know if such myocardial fibrosis occurs in the great apes in the wild. The second mystery is why the great apes do not often develop the severe forms of atherosclerosis and coronary thrombosis characteristic of humans. Chimpanzees in captivity have a life-span long enough to be permissive of atherogenesis, and as discussed, many have lipid profile levels that would be considered a significant risk for coronary cardiovascular disease in humans. Captive great apes are also not free of other human coronary risk factors such as hypertension exacerbated by high salt intake (Denton et al. 1995; Elliott et al. 2007) and obesity. They are also relatively sedentary when compared to their wild counterparts, a lifestyle similar to modern humans. However, despite such similarities, there is relative lack of severe atherosclerotic lesions in chimpanzees compared to humans, and myocardial infarctions in great apes are very rare.
Thus, it appears likely that there are other genetic and/or environmental factors that predispose humans to severe atherosclerosis. One possibility we have recently suggested is an immune response to the metabolic incorporation and accumulation of a dietary nonhuman sialic acid N-glycolylneuraminic acid (Neu5Gc) in large blood vessel endothelia, in the face of an ongoing antibody immune response against this foreign molecule (Nguyen et al. 2005; Padler-Karavani et al. 2008; Varki 2008). We hypothesize that this combination generates additional chronic inflammation in human blood vessels, accelerating the atherosclerotic process. Another contributing factor could be the hyper-reactive phenotype of human T cells, relative to those of chimpanzees (Nguyen et al. 2006; Varki 2008), potentially aggravating the chronic inflammation seen in atherosclerotic plaques. We are currently working towards a mouse model that is ‘humanized’ from the point of view of sialic acid biology. However, the resistance of the mouse to spontaneous atherosclerosis is a significant barrier, and standard mouse models of atherosclerosis require major perturbations of cholesterol homeostasis and inflammation to engender disease progression. Thus, studies in mice may or may not be completely relevant to the differences of human and chimpanzee pathology.
In search of a mechanism for IMF in great apes
We and others are currently unable to identify any specific marker or measurement that could predict the risk of the IMF cardiac disease that is common in chimpanzees, and the underlying etiology that remains unknown. As an initial attempt to search for mechanisms, we compared apparently normal-appearing myocardial sections from humans and great apes using histochemical methods. Paraffin sections of hearts from humans, chimpanzees, gorillas, and orangutans were stained using the Masson-Trichrome stain for collagen. As shown in Fig. 3, otherwise normal heart sections from all three great apes showed collagen bundles that appeared to divide the heart muscle along the planes where the larger blood vessels were situated. This pattern was not observed in the human heart sections. The sections of heart muscle were from cases not related to sudden deaths or heart failure, although some of the human cases had a clinical history of hypertension. Thus, even in otherwise healthy-appearing great ape hearts there is a pattern of increased collagen in the heart muscle, which is visible in the connective tissue surrounding blood vessels and extends for some distances, conferring an appearance of heart muscle in ‘bundles’. This was observed also in a chimpanzee as young as 13 years old. It remains uncertain whether this represents the normal condition of great apes, or incipient IMF.
Figure 3.
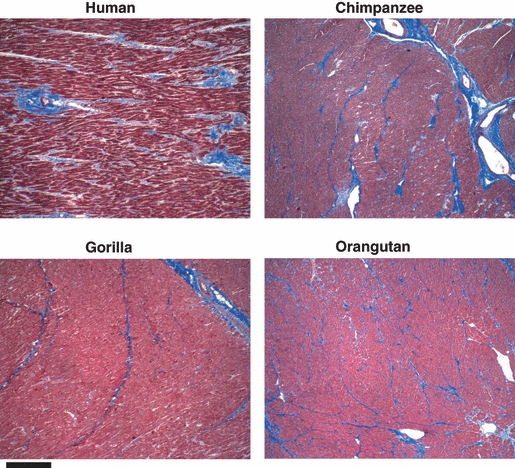
Masson's trichrome staining for collagen in histologically normal-appearing myocardial tissue sections from humans and great apes. ×40 magnification. Scale bar represents 50 microns. Examples are shown of the staining patterns in human and great apes. The results shown are typical of that seen with six human, six chimpanzee, three gorilla, and four orangutan samples.
Given the known differences in sialic acid biology between humans and great apes (Varki 2008), we also stained normal-appearing myocardial sections from humans and apes with two lectins that bind sialic acids: Sambucus nigra agglutinin (SNA) which recognizes terminal Siaα2-6Galβ1-4GlcNAcβ- units on N-linked glycan chains of glycoproteins, and Maackia Amurensis hemagglutinin (MAH), which recognizes Siaα2-3Gal termini on various glycoconjugates (Martin et al. 2002; Varki and Varki 2007). Both SNA and MAH strongly stained large areas of heart sections from chimpanzees, gorillas and orangutans, with MAH staining again showing evidence of encircling ‘bundles’ that were not seen in human heart sections (Fig. 4). These differences with SNA and MAH lectin staining imply that terminal sialic acids are much denser in the great ape heart. As terminal sialic acids are often the target for viral pathogens (Varki and Varki 2007; Varki 2008), one can speculate that this may render ape hearts more prone to infections by some viral pathogen that binds sialic acids and initiates the fibrotic process. In this regard, occasional lymphocytic infiltrates can occasionally surround the scarred areas, suggesting the possibility of precedent viral myocarditis in some cases (data not shown).
Figure 4.
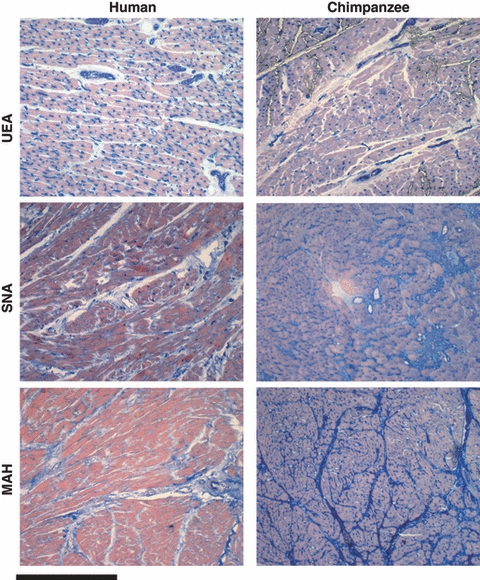
Lectin staining of histologically normal-appearing myocardial sections from humans and great apes. Examples are shown of the typical lectin staining patterns (blue) in human and chimpanzee myocardial sections with Ulex Europaeus Agglutinin (UEA), which recognizes terminal fucose residues in the sequence Fucα1-2Galβ1-4GlcNAcβ-; Sambucus nigra agglutinin (SNA) which recognizes terminal Siaα2-6Galβ1-4GlcNAcβ- units on N-linked glycan chains of glycoproteins; and, Maackia Amurensis hemagglutinin (MAH), which recognizes Siaα2-3Gal termini on various glycoconjugates (Martin et al. 2002; Varki and Varki 2007). The results shown are typical of that seen with 11 human and seven chimpanzee samples. Three gorilla and four orangutan samples gave results similar to that of the chimpanzee sections.
We also used the Ulex Europaeus agglutinin (UEA) lectin, which recognizes terminal fucose residues in the sequence Fucα1-2Galβ1-4GlcNAcβ-, and is very useful for the identification of blood vessels in human sections. Remarkably, we found that while the UEA lectin stained small coronary vessels as well as numerous capillary blood vessels in human hearts, sections from all three apes showed only staining of the larger vessels, with many fewer intermyocardial fiber capillaries. Endurance running is thought to be a derived capability of the genus Homo (Bramble and Lieberman 2004). Thus, the apparently larger number of human intermyocardial capillaries identified by the UEA lectin may imply a richer myocardial blood supply in humans, and this may possibly reduce the risk of developing myocardial fibrosis. Alternatively, such differences in glycosylation could by themselves alter interactions amongst extracellular matrix components, perhaps predisposing the apes to the fibrotic process. These and other possibilities deserve further investigation. A truly prospective, longitudinal study of captive chimpanzee cohorts using noninvasive techniques such as echocardiography during the natural course of an animal's lifetime would also be an improvement over the retrospective data currently available for analyses like ours. Regardless of the model or the study, answers to these questions would be of value not only to the medical care of both humans and other hominids, but also to our understanding of unique features of human evolution.
Acknowledgments
The authors wish to thank the numerous dedicated workers who cared for these great apes over their lifetimes, and assisted in the collection of necropsy tissues. This work was supported by NIH grants to A.V. and N.V. (R01GM32373 and R01CA38701), to the Yerkes National Primate Research Center (RR00165), and to the Primate Foundation of Arizona (2 U42 RR15090-07). We also acknowledge general support from the Mathers Foundation of New York.
Literature cited
- Abbott RD, Garrison RJ, Wilson PW, Epstein FH, Castelli WP, Feinleib M, LaRue C. Joint distribution of lipoprotein cholesterol classes. The Framingham study. Arteriosclerosis. 1983;3:260–272. doi: 10.1161/01.atv.3.3.260. [DOI] [PubMed] [Google Scholar]
- Altenburg M, Johnson L, Wilder J, Maeda N. Apolipoprotein E4 in macrophages enhances atherogenesis in a low density lipoprotein receptor-dependent manner. Journal of Biological Chemistry. 2007;282:7817–7824. doi: 10.1074/jbc.M610712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder CJ, Chang MK, Shaw PX, Miller YI, Hartvigsen K, et al. Innate and acquired immunity in atherogenesis. Nature Medicine. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- Binder CJ, Shaw PX, Chang MK, Boullier A, Hartvigsen K, et al. The role of natural antibodies in atherogenesis. Journal of Lipid Research. 2005;46:1353–1363. doi: 10.1194/jlr.R500005-JLR200. [DOI] [PubMed] [Google Scholar]
- Blaton V, Peeters H. The nonhuman primates as models for studying human atherosclerosis: studies on the chimpanzee, the baboon and the rhesus macacus. Advances in Experimental Medicine and Biology. 1976;67:33–64. doi: 10.1007/978-1-4614-4618-7_2. [DOI] [PubMed] [Google Scholar]
- Boden WE. Management of chronic coronary disease: is the pendulum returning to equipoise? American Journal of Cardiology. 2008;101:69D–74D. doi: 10.1016/j.amjcard.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Bramble DM, Lieberman DE. Endurance running and the evolution of Homo. Nature. 2004;432:345–352. doi: 10.1038/nature03052. [DOI] [PubMed] [Google Scholar]
- Caglayan E, Stauber B, Collins AR, Lyon CJ, F Yin, et al. Differential roles of cardiomyocyte and macrophage peroxisome proliferator-activated receptor gamma in cardiac fibrosis. Diabetes. 2008;57:2470–2479. doi: 10.2337/db07-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calara F, Silvestre M, Casanada F, Yuan N, Napoli C, et al. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein E-deficient and LDL receptor-deficient mice. Journal of Pathology. 2001;195:257–263. doi: 10.1002/path.915. [DOI] [PubMed] [Google Scholar]
- Corson MA, Jones PH, Davidson MH. Review of the evidence for the clinical utility of lipoprotein-associated phospholipase A2 as a cardiovascular risk marker. American Journal of Cardiology. 2008;101:41F–50F. doi: 10.1016/j.amjcard.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Cousins D. Mortality factors in captive gorillas (Gorilla gorilla. International Zoo News. 1983;30:5–17. [Google Scholar]
- Daniels SR, Greer FR. Lipid screening and cardiovascular health in childhood. Pediatrics. 2008;122:198–208. doi: 10.1542/peds.2008-1349. [DOI] [PubMed] [Google Scholar]
- Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. New England Journal of Medicine. 1984;310:1137–1140. doi: 10.1056/NEJM198405033101801. [DOI] [PubMed] [Google Scholar]
- Denton D, Weisinger R, Mundy NI, Wickings EJ, Dixson A, et al. The effect of increased salt intake on blood pressure of chimpanzees. Nature Medicine. 1995;1:1009–1016. doi: 10.1038/nm1095-1009. [DOI] [PubMed] [Google Scholar]
- Diez J. Mechanisms of cardiac fibrosis in hypertension. Journal of Clinical Hypertension (Greenwich) 2007;9:546–550. doi: 10.1111/j.1524-6175.2007.06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doane CJ, Lee DR, Sleeper MM. Electrocardiogram abnormalities in captive chimpanzees (Pan troglodytes. Comparative Medicine. 2006;56:512–518. [PubMed] [Google Scholar]
- Doucet C, Huby T, Chapman J, Thillet J. Lipoprotein[a] in the chimpanezee: relationship of apo[a] phenotype to elevated plasma Lp[a] levels. Journal of Lipid Research. 1994;35:263–270. [PubMed] [Google Scholar]
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- Eichner JE, Kuller LH, Orchard TJ, Grandits GA, McCallum LM, et al. Relation of apolipoprotein E phenotype to myocardial infarction and mortality from coronary artery disease. American Journal of Cardiology. 1993;71:160–165. doi: 10.1016/0002-9149(93)90732-r. [DOI] [PubMed] [Google Scholar]
- Elliott P, Walker LL, Little MP, Blair-West JR, Shade RE, et al. Change in salt intake affects blood pressure of chimpanzees: implications for human populations. Circulation. 2007;116:1563–1568. doi: 10.1161/CIRCULATIONAHA.106.675579. [DOI] [PubMed] [Google Scholar]
- Escobar E. Hypertension and coronary heart disease. Journal of Human Hypertension. 2002;16(Suppl. 1):S61–S63. doi: 10.1038/sj.jhh.1001345. [DOI] [PubMed] [Google Scholar]
- Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:996–1003. doi: 10.1161/ATVBAHA.106.131755. [DOI] [PubMed] [Google Scholar]
- Farb A, Tang AL, Burke AP, Sessums L, Liang Y, et al. Sudden coronary death. Frequency of active coronary lesions, inactive coronary lesions, and myocardial infarction. Circulation. 1995;92:1701–1709. doi: 10.1161/01.cir.92.7.1701. [DOI] [PubMed] [Google Scholar]
- Fernandez ML, Webb D. The LDL to HDL cholesterol ratio as a valuable tool to evaluate coronary heart disease risk. Journal of the American College of Nutrition. 2008;27:1–5. doi: 10.1080/07315724.2008.10719668. [DOI] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gay MA, Gonzalez-Juanatey C, Miranda-Filloy JA, Garcia-Porrua C, Llorca J, et al. Cardiovascular disease in rheumatoid arthritis. Biomedicine and Pharmacotherapy. 2006;60:673–677. doi: 10.1016/j.biopha.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Gray R, O'Neal RM, Jordan FB. Sudden death associated with atherosclerosis in a gorilla. Journal of the American Veterinary Medical Association. 1981;179:1306–1307. [PubMed] [Google Scholar]
- Hacia JG, Fan JB, Ryder O, Jin L, Edgemon K, et al. Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays [see comments] Nature Genetics. 1999;22:164–167. doi: 10.1038/9674. [DOI] [PubMed] [Google Scholar]
- Hainsey BM, Hubbard GB, Leland MM, Brasky KM. Clinical parameters of the normal baboons (Papio species) and chimpanzees (Pan troglodytes. Laboratory Animal Science. 1993;43:236–243. [PubMed] [Google Scholar]
- Hanlon CS, Rubinsztein DC. Arginine residues at codons 112 and 158 in the apolipoprotein E gene correspond to the ancestral state in humans. Atherosclerosis. 1995;112:85–90. doi: 10.1016/0021-9150(94)05402-5. [DOI] [PubMed] [Google Scholar]
- Hansen JF, Alford PL, Keeling ME. Diffuse myocardial fibrosis and congestive heart failure in an adult male chimpanzee. Veterinary Pathology. 1984;21:529–531. doi: 10.1177/030098588402100514. [DOI] [PubMed] [Google Scholar]
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. New England Journal of Medicine. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Stanford C. Periodontal disease and atherosclerosis: from dental to arterial plaque. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:1309–1311. doi: 10.1161/01.ATV.0000087144.24654.71. [DOI] [PubMed] [Google Scholar]
- Herndon JG, Tigges J. Hematologic and blood biochemical variables of captive chimpanzees: cross-sectional and longitudinal analyses. Comparative Medicine. 2001;51:60–69. [PubMed] [Google Scholar]
- Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends in Biochemical Sciences. 2007;32:71–77. doi: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell S, Hoffman K, Bartel L, Schwandt M, Morris J, et al. Normal hematologic and serum clinical chemistry values for captive chimpanzees (Pan troglodytes. Comparative Medicine. 2003;53:413–423. [PubMed] [Google Scholar]
- Hubbard GB, Lee DR, Eichberg JW. Diseases and pathologies at the Southwest Foundation for Biomedical Research. American Journal of Primatology. 1991;24:1–8. [Google Scholar]
- Huby T, Dachet C, Lawn RM, Wickings J, Chapman MJ, et al. Functional analysis of the chimpanzee and human apo(a) promoter sequences: identification of sequence variations responsible for elevated transcriptional activity in chimpanzee. Journal of Biological Chemistry. 2001;276:22209–22214. doi: 10.1074/jbc.M102204200. [DOI] [PubMed] [Google Scholar]
- Ilveskoski E, Perola M, Lehtimaki T, Laippala P, Savolainen V, et al. Age-dependent association of apolipoprotein E genotype with coronary and aortic atherosclerosis in middle-aged men: an autopsy study. Circulation. 1999;100:608–613. doi: 10.1161/01.cir.100.6.608. [DOI] [PubMed] [Google Scholar]
- John BT, Tamarappoo BK, Titus JL, Edwards WD, Shen WK, et al. Global remodeling of the ventricular interstitium in idiopathic myocardial fibrosis and sudden cardiac death. Heart Rhythm. 2004;1:141–149. doi: 10.1016/j.hrthm.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Lammey ML, Baskin GB, Gigliotti AP, Lee DR, Ely JJ, et al. Interstitial myocardial fibrosis in a captive chimpanzee (Pan troglodytes) population. Comparative Medicine. 2008a;58:389–394. [PMC free article] [PubMed] [Google Scholar]
- Lammey ML, Lee DR, Ely JJ, Sleeper MM. Sudden cardiac death in 13 captive chimpanzees (Pan troglodytes) Journal of Medical Primatology. 2008b;37(Suppl. 1):39–43. doi: 10.1111/j.1600-0684.2007.00260.x. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM. Inflammation and atherosclerosis: role of C-reactive protein in risk assessment. American Journal of Medicine. 2004;116(Suppl. 6A):9S–16S. doi: 10.1016/j.amjmed.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Lowenstine LJ. A primer of primate pathology: lesions and nonlesions. Toxicologic Pathology. 2003;31(Suppl):92–102. doi: 10.1080/01926230390177668. [DOI] [PubMed] [Google Scholar]
- Manning GW. Coronary disease in the ape. American Heart Journal. 1942;23:719–724. [Google Scholar]
- Martin LT, Marth JD, Varki A, Varki NM. Genetically altered mice with different sialyltransferase deficiencies show tissue-specific alterations in sialylation and sialic acid 9-O-acetylation. Journal of Biological Chemistry. 2002;277:32930–32938. doi: 10.1074/jbc.M203362200. [DOI] [PubMed] [Google Scholar]
- McNamara TE, Dolensek EP, Liu S, Dierenfeld ED. Cardiomyopathy associated with vitamin E deficiency in two mountain lowland gorillas. Proceedings of the First International Conference on Zoological and Avian Medicine. 1987;1:493. [Google Scholar]
- Meerarani P, Badimon JJ, Zias E, Fuster V, Moreno PR. Metabolic syndrome and diabetic atherothrombosis: implications in vascular complications. Current Molecular Medicine. 2006;6:501–514. doi: 10.2174/156652406778018680. [DOI] [PubMed] [Google Scholar]
- Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93:1137–1146. doi: 10.1136/hrt.2003.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson L, Montali RJ. Pathology and diseases of great apes at the National Zoological Park Zoo. Zoo Biology. 1990;9:99–105. [Google Scholar]
- Nguyen DH, Tangvoranuntakul P, Varki A. Effects of natural human antibodies against a nonhuman sialic acid that metabolically incorporates into activated and malignant immune cells. Journal of Immunology. 2005;175:228–236. doi: 10.4049/jimmunol.175.1.228. [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of Siglec expression on T lymphocytes during human evolution. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7765–7770. doi: 10.1073/pnas.0510484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padler-Karavani V, Yu H, Cao H, Chokhawala H, Karp F, et al. Diversity in specificity, abundance, and composition of anti-Neu5Gc antibodies in normal humans: potential implications for disease. Glycobiology. 2008;18:818–830. doi: 10.1093/glycob/cwn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Friday G, Furie K, Go A, et al. Heart disease and stroke statistics--2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–e171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- Schmidt RE. Systemic pathology of chimpanzees. Journal of Medical Primatology. 1978;7:274–318. doi: 10.1159/000459914. [DOI] [PubMed] [Google Scholar]
- Schulman FY, Farb A, Virmani R, Montali RJ. Fibrosing cardiomyopathy in lowland gorillas (Gorilla gorilla gorilla) in the United States: A retrospective study. Journal of Zoo and Wildlife Medicine. 1995;26:43–51. [Google Scholar]
- Seiler BM, Dick EJ, Jr, Guardado-Mendoza R, Vandeberg JL, Williams JT, et al. Spontaneous heart disease in the adult chimpanzee (Pan troglodytes. Journal of Medical Primatology. 2008 doi: 10.1111/j.1600-0684.2008.00307.x. Jul 30 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer Y, Shoenfeld Y. Mechanisms of disease: atherosclerosis in autoimmune diseases. Nature Clinical Practice. Rheumatology. 2006;2:99–106. doi: 10.1038/ncprheum0092. [DOI] [PubMed] [Google Scholar]
- Shiomi M, Ito T, Yamada S, Kawashima S, Fan J. Development of an animal model for spontaneous myocardial infarction (WHHLMI rabbit) Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:1239–1244. doi: 10.1161/01.ATV.0000075947.28567.50. [DOI] [PubMed] [Google Scholar]
- Soltesz P, Szekanecz Z, Kiss E, Shoenfeld Y. Cardiac manifestations in antiphospholipid syndrome. Autoimmunity Reviews. 2007;6:379–386. doi: 10.1016/j.autrev.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Srinivasan SR, Radhakrishnamurthy B, Smith CC, Wolf RH, Berenson GS. Serum lipid and lipoprotein responses of six nonhuman primate species to dietary changes in cholesterol levels. Journal of Nutrition. 1976;106:1757–1767. doi: 10.1093/jn/106.12.1757. [DOI] [PubMed] [Google Scholar]
- Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part V: the discovery of the statins and the end of the controversy. Journal of Lipid Research. 2006;47:1339–1351. doi: 10.1194/jlr.R600009-JLR200. [DOI] [PubMed] [Google Scholar]
- Steinberg D. The statins in preventive cardiology. New England Journal of Medicine. 2008;359:1426–1427. doi: 10.1056/NEJMp0806479. [DOI] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing, Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Traub O, Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arteriosclerosis, Thrombosis, and Vascular Biology. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- Varki A. Multiple changes in sialic acid biology during human evolution. Glycoconjugate Journal. 2008a doi: 10.1007/s10719-008-9183-z. Sep 7 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Sialic acids in human health and disease. Trends in Molecular Medicine. 2008b;14:351–360. doi: 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Nelson D. Genomic differences between humans and chimpanzees. Annual Review of Anthropology. 2007;36:191–209. [Google Scholar]
- Varki NM, Varki A. Diversity in cell surface sialic acid presentations: implications for biology and disease. Laboratory Investigation. 2007;87:851–857. doi: 10.1038/labinvest.3700656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Geschwind DH, Eichler EE. Explaining human uniqueness: genome interactions with environment, behaviour and culture. Nature Review. Genetics. 2008;9:749–763. doi: 10.1038/nrg2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan RS, Sullivan LM, Wilson PW, Sempos CT, Sundstrom J, et al. Relative importance of borderline and elevated levels of coronary heart disease risk factors. Annals of Internal Medicine. 2005;142:393–402. doi: 10.7326/0003-4819-142-6-200503150-00005. [DOI] [PubMed] [Google Scholar]
- Vastesaeger M, Peeters H, Blaton V, Petrovas C, Mortelmans J. Somes aspects of the chimpanzee as a model for experimental atherosclerosis. Advances in Experimental Medicine and Biology. 1975;63:359–369. doi: 10.1007/978-1-4684-3258-9_26. [DOI] [PubMed] [Google Scholar]
- Virmani R, Burke AP, Farb A. Sudden cardiac death. Cardiovascular Pathology. 2001;10:211–218. doi: 10.1016/s1054-8807(01)00091-6. [DOI] [PubMed] [Google Scholar]
- White HD, Chew DP. Acute myocardial infarction. Lancet. 2008;372:570–584. doi: 10.1016/S0140-6736(08)61237-4. [DOI] [PubMed] [Google Scholar]
- Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EF. Systemic effects of smoking. Chest. 2007;131:1557–1566. doi: 10.1378/chest.06-2179. [DOI] [PubMed] [Google Scholar]