

Abstract
Because of intrinsic differences in their genetic architectures, wild populations invaded by domesticated individuals could experience population-specific consequences following introgression by genetic material of domesticated origin. Expression levels of 16 000 transcripts were quantified by microarrays in liver tissue from farm, wild, and farm-wild backcross (i.e. F1 farm-wild hybrid × wild; total n = 50) Atlantic salmon (Salmo salar) raised under common environmental conditions. The wild populations and farm strain originated from three North American rivers in eastern Canada (Stewiacke, Tusket, and Saint John rivers, respectively). Analysis of variance revealed 177 transcripts with different expression levels among the five strains compared. Five times more of these transcripts were differentiated between farmed parents and Tusket backcrosses (n = 53) than between Stewiacke backcrosses and their farmed parents (n = 11). Altered biological processes in backcrosses also differed between populations both in number and in the type of processes impacted (metabolism vs immunity). Over-dominant gene expression regulation in backcrosses varied considerably between populations (23% in Stewiacke vs 44% in Tusket). Hence, the consequences of introgression of farm genetic material on gene expression depended on population-specific genetic architectures. These results support the need to evaluate impacts of farm-wild genetic interactions at the population scale.
Keywords: conservation, contemporary evolution, evolutionary applications, farmed salmon, gene expression, genetic architecture, hybridization, introgression
Introduction
Domestication has been closely linked to the rise of human history (Lev-Yadun et al. 2000; Zeder 2006). It is defined as artificial selection, over many generations, for traits deemed advantageous to humans in animal and plant species (Diamond 2002; Mignon-Grasteau et al. 2005; Taberlet et al. 2008). Morphology, behaviour, and life history are the most commonly selected traits, procuring benefits to humans (Diamond 2002). The mechanisms permitting improvement of domesticated strains lie in the heritable genetic basis of the phenotypic characteristics being selected (Jensen 2006). The process of domestication could lead to changes in the genetic architecture of the strains under selection, that is, changes in the inter-related genetic effects that are responsible for the development of phenotypic characters (Burger et al. 2008). Domesticated populations or species may be considered invasive wherever they are found in the wild, either as established feral populations or recurrent due to chronic immigration. Indeed, domesticated species are known to be responsible for a high proportion of invasion cases (Mack et al. 2000; Sakai et al. 2001).
Invasive species can be defined as species that proliferate in new environments or colonize them repeatedly (Mack et al. 2000; Lee 2002). They can pose ecological and evolutionary threats to local populations by competing for space and resources (Randi 2008), or by inducing shifts in ecosystem balance and selection regimes (Carroll 2007). In the case of invading domesticated species, which may readily hybridize with local wild populations, ecological consequences are compounded by possible genetic interactions (Suarez and Tsutsui 2008), which are thought to result in a reduction of genetic diversity and a loss of local adaptation (Mooney and Cleland 2001; Sakai et al. 2001).
The recent and ongoing domestication of Atlantic salmon (Salmo salar L.) through aquaculture activities offers a useful system to study the genetic consequences of hybridization between wild populations and invasive domesticated strains. Atlantic salmon farmed strains have been under selection for 5–12 generations in different parts of the globe. Traits under selection include growth rate, age at maturity, and pathogen resistance (Friars et al. 1995; Gjoen and Bentsen 1997; Glebe 1998; O'Flynn et al. 1999; Hindar et al. 2006). As a consequence, farmed salmon generally differ from their ancestral wild populations by having a higher growth rate and older age at maturity. Unintentional selection additionally leads to reduced genetic diversity, increased fat content, increased aggressiveness, and reduced response to predation, which may result in poorer survival and reproductive capacity of farmed salmon in the wild (Fleming and Einum 1997; Fleming et al. 2000; McGinnity et al. 2003; Skaala et al. 2005). Consequently, introgressive hybridization between farmed and wild fish may have major consequences for the fitness of individuals in wild populations (McGinnity et al. 2003; Castillo et al. 2008; Hutchings and Fraser 2008).
It is widely accepted that modulation of gene expression regulation plays an important role in evolution (King and Wilson 1975; Stern 2000; Oleksiak et al. 2002; Wittkopp 2007; Fay and Wittkopp 2008). For example, small changes in temporal and spatial gene expression can have dramatic consequences on development and phenotypic characteristics (Lindsey and Topping 1993; Cho et al. 1998; Yabuta et al. 2006). By measuring cDNA levels for thousands of transcripts simultaneously, DNA microarrays represent a powerful means of identifying evolutionarily important gene expression differences (Gibson 2002; Ranz and Machado 2006). Since the level of gene transcription is correlated with the presence of the protein that it encodes for, gene expression regulation is likely to be linked to physiological differences among individuals and populations (Schulte 2004). In Atlantic salmon, Roberge et al. (2006) showed that 5–7 generations of domestication were sufficient to induce important changes in patterns of gene expression between farmed and wild salmon. Recently, Roberge et al. (2008) demonstrated that introgressive hybridization between farmed and wild salmon sharing the same river of origin induced mis-regulation at numerous genes involved in a wide variety of physiological functions.
This project builds on these previous studies (Roberge et al. 2006, 2008) by comparing patterns of liver gene expression between backcross hybrids generated from crossing a strain of farmed salmon with two wild populations displaying distinct life history attributes. The main objective was to assess the differential consequences of introgressive hybridization in populations characterized by different genetic backgrounds. More specifically, salmon from two wild populations and one farm strain were firstly used to generate two F1 (farm-wild) hybrids. These wild and hybrid strains were raised under common environmental conditions for their entire lives and then crossed to produce a new generation of these strains and, additionally, two backcross hybrids. The liver transcriptomes of the three parental strains and two backcross hybrids were then compared to evaluate the genetic consequences of introgression of farm genetic material in the two different wild populations. In this way, it was possible to uncover genes differentially expressed between the five strains, which may be indicative of altered biological processes, as well as patterns of additive versus nonadditive expression regulation inheritance.
Our work therefore expands upon previous research efforts which paid little attention to the extent to which domesticated genomes might interact differently when mixed with distinct wild genomes. Indeed, to our knowledge, this project represents the first attempt to compare population-specific impacts of domestic-wild hybridization on gene expression for any vertebrate species. This represents a significant contribution to our current understanding of the impact of farm escapes on wild populations. Indeed, our results revealed how genetically distinct wild populations from a small geographical area can be differently affected by introgression from a domesticated strain. This in turns indicates that aquaculture accidental releases should be more tightly controlled and their potential genetic impacts be assessed on a smaller scale, ideally at the population level.
Methods
Sampling
Atlantic salmon genitors from the Stewiacke River (NS, 45°08′00″N, 63°22′57″W) and Tusket River (NS, 43°41′00″N, 65°56′57″W) populations, as well as from the main farm strain used in the Bay of Fundy aquaculture industry (20 individuals each), were used to generate our crosses. The farm strain originally comprised individuals from the Saint-John River population (NB, 45°16′00″N, 66°04′00″W) and had undergone four generations of selection in 2005 when the genitors were sampled. Both the Stewiacke and Saint-John rivers are part of the Bay of Fundy area, while the Tusket River flows in the Atlantic Ocean just outside the Bay of Fundy. About 200 km separate the Saint-John River from each of the Stewiacke and Tusket rivers. In this experiment, wild salmon from the Stewiacke and Tusket rivers were crossed with farmed individuals to generate two strains of F1 hybrids. The two hybrid strains were then crossed to their respective wild ancestor population to generate backcross hybrids. For each generation, the individuals were reared without intentional selection in a controlled environment to avoid environmental effects and biases in selecting the genitors for the next generation. Thus, salmon from five crosses were compared in this study: the farm strain, the two wild populations, and the two backcross strains. More details about how the crosses were made can be found in Fraser et al. (2008) and Houde et al. (2009).
RNA extraction, cDNA hybridization, and microarray experimental design
Individuals were killed prior to being weighed, measured, and sampled 7 months after hatching. Livers were then extracted and promptly frozen at −80°C. Great care was taken to ensure that no environmental influences, such as time of day at killing or weight of fish, had any bias within the different strains. Total RNA was extracted from the livers of 10 individuals randomly drawn from each strain, for a total of 50 individuals. All steps that involved RNA were performed at 4°C, unless stated otherwise. The extractions followed a standard phenol-chloroform protocol. The livers (50–200 μg) were mixed in 1 mL of TRIzol® Reagent, using a Quiagen TissuLyser homogenizer; 200 μL of chloroform was added. After mixing and centrifuging (12 000 g, 10 min), the aqueous layer was transferred and 500 μL of isopropanol (Sigma, Saint-Louis, Missouri, US) was added. The samples were mixed and stored at −20°C for an hour and then centrifuged (12 000 g, 15 min) before discarding the isopropanol. The RNA pellets were washed in 1 mL 70% ethanol, left to stand for 10 min, and centrifuged (12 000 g, 10 min). Ethanol was removed. The samples were dried for 5 min at room temperature, and the ethanol wash steps were repeated twice. The pellets were then resuspended in 100 μL non-DEPC treated nuclease-free water (Ambion) spiked with 1 μL RNase inhibitor (Ambion). The samples were filtered using Millipore microcons and the RNA resuspended in 50 μL non-DEPC treated nuclease-free water (Ambion, Austin, Texas, US) spiked with 1 μL RNase inhibitor (Ambion). The samples were then treated with DNase I. For each μg of RNA treated, a solution of 10 μL was made, containing: 1 μL of 10X DNase I Reaction Buffer [200 μm Tris–HCl (pH 8.4), 20 mm MgCl2, and 500 μm KCl], 1 μL DNase I, amplification grade (Invitrogen, Carlsbad, California, US), and completed to 10 μL using DEPC-treated water. The mix was incubated at room temperature for 30 min and the DNase inactivated by adding 1 μL of 25 mm EDTA and heating to 65°C for 10 min. The samples were then concentrated by a factor of 25 with Millipore microcons. RNA was quantified using a GeneQuant capillary spectrometer from Pharmacia and its integrity assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California, US). Fifteen micrograms RNA from each individual was retro-transcribed and labelled using Genisphere 3DNA Array 50 kit, Invitrogen's Superscript II retro-transcriptase, and Genisphere Cy3 and Alexa 647 dyes. A modified protocol, adapted from the Genisphere Array 50 Protocol, was used and can be found at http://web.uvic.ca/cbr/grasp/.
Briefly, 15 μg total RNA was reverse-transcribed by using special oligo-d(T) primers with 5′ unique sequence overhangs for the labelling reactions. Microarray slides were prepared for hybridization by washing twice for 5 min in 0.1% SDS, washing five times for 1 min in MilliQ H2O, and drying by centrifugation (5 min at 514 g in 50 mL conical tubes). The cDNA was hybridized to the microarrays in a formamide-based buffer (25% formamide, 4X SSC, 0.5% SDS, 2X Denhardt's solution) with competitor DNA [LNA dT bloker (Genisphere, Hatfield, Pennsylvania, US), human COT-1 DNA (Sigma)] for 16 h in hybridization chambers (Corning, New York, US) placed in a 51°C water bath. The arrays were washed once for 5 min at 45°C in 2X SSC, 0.1% SDS, twice for 3 min in 2X SSC, 0.1% SDS at room temperature (RT), twice for 3 min in 1X SSC at RT, twice for 3 min in 0.1X SSC at RT, and dried by centrifugation. The Cy3 and Alexa 647 fluorescent dyes attached to DNA dendrimer probes (3DNA capture reagent, Genisphere) were then hybridized to the cDNA bound on the microarray using the same hybridization solution as earlier. The 3DNA capture reagents bound to their complementary cDNA capture sequences on the oligo d(T) primers. This second hybridization was carried over a two hour period in hybridization chambers (Corning) placed in a 51°C water bath. The arrays were then washed and dried by centrifugation as before.
The cDNA microarrays used in this study were obtained from the consortium for Genomics Research on All Salmonids Project (cGRASP) and are printed with 16 006 salmonid cDNA sequences. The sequences were obtained from over 175 Atlantic salmon and rainbow trout cDNA libraries of various tissues at various development stages. This effort yielded more than 300 000 salmonid cDNA sequence reads, which were assembled into over 40 000 unique contigs using PHRAP (minimum overlap score: 100, repeat stringency: 0.99). The contigs were aligned with GenBank nucleotide and amino acid sequence databases using BLASTN and BLASTX, respectively. Significance threshold for a significant BLAST hit was fixed at E = 1 × 10−15 (von Schalburg et al. 2005, 2008). In the text, the word ‘transcript’ refers to detected expression for one of the cDNA features printed on the microarrays, whereas ‘gene’ will refer to single genes, sometimes represented by many transcripts, for which there is available annotation information.
The microarray design for this experiment consisted of a double loop equilibrated design with dye swap. With this design, each strain was compared directly (on the same microarray) to every other strain for exactly five individuals, which made the design equilibrated. Every individual of the five strains was used twice and contrasted against two individuals from different strains, for a total of 10 individuals per strain. For each individual, the two technical replicates were made using both fluorochromes (dye swap). Finally, the two individuals compared together on one microarray were linked to those on other microarrays by sharing a common individual, thus forming closed chains, or loops. In this experiment, two complementary sets of loops, with five repetitions each, were used. This design grants equilibrated statistical power for all the possible inter-strain comparisons.
Signal detection, normalization and statistical testing
On each microarray slide, the expression level of the 16 000 transcripts was measured for two individuals, using different dyes. Hereafter, the measure of the global expression level of all these transcripts for one individual in one microarray will be referred to as a replicate. Fluorescence signals were detected using a ScanArray scanner (Perkin Elmer, Waltham, Massachusetts, US). Spots were located and quantified with the QuantArray 3.0 software (Packard BioScience) using the histogram quantification method to determine the mean intensity of each spot. Local background was subtracted for each spot. In order to use only transcripts expressed above the noise level in the analysis, a threshold of minimum expression was fixed as the mean plus two times the standard deviation of pseudo-spots on the array that contained no cDNA sequences. Further analyses thus included only transcripts that were expressed above this threshold in at least one of the five strains, for at least 19 replicates out of 20 (10 fish per strain with two measures each). A total of 3715 transcripts, expressed in the livers, were thus retained for analysis. Data were normalized by dividing all replicates by their mean transcript expression level and multiplying by 1000, and then loaded in the R/MAANOVA package (http://research.jax.org/faculty/churchill/software/Rmaanova/index.html). All subsequent statistical analyses were done within this package, unless stated otherwise. Missing data were imputed using the K nearest neighbour algorithm with 10 neighbours and then transformed by a base 2 logarithm. Imputed missing data represented 0.3% of all analysed data (1134 missing data points for 3715 transcripts each with 100 measures, representing a total of 371 500 data points). In order to correct for intensity-linked distortions, a regional LOWESS correction was used. The data were then normalized again by dividing each replicate by its mean transcript expression level and multiplying by the mean expression level of all the replicates. This procedure ensured that the data could afterward be used to compute the intra-strain mean expression levels for each transcript without global patterns of expression showing any individual or strain biases. As the same amounts of mRNA were used in each replicate, the total quantity of expressed transcripts should be the same in all of them. The significance of the observed differences in transcription level between the five strains was then assessed using a mixed ANOVA model with ‘Dye’, ‘Loop’ and ‘Strain’ as fixed terms and ‘Array’ and ‘Sample’ as random terms. A permutation-based F-test (Fs, with 1000 sample ID permutations) was used to solve the mixed-model equations. A False Discovery Rate (FDR) procedure was then used, as implemented in the Q-value R package (Storey et al. 2004), to generate a list of transcripts containing an estimated 20% of false positives (FDR = 0.2), which in our case corresponded to a maximum P-value of 0.013 (see Results). This provided a list of candidate transcripts on which contrasts were performed for the eight comparisons of interest between the five strains. Expression fold changes were obtained by dividing the mean expression level of a transcript in one strain by its mean expression level in another one for all the possible two by two strains combinations.
Gene ontology
Using a significant threshold of P < 0.05 for the contrasts, genes possessing Unigene annotation information and being differentially expressed in four comparisons were selected to evaluate the over-representation of biological processes in these comparisons. The first comparison was between the pure Stewiacke and Tusket populations, to uncover any biological function differentiation between these two wild populations. The second comparison included genes that were differentially expressed between the farm strain and either or both of the wild strains, with the purpose of revealing any differences between the farm strain and the wild populations. The third comparison comprised the genes that were differentially expressed between the Stewiacke backcross strain and either or both of pure Stewiacke and pure farm strains. The fourth comparison comprised the genes that were differentially expressed between the Tusket backcross strain and either or both of pure Tusket and pure farm strains. These last two comparisons served to discover the altered biological functions in the backcrosses. Expected and observed presence of annotated genes in different biological process categories were contrasted for these four comparisons. The proportional representation of all the biological processes from the analysed genes (3715 transcripts, 515 Unigenes, see Results) was used as an expected proportion under a random sampling hypothesis. Unigene numbers were converted to Entrez GeneID numbers using the online David Gene ID Conversion tool available on line at: http://david.abcc.ncifcrf.gov/conversion.jsp. The same procedure was used to convert the Unigene numbers associated with each of the four comparisons. The online Panther Classification System gene list comparison tool (http://www.pantherdb.org/tools/compareToRefListForm.jsp) was then used to evaluate the over-representation of biological processes in the four comparisons. Biological processes represented by only one transcript were discarded from the analysis in order to minimize the occurrence of false positives. The biological processes presented in the results are those that were more differently represented between the strains than expected by chance, within at least one of the four comparisons.
Gene expression inheritance
The prevalence of additivity and nonadditivity of gene expression regulation was measured in the two backcross strains, using the distribution of dominance effects, calculated as d/a ratios, where a is half the difference in expression level between the wild and farm strains [a = (Wild − Farm)/2] and d is the difference in expression level between the backcross individual and the average of the parental strains [d = BC − mean (parents)] (Falconer and Mackay 1996). Transcripts with expression levels in backcross individuals that were closer to the wild parents had positive dominance effect values, while those closer to the farm parent had negative values. This allowed us to determine how many transcripts displayed farm versus wild dominance or over-dominance by presenting the distribution of d/a ratios. In the calculation of d/a ratios, only transcripts that were significantly different in expression level between the pure farm and the respective pure wild strains were used (contrast, P < 0.05). In this study, the farm and wild strains represented the grandparents of the backcross strains rather than the parents. Also, we had no information concerning the F1 hybrids’ level of expression. Consequently, as a measure of ‘mean (parents)’ in the calculation of the d-value, we assumed that, under an additive model, the mean of the parents of the backcross progeny should correspond to the sum of three-fourth of the wild and one-fourth of the farm transcript expression levels. Here, the main objective was to evaluate gene expression inheritance patterns corresponding to, or diverging from, additivity. To this end, it was assumed that the mode of inheritance of gene expression levels from the grandparents to the hybrid parents was additive.
Additivity
A d/a ratio of 0 (i.e. d = 0) corresponds to perfect additivity of gene expression. We set an arbitrary range of −0.5 to +0.5 to include transcripts displaying additivity of gene expression inheritance.
Dominance
Ratios (d/a) of −1 or +1 correspond to complete dominance. In order to include transcripts showing a gene expression inheritance behaviour closer to dominance relative to additivity, we used the arbitrary d/a ratio thresholds of −1.5 to −0.5 (farm dominance) and +0.5 to +1.5 (wild dominance).
Nonadditivity
A d/a ratio outside the range of the mean values of the parents correspond to nonadditivity of gene expression inheritance. Therefore, d/a ratios smaller than −1.5 or greater than +1.5 were considered to represent transcripts showing nonadditive expression inheritance. It should be noted that negative and positive values are not linked to under- and over-expression, respectively. They merely reflect that the transcript displays a nonadditive expression inheritance and that its expression level is outside the range observed in the parents, while being closer either to the farm (negative value) or wild strain (positive value).
Tests of variance homoscedasticity of the two d/a ratio distributions (Ansari-Bradley test, P-value = 0.041) and normality (Shapiro test: Stewiacke P-value = 5.7 × 10−11, Tusket P-value = 6.4 × 10−11) were all rejected. Consequently, potential deviations from the expected distribution around zero were explored with the Wilcoxon signed rank test, a nonparametric alternative to the t-test. Lastly, it is important to note that these models are based on diploid organisms and that salmonids are residual tetraploids. Since no model accounts for tetraploidy, such an analysis is more exploratory by nature. Nevertheless, it remains useful in classifying the various genes in terms of patterns of gene expression inheritance observed in hybrids relative to pure strains.
Results
Anova
The results of the ANOVA revealed 479 transcripts that were differentially expressed in the livers of at least one of the five strains relative to all others. This included 12.9% of all significantly expressed transcripts (n = 3715) and represented 2.6 times more transcripts than expected by chance (186 transcripts, α = 0.05). These genes were used to estimate the gene expression level at which there was an empirical 50% probability of a significant call, GEL50, as described in Townsend (2004). The GEL50 value obtained was 1.4, meaning that there was a 50% probability of calling significant a difference in expression levels of a factor of 1.4 times. This value is somewhat lower than what is usually found in the literature for various species including fish, flies, yeast, fungus, and plants (Meiklejohn et al. 2003; Johannesson et al. 2006; Clark and Townsend 2007; Hutter et al. 2008). After FDR correction, 177 transcripts were significant, corresponding to a maximum P-value of 0.013. This amounts to 4.76% of all analysed transcripts and represents five times more transcripts than expected by chance. The 177 significant transcripts are presented in supplementary Table S1, along with their normalized expression levels in the five strains, inter-strain fold changes, ANOVA P-values, FDR q-values, and gene annotation, when available. These transcripts included 100 unique candidate genes as well as 30 transcripts with no annotation (BLAST e-values higher than 1 × 10−15). Finally, 23 of the 100 sequences with known homologues were each represented by two to up to seven transcripts.
Patterns of gene expression
The contrast results (α = 0.01) were used to assess the proportion of transcripts out of 177 that differed significantly in expression level between the five studied strains (Fig. 1). These results revealed that the two wild populations differed in their transcriptome profiles and that farm-wild introgression resulted in a reduction of inter-population differentiation. Specifically, in the wild-wild comparison (Stewiacke-Tusket), 54 transcripts showed significant differences in expression level. However, when comparing the two backcross strains, the number of differentially expressed transcripts decreased to 35. This reduction of the differences between the wild and backcross comparisons was statistically significant under Fisher's test for equality of proportions (P-value = 0.027).
Figure 1.
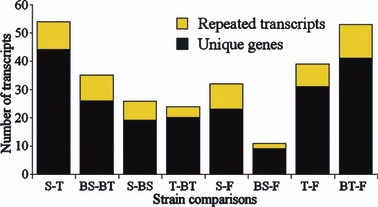
Number of transcripts showing significantly different expression in pair-wise strain comparisons (contrast, maximum P-value of 0.01). The black portions of the bars show the number of unique significant genes, while the yellow portions represent the number of repeated transcripts from the unique genes. (S = Stewiacke, T = Tusket, F = Farm, BS = Backcross Stewiacke, BT = Backcross Tusket).
The two wild populations were about equally distinct from the farm strain, but changes in gene expression following introgression were largely different in the two backcross strains. For instance, the pure Stewiacke and Tusket strains differed from the farm strain for 32 and 39 transcripts, respectively; the difference between these two numbers was not significant (Fisher's test for equality of proportions: P = 0.426). Similarly, when comparing the wild strains to their respective farm-wild backcrosses, Stewiacke and Tusket strains showed, respectively, 26 and 24 differentially expressed transcripts (Fisher's test for equality of proportions: P-value = 0.880). However, when comparing each backcross to the pure farm strain, the Tusket backcross strain showed five times more differences in expression (53 transcripts) than the Stewiacke backcross strain (11 transcripts). This pattern was also reflected in the fold-change distributions for these two comparisons (Fig. 2); the Tusket backcross versus farm comparison showed higher absolute fold-changes than the Stewiacke backcross versus farm comparison (t-test P-value = 3.5 × 10−6).
Figure 2.
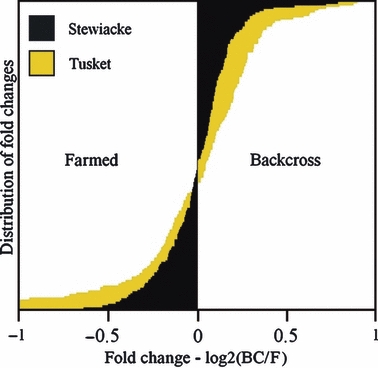
Fold change distributions for Backcross-Stewiacke vs Farm (black) and Backcross-Tusket vs Farm (yellow). The x-axis represents the log2 of the backcross (BC) to Farm (F) ratio for each transcript, while the y-axis shows the distributions of fold-changes for the 177 candidate transcripts.
Gene expression inheritance
The patterns of dominance effects (d/a) further suggested that the wild populations experienced different and unpredictable consequences following introgression with farm salmon (Fig. 3). Proportions of transcripts showing additive genetic control in the two backcross strains were relatively low, similar, and not statistically different (Stewiacke backcross: 21%, Tusket backcross: 18%, P-value = 0.647). However, the transcripts displaying a dominant control were disproportionally represented in the Stewiacke backcross strain, and this difference was nearly significant (Stewiacke: 56%, Tusket: 39%, P-value = 0.067). Furthermore, the transcripts behaving in a nonadditive manner were significantly more represented in the Tusket strain (Stewiacke: 23%, Tusket: 44%, P-value = 0.022). The proportion of transcripts with extreme d/a ratios in the backcrosses (lower than −4 or higher than 4) in the Tusket backcross strain was also two times higher than in the Stewiacke backcross strain, although this trend was not significant (Stewiacke: 6.3%, 3 transcripts, Tusket: 16%, 13 transcripts, P-value = 0.167). Moreover, all three cases of extreme nonadditivity in the Stewiacke backcross strain were cases of under-expression, while they were all cases of over-expression (13 transcripts) in the Tusket backcross strain (data not shown). Altogether, 80% of all analysed transcripts in the backcross strains departed from an additive model of gene expression control (Stewiacke: 79.2%, Tusket: 82.6%). Another striking result was that both distributions of d/a ratios were significantly shifted towards farm dominance (negative values), as revealed by the Wilcoxon signed rank test, with the effect being more profound in the Stewiacke strain (Stewiacke median: −0.78, P-value = 0.001; Tusket median: −0.62, P-value = 0.048).
Figure 3.
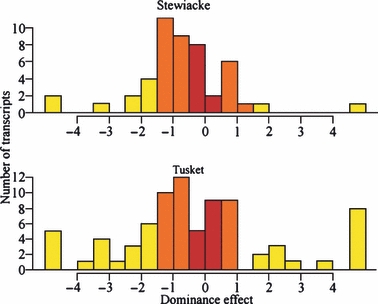
Patterns of gene expression regulation inheritance. This figure plots the number of transcripts (y-axis) as a function of their dominance effect (d/a), as explained in the Materials and Methods. Bars in dark red represent additive control of gene expression regulation, bars in orange show dominance effects, and bars in light yellow show nonadditivity (either under- or over-dominance). The light yellow bars lying outside the scale regroup all the transcripts with extreme d/a values outside the −4 to 4 range. Positive values represent wild dominance or over-dominance, while negative values show farm dominance or over-dominance.
Gene ontology
The gene ontology analysis identified six biological processes that were over-represented in the four comparisons (Table 1). These six processes can be grouped into the more general categories of metabolism (including other carbohydrate metabolism, lipid and fatty acid transport, amino acid catabolism, and carbohydrate metabolism) and immunity (including immunity and defence, as well as macrophage-mediated immunity). Among the genes associated with metabolism were malic enzyme, DGP-L-fucose synthetase, apolipoprotein, and alpha amylase. Genes associated with immunity included CD63 and CD209 antigenes, and D-dopachrome tautomerase. Here, over-representation means that the strains compared were more different for a given biological process than expected by chance, in terms of the number of genes being differentially expressed. The results revealed wild-wild and farm-wild differentiation, as well as population-specific consequences following introgression. The first comparison revealed an over-representation of three biological processes, all part of the more general metabolism category, involved in the differentiation of the Stewiacke and Tusket wild populations. These processes were: carbohydrate metabolism, lipid and fatty acid transport, and amino acid catabolism.
Table 1.
Biological processes showing differentiation of gene expression in four comparisons
| Biological process | Unigenes (515) | S vs T (12) | F vs S + T (16) | BS vs S + F (12) | BT vs T + F (19) |
|---|---|---|---|---|---|
| Other carbohydrate metabolism | 0.58 (3) | 16.7 (2) | 12.5 (2) | 8.3 (1) | 10.5 (2) |
| Lipid and fatty acid transport | 1.17 (6) | 16.7 (2) | 12.5 (2) | 0.0 (0) | 10.5 (2) |
| Amino acid catabolism | 1.55 (8) | 16.7 (2) | 12.5 (2) | 16.7 (2) | 10.5 (2) |
| Carbohydrate metabolism | 4.85 (25) | 16.7 (2) | 18.8 (3) | 8.3 (1) | 15.8 (3) |
| Immunity and defense | 10.9 (56) | 8.3 (1) | 25.0 (4) | 50.0 (6) | 21.1 (4) |
| Macrophage-mediated immunity | 0.97 (3) | 0.0 (0) | 6.3 (1) | 16.7 (2) | 10.5 (2) |
(S = Stewiacke, T = Tusket, F = Farmed, BS = Backcross Stewiacke, BT = Backcross Tusket). The first column lists the biological processes, as defined by the Panther online gene classification tool. The next column gives the expected percentages from the representation of each biological process in the whole set of significantly expressed genes. The number of unique genes used in the calculations are given in parentheses. The remaining four columns give the actual representation (in percent) of the genes for the listed biological processes in the four comparisons. Values in bold indicate that a biological process showed significantly more differences than expected by chance. The first comparison (S vs T) contrasts the Stewiacke and Tusket populations. The second comparison (F vs S + T) includes the genes that were differentially expressed between the farm strain and either or both of the wild populations. The third comparison (BS vs S + F) comprised the genes that were differentially expressed between the Stewiacke backcross strain and either or both of pure Stewiacke and pure farm strains. The fourth comparison (BT vs T + F) comprised the genes that were differentially expressed between the Tusket backcross strain and either or both of pure Tusket and pure farm strains.
When contrasting the farm strain to the two wild strains (second comparison), four biological processes, also part of the metabolism category, showed over-representation. The three over-represented categories from the Stewiacke versus Tusket comparison were still present, along with the carbohydrate metabolism category. This category overlaps with the ‘other carbohydrate metabolism’ category. The farm strain thus showed no new differences in terms of biological functions when contrasted to the wild populations that the wild populations did not already show when compared together.
The third and fourth comparisons, contrasting the Stewiacke and Tusket backcrosses to their pure parental strains, respectively, revealed how different physiological processes were affected in the two wild populations following introgression with farm genetic material. The Stewiacke backcross strain differed from its parental strains at three biological processes, one of which fell into the metabolism category (amino acid catabolism), and two of which regrouped into the immunity category (immunity and defense, and macrophage-mediated immunity). Half of the annotated genes showing differential expression between the Stewiacke backcross and its parental strain played a role in the immunity and defense process, while only 10.9% were expected by chance. The macrophage-mediated immunity process included 16.7% of the differentially expressed genes, yet less than 1% was expected by chance. Although the three other comparisons all revealed over-representation of lipid and fatty acid transport, the Stewiacke backcross did not differ from parental strains for this function. Contrary to the Stewiacke backcross, the Tusket backcross did not differ as much from its parental strains in the general immunity category (only the macrophage-related immunity process differed), but showed more differentiation in the metabolism category (three significant processes: other carbohydrate metabolism, lipid and fatty acid transport, and amino acid catabolism).
Discussion
The main objective of this study was to investigate the population specificity of changes to gene expression following farm-wild hybridization in backcrosses. Specifically, the level of gene expression differentiation between two wild Atlantic salmon populations and a farm strain was measured to evaluate the consequences of introgression in backcross strains (hybrid × wild) on gene expression regulation for two wild populations. Our results provided evidence of significant differentiation in liver gene expression profiles between the two wild populations and the farm strain reared in identical, controlled conditions. Most importantly, we showed that the impacts of farm-wild introgression on gene expression regulation were largely unpredictable and depended partly on the genetic architecture of the introgressed wild population. The concept of genetic architecture refers to the inter-related genetic effects that are responsible for the development of a phenotypic character, and is also referred to as the genotype-phenotype map (Hansen 2006). The interacting properties of this map – including epistasis, polygeny, pleiotropy, and plasticity – make it difficult to anticipate what the phenotypic product of a given genotype might be (Lynch 2007). The interpretation of our results and their potential consequences are discussed below.
Differentiation between the wild populations
The two wild Atlantic salmon populations studied here originate from two distinct geographic areas in eastern Canada: the Stewiacke River within the Inner Bay of Fundy (New Brunswick, Nova Scotia), and the Tusket River from the Southern Uplands (Nova Scotia). Populations found in rivers within these respective regions generally exhibit more similarities in their life histories, as well as phenotypic and neutral genetic differentiation, than between regions (Verspoor 2005; COSEWIC 2006; Fraser et al. 2007). The rivers harbouring the studied wild populations differ in a number of physico-chemical properties, including the length of their respective estuaries (the Stewiacke River estuary is much longer than the Tusket estuary) and the acidity of the rivers (Tusket River is much more acidic: pH = 4.6–5.2 vs Stewiacke River 6.0–6.5) due to the geological properties of the region and acid rainfall (Lacroix and Knox 2005; Ginn et al. 2007). These populations also differ in the distances that subadults/adults migrate to and from marine feeding areas (Tusket: 2500–3000 km vs Stewiacke 500–1500 km) (Jessop 1986; Mills 1989; Lacroix et al. 2005; COSEWIC 2006). It is noteworthy that the populations of both the entire Bay of Fundy and the Atlantic coast of Nova Scotia are critically depleted, the populations of the Inner Bay of Fundy having been listed as endangered under Canada's Species At Risk Act (COSEWIC 2006; NASCO 2007).
To our knowledge, the results of this study provide the first evidence of gene expression differentiation between wild populations of Atlantic salmon. We found that 54 transcripts showed significantly different expression between the Stewiacke and Tusket populations, which corresponded to the highest number of differentially expressed transcripts observed between any of the five strains compared in this study. These results also correspond to neutral microsatellite data (P. T. O'Reilly, unpublished data), revealing significant genetic differentiation between these two populations (Fst value of 0.0585, P < 0.001). Since the microsatellite data rely on putatively neutral markers, part of the genetic differentiation may be explained by mutations and drift. However, differences in transcription patterns between populations may also reflect local adaptation (Taylor 1991). In an extensive review surveying well over 400 published studies, Garcia de Leaniz et al. (2007) concluded that there was an impressive quantity of circumstantial evidence suggesting that populations of Atlantic salmon show inherited adaptive variation. Although they warned that the nature and extent of Atlantic salmon local adaptation remains poorly defined, mainly because very few reciprocal transfers and common-garden field experiments are available, they also found support for the hypothesis of local adaptation compelling. According to their meta-analysis, local selective pressures appear to be strongly influenced, among other factors, by stream morphology and migration distance. Also, genotype-by-environment interactions were detected for traits like body size, growth, tolerance to pH, as well as resistance to various diseases (Garcia de Leaniz et al. 2007), suggesting that different genotypes could be optimal in different environmental settings, a condition for local adaptation. Past and ongoing studies have also provided evidence that the salmon populations inhabiting the Stewiacke and Tusket rivers exhibit genetic differences in body size and growth (D.J. Fraser, A.S. Houde, P.V. Debes, J.D. Eddington, J.A. Hutchings, unpublished data), tolerance to pH (Fraser et al. 2008), and disease resistance (Lawlor et al. 2009). The case for adaptation to local conditions certainly seems plausible in the Atlantic salmon, and there are compelling indications supporting the claim that the wild populations used in the present study may display local adaptation.
This study's findings are also consistent with the hypothesis that gene expression differences observed between wild populations may be adaptive. Indeed, the genes representing the transcripts that differed between the Stewiacke and Tusket populations tended to fall into three biological process categories, all playing an evident role in metabolism and growth, namely lipid and fatty acid transport, carbohydrate metabolism, and amino acid catabolism. An ongoing study by Fraser et al. (unpublished data) showed that, in a controlled environment, Tusket individuals grew faster than their Stewiacke counterparts. It has also been suggested that lipid metabolism and transport could be linked to migration-related osmoregulatory changes associated with transition from fresh water to salt water (Sheridan et al. 1985; Li and Yamada 1992). Given the variation between Stewiacke and Tusket rivers estuary lengths, and thus in the duration of the acclimatizing period to the saline environment, the variation observed in the transcription level of lipid metabolism and transport associated genes could be a consequence of natural selection. Thus, while specific studies using common garden and reciprocal transplant designs would be greatly needed to support this assumption, it is our contention that some of the gene expression differences observed in this study is linked to local adaptation.
Differentiation between farm and wild strains
Before establishing the impacts of farm-wild introgression on wild populations, the level of differentiation between the farm strain and the two wild populations was explored. The Stewiacke and Tusket populations showed differentiation from the farm strain at 32 and 39 transcripts, respectively. Therefore, from a gene expression standpoint, both wild populations were about equally distinct from the farm strain. The microsatellite analysis revealed a similar pattern when comparing the two studied wild populations to the Saint John River population that was used to generate the farm strain. The magnitude of genetic differentiation between Stewiacke and Saint John salmon, as reflected by the mean Fst, was 0.035, whereas that between Tusket and Saint John salmon was 0.033 (these mean Fst values are based on data from comparison of the two wild populations and fish from three Saint John River tributaries; all P-values are lower than 0.002; O'Reilly, unpublished data). These values show that the two wild populations have a very similar level of neutral genetic differentiation when compared to the founding population of the farm strain.
The implications of these microsatellite results are twofold. First, even though they compare the studied wild populations to the ancestors of the farm strain rather than to the farm strain itself, they support the finding that the two wild populations should be about equally differentiated from the farm strain. Second, when comparing wild-wild differentiation to farm-wild differentiation, the microarray and microsatellite data were congruent in suggesting that the wild-wild differentiation (54 transcripts, Fst = 0.058) was greater than the farm-wild distance (32 and 39 transcripts) or farm ancestor-wild distance (Fst values = 0.035 and 0.033). As a result, the farm strain appears to be a slightly modified lineage descending from a wild population that was intermediate to the Stewiacke and Tusket populations. Thus, under an additive model of gene expression regulation, and given that the two wild populations are about equally differentiated from the farm strain, one would expect similar quantitative and qualitative impacts following introgression. Clearly, this was not the case, as we discuss further below.
Effects of farm-wild hybridization
One concern about natural Atlantic salmon populations being introgressed by farm genetic material is that the wild populations might lose some of their local adaptations, making them less fit in their environment (McGinnity et al. 2003; Castillo et al. 2008; Hutchings and Fraser 2008). This would in turn render introgressed wild populations more vulnerable to the different causes that have been alleged to explain their decline, ranging from overfishing and pollution to human changes in freshwater environments and reduced at-sea survival (Parrish et al. 1998; Friedland et al. 2003; Lage and Kornfield 2006). Farm-wild hybridization might also lead to homogenization across introgressed populations, thus eroding population structure (Tufto and Hindar 2003; Hindar et al. 2006). The results of this study corroborate these expectations at the level of gene expression. Indeed, while the number of differentially expressed transcripts between the wild Stewiacke and Tusket populations was 54, this number decreased to 35 when comparing the two backcross strains, representing a loss of a third of the inter-population differentiation. In this study, 25% of every backcross individual's genome came from farm individuals. While such a scenario might seem extreme, many rivers in North America within 300 km of extensive salmon aquaculture now harbour exclusively farm spawning populations or populations composed of a mix of spawners that include a very high proportion of farmed individuals (Morris et al. 2008). As a result, there is a good chance of encountering rivers that contain a high proportion of hybrid individuals with substantially altered gene expression profiles relative to pure wild fish. Lastly, it should be noted that backcross individuals differed from their wild ancestors (Stewiacke: 26 differentially expressed transcripts; Tusket: 24 transcripts) and that such a level of differentiation represents about half of the difference observed between the two wild populations. As a result, there seems to be a significant impact in introgressed populations, as suggested here by changes in gene expression in backcross individuals relative to their wild ancestors. Such changes in gene expression can have a direct influence on the fitness of introgressed individuals within wild populations of Arabidopsis thaliana and fruit fly strains (Holloway et al. 2007; Swindell et al. 2007). If this is also the case for Atlantic salmon, changes to gene expression following farm-wild introgression could result in the erosion of local adaptation. Admittedly, the link between gene expression regulation and fitness has not yet been shown in an entirely satisfactory manner, and this identifies a potentially neglected field of research in evolutionary biology.
Population specificity of introgression effects
The present study finds that the consequences of introgression on the patterns of gene expression were substantially different in the two wild populations. Moreover, these consequences did not match expectations based on a purely additive model of gene expression regulation and on the extent of population divergence between the wild populations and the farm strain. Indeed, while both pure wild populations were about equally differentiated from the farm strain, the number of transcripts differing in expression between the backcross individuals and the pure farm strain was five times higher for the Tusket than for the Stewiacke population. Moreover, five transcripts showed extreme fold-change differences (expressed two times higher in one of the strains) between the Tusket backcross and the farm strain, which was not observed for the Stewiacke comparison. Instead, the Stewiacke backcross displayed extreme fold-change differences from the pure Stewiacke strain for seven transcripts.
The altered biological processes in backcross individuals also showed distinct patterns between the Stewiacke and Tusket populations. Thus, our results revealed substantial differences in introgression effects from the same farm strain, with the Stewiacke population apparently more affected in terms of mis-expression of immunity-related genes, while the Tusket population seemed mostly affected for metabolism related genes. It is noteworthy that another study evaluating disease resistance revealed that pure Stewiacke individuals had a lower capacity to resist bacterial infections than pure Tusket individuals (Lawlor et al. 2009) and that, as stated above, Tusket individuals displayed faster growth than Stewiacke individuals in the first three years after hatching (Fraser et al., unpublished data).
The patterns of gene expression inheritance (d/a ratio) observed in the introgressed backcross strains relative to the pure parental strains provided further evidence that the consequences of introgression are both unpredictable and population specific. First, there were relatively few transcripts displaying an additive control of expression (Stewiacke: 21%, Tusket: 18%), leaving approximately 80% of transcripts with either dominant or nonadditive patterns of gene expression regulation inheritance. In addition, the backcross individuals also tended to be closer to their farm ancestors, a trend that was much clearer in the Stewiacke strain. These results clearly showed a tendency for backcross individuals not to be intermediate to their parents for the expression level of a majority of transcripts. Moreover, the proportion of nonadditivity was about twice as high in the Tusket backcross strain when compared with the Stewiacke backcross strain (44% vs 23%, P = 0.022). As a consequence, not only were the backcross individuals not intermediate to their parents, they also differed in a population-specific manner for nonadditive patterns of gene expression inheritance.
A recent study evaluated the consequences of introgression between farm and wild salmon, both originating from the Saint-John River, New-Brunswick (Roberge et al. 2008). By comparing backcross individuals to their parental strains, these authors found that inheritance patterns of gene expression were to a large extent nonadditive. However it remained unclear if this observation could be generalized to other wild salmon populations. Thus, our results add to the study of Roberge et al. (2008) by clearly showing that the outcomes of introgressive hybridization are highly variable and population specific, perhaps as a consequence of locally adapted genomes and diverging genetic architectures, in accordance with theory about the complex properties of the genetic architecture (Mackay 2001; Rieseberg et al. 2003; Lynch 2007). Hence, given the increasing evidence that genetic architectures vary among populations within a given species (Merila et al. 2004; Lavagnino et al. 2008), it appears difficult to predict with any degree of certainty what the consequences of hybridization between wild and farm strains might be at the population level. This model thus supports the hypothesis that the consequences of genomic interactions induced by the admixture of farm and wild genomes will vary among populations in an unpredictable manner, given that the hybrid genomes represent a mix of two potentially divergent and independently functional genetic architectures (Fenster and Galloway 2000; Burke and Arnold 2001; Mackay 2001; Roberge et al. 2008). This effect is potentially more accute in backcross individuals, given the genetic recombination occurring during meiosis in F1 hybrids and the high prevalence of nonadditive interactions.
Limitations of the study
Potential shortcomings should be considered when interpreting the results of our study. Firstly, genetic drift and sampling biases over two generations of crosses in the laboratory could have led to some of the differences observed when crossing farm and wild individuals. It is difficult to gauge how much drift may have occurred between the wild F2 and the original wild populations. However, it seems highly improbable that any differences potentially attributable to drift would be sufficient to explain by itself the magnitude of differences that we observed between populations, especially in terms of additive versus nonadditive mode of regulation. Instead, we propose that a possible genetic drift effect would add to the variable genetic interactions associated with population-specific genetic architecture, and thus contribute to unpredictable outcomes of farm-wild introgressive hybridization for different wild salmon populations.
Secondly, due to the limited space available, families within each of the five strains were pooled and reared in mixed groups across four to five different tanks per strain. As a result, information concerning the family provenance of each individual is unknown. Thus, some strains could be represented by fewer families and the number of individuals sampled per family could differ. This could potentially lead to differences in heterogeneity of the five strains studied and, thus, to differences in the level of variances within these strains. While this possibility cannot be entirely discounted, all possible care was taken to randomize rearing, sampling, and experimental procedures. Also, this potential bias could not account for some of our main findings, such as the predominance of nonadditive control in gene regulation.
Lastly, a false discovery rate (FDR) of 0.2 was used with the ANOVA results in order to generate the list of significant transcripts used in this study. It could be argued that a relatively high FDR value would reduce the confidence in the individual significant transcripts. However, the P-values associated with these ANOVA results varied between 0.013 and 1.8 × 10−5 (mean P-value 0.005). Moreover, this FDR threshold level retained five times more significant transcripts than expected by chance. For the analysis of the number of transcripts differing between the five strains, contrasts with a threshold P-value of 0.01 were used. Thus, while we cannot rule out that a certain proportion of the genes retained are false positives, we are confident that this does not alter the main conclusions of this study, which are primarily based on global patterns of strain-specific gene expression rather than on individual transcripts. Finally, FDR thresholds up 0.15–0.2 have been applied in previous studies (Hughes et al. 2006; Swanson-Wagner et al. 2006).
Concluding remarks
Overall, the present study showed that wild Atlantic salmon populations can experience substantially divergent genomic consequences following hybridization and backcrossing (wild × hybrid) with a farm strain. The impacts of introgression were both unpredictable and population-specific; each wild population experienced impacts of a different qualitative and quantitative order when introgressed with farm genetic material. To our knowledge, this is the first study to attempt assessment of population-specific impacts of farm introgression on patterns of gene expression in wild populations for a vertebrate species. In light of these results, we suggest that aquaculture accidental releases and stocking activities using strains originating from remote regions should be minimized and more controlled, as these activities have potentially negative and unpredictable consequences on local populations. Our results also suggest that their impacts should also be evaluated at a smaller regional scale than previously considered, and ideally at a population-specific level. This study also contributes to the growing realization that much remains to be learned about the complex functioning of the genetic architecture. This implies that simple models that assume additivity of character inheritance may have little power for predicting phenotypes, which is of special interest in the context of conservation-related risk assessment. Ideally, similar work should be accomplished using populations within different species to explore the degree to which these results can be generalized.
Acknowledgments
We thank the staff of the Atlantic salmon broodstock program (St. Andrew's, New Brunswick) and the Department of Fisheries and Oceans Canada Coldbrook Biodiversity Facility (Coldbrook, Nova Scotia) for providing the initial salmon gametes used to generate spawning adults for this study. Thanks as well to Dalhousie University's Aquatron Laboratory for their assistance during the experimental portion of our study, to J. Eddington and V. Albert for assistance with raising and sampling the fish, and to B. Bougas, G. Côté, J. Mavarez, A. Nolte, L. Papillon, S. Renaut, and C. Roberge for assistance and support in the laboratory. P. O'Reilly kindly provided the microsatellite data and analysis. We would especially like to thank A. Nolte, S. Renaut, C. Roberge, as well as associate editor C.E. Lee and three anonymous referees for their valuable comments on a previous version of this manuscript. This research was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Strategic Project Grant awarded to J. Hutchings and L. Bernatchez. Experiments undertaken with Atlantic salmon in this study comply with the requirements of the Canadian Council on Animal Care (CCAC).
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Significant transcripts from the ANOVA (FDR<0.2) including all the possible fold-changes (presented in percentage) between the five strains.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Literature cited
- Burger JC, Chapman MA, Burke JM. Molecular insights into the evolution of crop plants. American Journal of Botany. 2008;95:113–122. doi: 10.3732/ajb.95.2.113. [DOI] [PubMed] [Google Scholar]
- Burke JM, Arnold ML. Genetics and the fitness of hybrids. Annual Review of Genetics. 2001;35:31–52. doi: 10.1146/annurev.genet.35.102401.085719. [DOI] [PubMed] [Google Scholar]
- Carroll SP. Brave New World: the epistatic foundations of natives adapting to invaders. Genetica. 2007;129:193–204. doi: 10.1007/s10709-006-9014-8. [DOI] [PubMed] [Google Scholar]
- Castillo AGF, Ayllon F, Moran P, Izquierdo JI, Martinez JL, Beall E, Garcia-Vazquez E. Interspecific hybridization and introgression are associated with stock transfers in salmonids. Aquaculture. 2008;278:31–36. [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Molecular Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Clark TA, Townsend JP. Quantifying variation in gene expression. Molecular Ecology. 2007;16:2613–2616. doi: 10.1111/j.1365-294X.2007.03354.x. [DOI] [PubMed] [Google Scholar]
- COSEWIC. COSEWIC Assessment and Update Status Report on the Atlantic Salmon Salmo salar (Inner Bay of Fundy Populations) in Canada. Ottawa: Committee on the Status of Endangered Wildlife in Canada; 2006. [Google Scholar]
- Diamond J. Evolution, consequences and future of plant and animal domestication. Nature. 2002;418:700–707. doi: 10.1038/nature01019. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. Essex, UK: Longman; 1996. [Google Scholar]
- Fay JC, Wittkopp PJ. Evaluating the role of natural selection in the evolution of gene regulation. Heredity. 2008;100:191–199. doi: 10.1038/sj.hdy.6801000. [DOI] [PubMed] [Google Scholar]
- Fenster CB, Galloway LF. Population differentiation in an annual legume: genetic architecture. Evolution. 2000;54:1157–1172. doi: 10.1111/j.0014-3820.2000.tb00551.x. [DOI] [PubMed] [Google Scholar]
- Fleming IA, Einum S. Experimental tests of genetic divergence of farmed from wild Atlantic salmon due to domestication. ICES Journal of Marine Science. 1997;54:1051–1063. [Google Scholar]
- Fleming IA, Hindar K, Mjolnerod IB, Jonsson B, Balstad T, Lamberg A. Lifetime success and interactions of farm salmon invading a native population. Proceedings of the Royal Society of London Series B-Biological Sciences. 2000;267:1517–1523. doi: 10.1098/rspb.2000.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DJ, Weir LK, Darwish TL, Eddington JD, Hutchings JA. Divergent compensatory growth responses within species: linked to contrasting migrations in salmon? Oecologia. 2007;153:543–553. doi: 10.1007/s00442-007-0763-6. [DOI] [PubMed] [Google Scholar]
- Fraser DJ, Cook AM, Eddington JD, Bentzen P, Hutchings JA. Mixed evidence for reduced local adaptation in wild salmon resulting from interbreeding with escaped farmed salmon: complexities in hybrid fitness. Evolutionary Applications. 2008;1:501–512. doi: 10.1111/j.1752-4571.2008.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friars GW, Bailey JK, Oflynn FM. Applications of selection for multiple traits in cage-reared Atlantic salmon (Salmo salar. Aquaculture. 1995;137:213–217. [Google Scholar]
- Friedland KD, Reddin DG, Castonguay M. Ocean thermal conditions in the post-smolt nursery of North American Atlantic salmon. ICES Journal of Marine Science. 2003;60:343–355. [Google Scholar]
- Garcia de Leaniz CG, Fleming IA, Einum S, Verspoor E, Jordan WC, Consuegra S, Aubin-Horth N, et al. A critical review of adaptive genetic variation in Atlantic salmon: implications for conservation. Biological Reviews. 2007;82:173–211. doi: 10.1111/j.1469-185X.2006.00004.x. [DOI] [PubMed] [Google Scholar]
- Gibson G. Microarrays in ecology and evolution: a preview. Molecular Ecology. 2002;11:17–24. doi: 10.1046/j.0962-1083.2001.01425.x. [DOI] [PubMed] [Google Scholar]
- Ginn BK, Cumming BF, Smol JP. Assessing pH changes since pre-industrial times in 51 low-alkalinity lakes in Nova Scotia, Canada. Canadian Journal of Fisheries and Aquatic Sciences. 2007;64:1043–1054. [Google Scholar]
- Gjoen HM, Bentsen HB. Past, present, and future of genetic improvement in salmon aquaculture. ICES Journal of Marine Science. 1997;54:1009–1014. [Google Scholar]
- Glebe BD. East coast salmon aquaculture breeding programs: history and future. Canadian Stock Assessment Secretariat Research Document. 1998 98/157. [Google Scholar]
- Hansen TF. The evolution of genetic architecture. Annual Review of Ecology Evolution and Systematics. 2006;37:123–157. [Google Scholar]
- Hindar K, Fleming IA, McGinnity P, Diserud A. Genetic and ecological effects of salmon farming on wild salmon: modelling from experimental results. ICES Journal of Marine Science. 2006;63:1234–1247. [Google Scholar]
- Holloway AK, Lawniczak MKN, Mezey JG, Begun DJ, Jones CD. Adaptive gene expression divergence inferred from population genomics. PLoS Genetics. 2007;3:2007–2013. doi: 10.1371/journal.pgen.0030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde AS, Fraser DJ, Hutchings JA. Reduced anti-predator responses in multi-generational hybrids between farmed and wild Atlantic salmon (Salmo salar L.) Conservation Genetics. 2009 DOI 10.1007/s10592-009-9892-2. [Google Scholar]
- Hughes KA, Ayroles JF, Reedy MM, Drnevich JM, Rowe KC, Ruedi EA, Caceres CE, et al. Segregating variation in the transcriptome: Cis regulation and additivity of effects. Genetics. 2006;173:1347–1364. doi: 10.1534/genetics.105.051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchings JA, Fraser DJ. The nature of fisheries- and farming-induced evolution. Molecular Ecology. 2008;17:294–313. doi: 10.1111/j.1365-294X.2007.03485.x. [DOI] [PubMed] [Google Scholar]
- Hutter S, Saminadin-Peter SS, Stephan W, Parsch J. Gene expression variation in African and European populations of Drosophila melanogaster. Genome Biology. 2008;9:R12. doi: 10.1186/gb-2008-9-1-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P. Domestication – from behaviour to genes and back again. Applied Animal Behaviour Science. 2006;97:3–15. [Google Scholar]
- Jessop B. Tech. Rep. Fish. Aquat. Sci. New Brunswick: Department of Fisheries and Oceans; 1986. Atlantic Salmon (Salmo salar) of the Big Salmon River. Pages 1–50, Can. [Google Scholar]
- Johannesson H, Kasuga T, Schaller RA, Good B, Gardner MJ, Townsend JP, Cole GT, et al. Phase-specific gene expression underlying morphological adaptations of the dimorphic human pathogenic fungus, Coccidioides posadasii. Fungal Genetics and Biology. 2006;43:545–559. doi: 10.1016/j.fgb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at 2 levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Lacroix GL, Knox D. Acidification Status within Some Atlantic Salmon Rivers of Nova Scotia. St Andrews, NB: Department of Fisheries and Oceans, Biological Station; 2005. Pages 72. [Google Scholar]
- Lacroix GL, Knox D, Stokesbury MJW. Survival and behaviour of post-smolt Atlantic salmon in coastal habitat with extreme tides. Journal of Fish Biology. 2005;66:485–498. [Google Scholar]
- Lage C, Kornfield I. Reduced genetic diversity and effective population size in an endangered Atlantic salmon (Salmo salar) population from Maine, USA. Conservation Genetics. 2006;7:91–104. [Google Scholar]
- Lavagnino NJ, Anholt RRH, Fanara JJ. Variation in genetic architecture of olfactory behaviour among wild-derived populations of Drosophila melanogaster. Journal of Evolutionary Biology. 2008;21:988–996. doi: 10.1111/j.1420-9101.2008.01546.x. [DOI] [PubMed] [Google Scholar]
- Lawlor JL, Dacanay A, Hutchnings JA, Brown LL, Sperker SA. Differences in pathogen resistance within and among cultured, conservation-dependent, and endangered populations of Atlantic salmon, Salmo salar L. Environmental Biology of Fishes. 2009;84:69–78. [Google Scholar]
- Lee CE. Evolutionary genetics of invasive species. Trends in Ecology & Evolution. 2002;17:386–391. [Google Scholar]
- Lev-Yadun S, Gopher A, Abbo S. Archaeology – the cradle of agriculture. Science. 2000;288:1602–1603. doi: 10.1126/science.288.5471.1602. [DOI] [PubMed] [Google Scholar]
- Li HO, Yamada J. Changes of the fatty-acid composition in smolts of masu salmon (Oncorhynchus masou), associated with desmoltification and sea-water transfer. Comparative Biochemistry and Physiology A, Comparative Physiology. 1992;103:221–226. [Google Scholar]
- Lindsey K, Topping JF. Embryogenesis – a question of pattern. Journal of Experimental Botany. 1993;44:359–374. [Google Scholar]
- Lynch M. The Origins of Genome Architecture. Sunderland, MA: Sinauer Associates Inc; 2007. [Google Scholar]
- Mack RN, Simberloff D, Lonsdale WM, Evans H, Clout M, Bazzaz FA. Biotic invasions: causes, epidemiology, global consequences, and control. Ecological Applications. 2000;10:689–710. [Google Scholar]
- Mackay TFC. The genetic architecture of quantitative traits. Annual Review of Genetics. 2001;35:303–339. doi: 10.1146/annurev.genet.35.102401.090633. [DOI] [PubMed] [Google Scholar]
- McGinnity P, Prodohl P, Ferguson K, Hynes R, O'Maoileidigh N, Baker N, Cotter D, et al. Fitness reduction and potential extinction of wild populations of Atlantic salmon, Salmo salar, as a result of interactions with escaped farm salmon. Proceedings of the Royal Society of London Series B-Biological Sciences. 2003;270:2443–2450. doi: 10.1098/rspb.2003.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Parsch J, Ranz JM, Hartl DL. Rapid evolution of male-biased gene expression in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9894–9899. doi: 10.1073/pnas.1630690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merila J, Soderman F, O'Hara R, Rasanen K, Laurila A. Local adaptation and genetics of acid-stress tolerance in the moor frog, Rana arvalis. Conservation Genetics. 2004;5:513–527. [Google Scholar]
- Mignon-Grasteau S, Boissy A, Bouix J, Faure JM, Fisher AD, Hinch GN, Jensen P, et al. Genetics of adaptation and domestication in livestock. Livestock Production Science. 2005;93:3–14. [Google Scholar]
- Mills D. Ecology and Management of Atlantic Salmon. London: Chapman & Hall; 1989. [Google Scholar]
- Mooney HA, Cleland EE. The evolutionary impact of invasive species. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5446–5451. doi: 10.1073/pnas.091093398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MRJ, Fraser DJ, Heggelin AJ, Whoriskey FG, Carr JW, O'Neil SF, Hutchings JA. Prevalence and recurrence of escaped farmed Atlantic salmon (Salmo salar) in eastern North American rivers. Canadian Journal of Fisheries and Aquatic Sciences. 2008;65:2807–2826. [Google Scholar]
- NASCO. Implementation Plan. Ottawa: North Atlantic Salmon Conservation Organization; 2007. Pages 51. [Google Scholar]
- O'Flynn FM, Bailey JK, Friars GW. Responses to two generations of index selection in Atlantic salmon (Salmo salar. Aquaculture. 1999;173:143–147. [Google Scholar]
- Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nature Genetics. 2002;32:261–266. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- Parrish DL, Behnke RJ, Gephard SR, McCormick SD, Reeves GH. Why aren't there more Atlantic salmon (Salmo salar)? Canadian Journal of Fisheries and Aquatic Sciences. 1998;55:281–287. [Google Scholar]
- Randi E. Detecting hybridization between wild species and their domesticated relatives. Molecular Ecology. 2008;17:285–293. doi: 10.1111/j.1365-294X.2007.03417.x. [DOI] [PubMed] [Google Scholar]
- Ranz JM, Machado CA. Uncovering evolutionary patterns of gene expression using microarrays. Trends in Ecology & Evolution. 2006;21:29–37. doi: 10.1016/j.tree.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Widmer A, Arntz AM, Burke JM. The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philosophical Transactions of the Royal Society of London Series B-Biological Sciences. 2003;358:1141–1147. doi: 10.1098/rstb.2003.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberge C, Einum S, Guderley H, Bernatchez L. Rapid parallel evolutionary changes of gene transcription profiles in farmed Atlantic salmon. Molecular Ecology. 2006;15:9–20. doi: 10.1111/j.1365-294X.2005.02807.x. [DOI] [PubMed] [Google Scholar]
- Roberge C, Normandeau E, Einum S, Guderley H, Bernatchez L. Genetic consequences of interbreeding between farmed and wild Atlantic salmon: insights from the transcriptome. Molecular Ecology. 2008;17:314–324. doi: 10.1111/j.1365-294X.2007.03438.x. [DOI] [PubMed] [Google Scholar]
- Sakai AK, Allendorf FW, Holt JS, Lodge DM, Molofsky J, With KA, Baughman S, et al. The population biology of invasive species. Annual Review of Ecology and Systematics. 2001;32:305–332. [Google Scholar]
- Von Schalburg KR, Rise ML, Cooper GA, Brown GD, Gibbs AR, Nelson CC, Davidson WS, et al. Fish and chips: various methodologies demonstrate utility of a 16,006-gene salmonid microarray. BMC Genomics. 2005;6:126. doi: 10.1186/1471-2164-6-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Schalburg KR, Cooper GA, Leong J, Robb A, Lieph R, Rise ML, Davidson WS, et al. Expansion of the genomics research on Atlantic salmon Salmo salar L. project (GRASP) microarray tools. Journal of Fish Biology. 2008;72:2051–2070. [Google Scholar]
- Schulte PM. Changes in gene expression as biochemical adaptations to environmental change: a tribute to Peter Hochachka. Comparative Biochemistry and Physiology B-Biochemistry & Molecular Biology. 2004;139:519–529. doi: 10.1016/j.cbpc.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Sheridan MA, Woo NYS, Bern HA. Changes in the rates of glycogenesis, glycogenolysis, lipogenesis, and lipolysis in selected tissues of the coho salmon (Oncorhynchus kisutch) associated with parr-smolt transformation. Journal of Experimental Zoology. 1985;236:5:54. doi: 10.1002/jez.1402360106. [DOI] [PubMed] [Google Scholar]
- Skaala O, Taggart JB, Gunnes K. Genetic differences between five major domesticated strains of Atlantic salmon and wild salmon. Journal of Fish Biology. 2005;67:118–128. [Google Scholar]
- Stern DL. Perspective: evolutionary developmental biology and the problem of variation. Evolution. 2000;54:1079–1091. doi: 10.1111/j.0014-3820.2000.tb00544.x. [DOI] [PubMed] [Google Scholar]
- Storey JD, Taylor JE, Siegmund D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2004;66:187–205. [Google Scholar]
- Suarez AV, Tsutsui ND. The evolutionary consequences of biological invasions. Molecular Ecology. 2008;17:351–360. doi: 10.1111/j.1365-294X.2007.03456.x. [DOI] [PubMed] [Google Scholar]
- Swanson-Wagner RA, Jia Y, DeCook R, Borsuk LA, Nettleton D, Schnable PS. All possible modes of gene action are observed in a global comparison of gene expression in a maize F-1 hybrid and its inbred parents. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6805–6810. doi: 10.1073/pnas.0510430103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindell WR, Huebner M, Weber AP. Plastic and adaptive gene expression patterns associated with temperature stress in Arabidopsis thaliana. Heredity. 2007;99:143–150. doi: 10.1038/sj.hdy.6800975. [DOI] [PubMed] [Google Scholar]
- Taberlet P, Valentini A, Rezaei HR, Naderi S, Pompanon F, Negrini R, Ajmone-Marsan P. Are cattle, sheep, and goats endangered species? Molecular Ecology. 2008;17:275–284. doi: 10.1111/j.1365-294X.2007.03475.x. [DOI] [PubMed] [Google Scholar]
- Taylor EB. A review of local adaptation in Salmonidae, with particular reference to Pacific and Atlantic salmon. Aquaculture. 1991;98:185–207. [Google Scholar]
- Townsend JP. Resolution of large and small differences in gene expression using models for the Bayesian analysis of gene expression levels and spotted DNA microarrays. BMC Bioinformatics. 2004;5:54. doi: 10.1186/1471-2105-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufto J, Hindar K. Effective size in management and conservation of subdivided populations. Journal of Theoretical Biology. 2003;222:273–281. doi: 10.1016/s0022-5193(03)00018-3. [DOI] [PubMed] [Google Scholar]
- Verspoor E. Regional differentiation of North American Atlantic salmon at allozyme loci. Journal of Fish Biology. 2005;67:80–103. [Google Scholar]
- Wittkopp PJ. Variable gene expression in eukaryotes: a network perspective. Journal of Experimental Biology. 2007;210:1567–1575. doi: 10.1242/jeb.002592. [DOI] [PubMed] [Google Scholar]
- Yabuta Y, Kurimoto K, Hinata Y, Seki Y, Saitou M. Gene expression dynamics during germline specification in mice identified by quantitative single-cell gene expression profiling. Biology of Reproduction. 2006;75:705–716. doi: 10.1095/biolreprod.106.053686. [DOI] [PubMed] [Google Scholar]
- Zeder MA. Central questions in the domestication of plants and animals. Evolutionary Anthropology. 2006;15:105–117. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.