

Abstract
Vector organisms are implicated in the transmission of close to a third of all infectious diseases. In many cases, multiple vectors (species or populations) can participate in transmission but may contribute differently to disease ecology and evolution. The presence of cryptic vector populations can be particularly problematic as differences in infection can be difficult to evaluate and may lead to erroneous evolutionary and epidemiological inferences. Here, we combine site-occupancy modeling and molecular assays to evaluate patterns of infection in the marine cycle of Lyme borreliosis, involving colonial seabirds, the tick Ixodes uriae, and bacteria of the Borrelia burgdorferi s.l. complex. In this cycle, the tick vector consists of multiple, cryptic (phenotypically undistinguishable but genetically distinct) host races that are frequently found in sympatry. Our results show that bacterial detection varies strongly among tick races leading to vector-specific biases if raw counts are used to calculate Borrelia prevalence. These differences are largely explained by differences in infection intensity among tick races. After accounting for detection probabilities, we found that overall prevalence in this system is higher than previously suspected and that certain vector–host combinations likely contribute more than others to the local dynamics and large-scale dispersal of Borrelia spirochetes. These results highlight the importance of evaluating vector population structure and accounting for detection probability when trying to understand the evolutionary ecology of vector-borne diseases.
Keywords: Lyme disease bacteria, pathogen detection, seabirds, site-occupancy models, transmission ecology
Introduction
Vectors are key components in the transmission of many important infectious diseases, but the spatial and temporal dynamics of many vector-borne pathogens still remain poorly understood. This is particularly true for vector-borne diseases with spatially extensive distributions and where multiple vector species participate in transmission (e.g. malaria, Lyme disease, dengue, etc.). In these systems, different vector types can vary in their contribution to local infection dynamics. Indeed, the relative role of each vector will depend on its capacity to acquire, maintain, and transmit the pathogen (i.e. vector competence; Lane 1994; Edman 2002). Likewise, the distribution, diversity, and abundance of vector species will strongly affect disease risk (Kurtenbach et al. 2006). For example, at the within-species scale, the subdivision of the vector into discrete populations (i.e. population structure) can affect the temporal and spatial scale at which transmission occurs (McCoy 2008). Although rarely tested, several cases of cryptic fine scale population structure in vector organisms have now been demonstrated (e.g. (Ravel et al. 2002; McCoy et al. 2003; Ravel et al. 2007; Kempf et al. 2009a,b), as has population-specific variation in vector competence (e.g. Lambrechts et al. 2005; Joy et al. 2008). In some cases, this structure can involve different sympatric vector populations that exploit different local hosts (i.e. host races; McCoy et al. 2005; Whiteman et al. 2006; Johannesen et al. 2008). As vector population structure can influence disease dynamics and evolution, patterns of infection need to be considered explicitly in such cases.
Lyme disease or Lyme borreliosis (LB), caused by spirochetes of the Borrelia burgdorferi species complex, is the most common vector-borne disease in the northern hemisphere and has major medical and economic impacts (Stanek and Strle 2003; Steere et al. 2004). Lyme disease spirochetes are maintained in complex natural transmission cycles including numerous tick vector and reservoir host species. Previous work suggested that the generalist nature of the common tick vectors (Ixodes spp.) acts as a homogenizing mechanism linking the different ecological niches of the bacteria (Kurtenbach et al. 2006). However, recent evidence shows that host specialization and cryptic divergence (i.e. hidden genetic structure) may frequently occur in ticks (McCoy et al. 2001, 2005; Kempf et al. 2009b; De Meeûs et al. unpublished data). Whereas much work has focused on the reservoir hosts of LB bacteria (e.g. LoGiudice et al. 2003, 2008; Brisson et al. 2008), less is known about tick population structure and associated dynamics, or the importance of this structure for the evolutionary ecology and epidemiology of Lyme disease (Qiu et al.2002; De Meeûs et al. 2004).
The marine cycle of LB was discovered more than a decade ago when Olsen et al. (1993) demonstrated the circulation of one of the pathogenic species of the LB complex, Borrelia garinii, among seabird reservoirs via the tick Ixodes uriae. Although current evidence suggests a role for seabirds in the global circulation of Lyme disease bacteria (Olsen et al. 1995; Olsen 2003, 2007; Duneau et al. 2008; Staszewski et al. 2008), the importance of the marine cycle in Lyme disease epidemiology has yet to be explicitly considered (e.g. Kurtenbach et al. 2006). As is still the case for the terrestrial tick vectors of Lyme disease bacteria (Ixodes ricinus species complex), I. uriae was traditionally considered as a generalist ectoparasite exploiting numerous species of colonial seabirds (Rothschild and Clay 1961; Wilson 1970; Guiguen 1988). However, genetic work has revealed that this tick consists of locally distinct seabird species-specific host races, which are often found in sympatry (McCoy et al. 2001, 2005). That is, McCoy et al. (2001, 2005) have demonstrated higher genetic differentiation between ticks from different sympatric seabird host species than between ticks from allopatric populations of the same seabird species. Furthermore, host-associated divergence in I. uriae seems to be a recurrent and spatially dynamic process, such that tick races have evolved recently and independently over different geographic regions (Kempf et al. 2009a). This system therefore provides a pertinent biological model to examine differences among vector races in pathogen transmission and the consequences of race evolution for the evolutionary ecology of Lyme disease bacteria.
Here, we examine the implications of host-associated genetic structure in this vector system by examining differences in local pathogen infection among different sympatric host races. Evaluating prevalence is often challenging due, in part, to difficulties in diagnosing infection, i.e. imperfect detection. Moreover, pathogen detection probabilities may be population-specific leading to vector-specific biases in estimated parameters and thus to erroneous epidemiological inferences. We therefore employ recently developed site-occupancy models (Mackenzie et al. 2002), to explicitly test for differences in pathogen detection probabilities among different vector races and to estimate vector-specific prevalence taking into account these detection probabilities. We evaluate the potential source of imperfect detection by combining statistical modeling with a quantitative PCR assay. Finally, we discuss these results in terms of their importance for the disease dynamics of this pathogen and for other vector-borne systems.
Materials and methods
Tick sampling
The tick sample included 58 adult ticks of unknown infection status collected from three different host races (ticks associated with Black-legged kittiwakes Rissa tridactyla, n = 25; Atlantic puffins Fratercula arctica, n = 15; and Common murres Uria aalge, n = 18) in four geographic locations [three large seabird colonies in Iceland: Skrudur (64°85′40″N, 13°83′80″W), Grimsey (66°83′30″N, 18°80′00″W), and Breidafjordur (65°82′30″N, 22°85′40″W), and one in Norway: Hornøya (70°82′20″N, 31°81′00″E)] (Table S1). In addition, we included 37 adult ticks that were ‘known positives’ from prior serological tests and that were sampled from the same colonies and host species (kittiwake ticks = 7, puffin ticks = 16, murre ticks = 14; Staszewski et al. 2008). These 37 ‘known positives’ are assumed to have the same detection probabilities as the other 58 ticks, and thus were used to improve the estimation of detection probabilities, but not in estimating prevalence. All ticks were sampled from as many individual host birds as possible (average 1.3 ± 0.1 ticks per bird) and were stored using the same procedures. We assumed that these ticks were representative samples from their populations and that their health-status did not affect their probability of being sampled (i.e. infected ticks were as likely to be collected as uninfected ticks).
PCR detection of Borrelia
Borrelia prevalence was estimated by a standard nested PCR procedure that involves amplifying a portion of the flagellin B (flaB) gene using DNA extractions of individual ticks (Johnson et al. 1992; Clark 2004). A positive infection was revealed by the presence of an amplification product on an agarose gel. All amplified products were subsequently sent for direct sequencing (see Duneau et al. 2008 for protocols).
Quantification of Borrelia infection
We applied a highly sensitive target-specific qPCR procedure to quantify relative differences in infection among the ticks collected from the three different seabird hosts. The quantitative real-time PCR assay utilized the LightCycler platform and SYBR Green I detection system (Applied Biosystems, Courtaboeuf, France). The tick sample included the same set of 95 tick extracts analyzed by nested PCR. Given that ticks were not homogeneously engorged and DNA concentrations differed among tick extracts, DNA templates were adjusted to a stock concentration of 40–50 ng/μL. For this analysis, we designed forward and reverse primers to amplify a conserved 214-pb region within the flagellin gene (qFlaB-F 5′-GGAATGCAACCTGCAAAAAT-3′ and qFlaB-R 5′-GGCTGTTGAGCTCCTTCTTG-3′). Based on the genome sequence of B. burgdorferi type strain B31, it was assumed that only a single copy of flaB was present per spirochete (Fraser et al. 1997). Reactions were performed in a 10 μL volume containing 1× FastStart Taq DNA polymerase mixture (Applied Biosystems), 4 mm MgCl2, 1 μm concentrations of each primer, and 1 μL of tick DNA (stock dilution of 40–50 ng/μL). The plasmid pB31/41-9 (kindly provided by Dr R. Wallich, Department of Applied Immunology, Heidelberg, Germany), containing the flagellin gene of B. burgdorferi B31 (X16833), was used as a quantification standard. A standard curve was generated with serial dilutions from 5 to 500 000 copies of the reference plasmid pB31/41-9. Two negative controls were included each time. The amplification program included the initial activation step of the FastStart Taq polymerase at 95°C for 10 min and 50 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 15 s, and extension at 72°C for 30 s. Fluorescence was measured at the end of each extension step. After amplification, a melting curve was acquired by heating the product at 20°C/s to 95°C, cooling it at 20°C/s to 60°C, keeping it at 60°C for 20 s, and then slowly reheating it at 0.1°C/s to 95°C. Fluorescence was measured through the slow reheating phase.
The relative number of spirochetes in each PCR mixture was calculated by comparing the crossing points of the samples with those of the standards using the Lightcycler software. Specificity of amplification products were tested by comparing the melting curves and the mean Tm values of the samples with those of different reference strains [B. garinii 20047 (GenBank accession no. D82846), B. burgdorferi B31 (X15661), Borrelia lusitaniae PotiB1 (DQ111035), and Borrelia afzelii VS461 (L29237)]. Post-PCR analyses were also performed using conventional gel electrophoresis followed by direct sequencing of all amplification products (Cogenics, Meylan, France).
Reliable and reproducible quantification of infection was achieved over five orders of magnitude, from 5 to 500 000 plasmid copies. Each sample was run up to four times. Fifty-four of the 95 adult ticks tested positive by qPCR and all runs were highly repeatable (Pearson R2 = 0.98, P < 0.001). We averaged the calculated number of spirochetes from each run for each sample and used this number as an index of the quantity of bacteria in each individual tick (i.e. infection intensity: Margolis et al. 1982).
Site-occupancy models: ‘naive’ versus ‘estimated’ prevalence
To determine the influence of imperfect detection, i.e. false negatives, on PCR prevalence estimates, we applied a site-occupancy modeling framework to the dataset (Mackenzie et al. 2002, 2006). Although more frequently applied in ecological modeling (e.g. Bailey et al. 2004; Altwegg et al. 2008; Weller 2008; Winchell and Doherty 2008), site-occupancy, and related, models are rarely incorporated into parasitological or epidemiological studies (Jennelle et al. 2007; Thompson 2007; Conn and Cooch 2009). Site-occupancy studies rely on a re-sampling of sites that are likely to harbor the species of interest to estimate the proportion of sites occupied, where it is explicitly acknowledged that species may often be missed during a survey even when they are present. Detection probabilities are estimated from the repeated visits. In our study, each tick is a ‘site’ that the pathogen may occupy and re-sampling entails testing an individual vector several times for the presence of infection. Patterns of presence and apparent absence over multiple tests are used to estimate the probability of detection (p) of the pathogen or, in other words, the sensitivity of the test. Essentially the apparent (or ‘naive’) prevalence is corrected for the detection probability and an unbiased estimate of the number of vector individuals occupied by the pathogen (ψ), i.e. estimated prevalence, is obtained.
Using the samples described above, we repeated nested PCRs five times on all 95 individuals, one PCR plate per day for 5 days using the same tick DNA extractions, PCR reagents, and thermocycler. From this data, we constructed histories for each sampled tick where each tick was analogous to a single site and each nested PCR test for the presence of Borrelia to a site visit in a standard site-occupancy analysis. For instance, a tick with a history (h) of 10101 indicates that the bacterium was detected on the first, third, and fifth PCR, but not on the second and fourth PCR. Thus, this tick was occupied by the bacterium (ψ), but the probability of detection was not equal to 1. A probability statement associated with this history would be
If no bacterium was detected in the five PCRs, then the history would be 00000. In this case, either the tick could be occupied by the bacterium, but it was not detected, or the tick could be unoccupied by the bacterium (i.e. uninfected). Thus, the probability statement associated with this history is
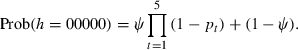 |
Once the probability statement for each of the s observed histories is formed, the model likelihood for the observed data can be constructed, such that
and parameters can be estimated using maximum likelihood methods. Parameters can also be modeled as a function of covariates using a logit link. For example, occupancy can be modeled as a function of an index of bacteria quantity such that logit(ψs) = β0 + β1(quantitys), where β0 is the y-intercept and β1 is the slope parameter associated with the quantity of bacteria. In our model set below, we use notation such that this structure would be designated as ψquantity. If we consider a parameter to be constant (equivalent to having only a y-intercept term in a model), then we use the label ‘constant’. Because we had a sample of known positive ticks, we were able to share information on the detection probability from this sample with the unknown prevalence sample by fixing prevalence for these ticks to 1 and modeling the detection parameters in common across the two groups of ticks. We constructed a number of models (see Model set section) to test hypotheses and explain variation in the detection and prevalence parameters.
Model set
We modeled prevalence (ψ) as constant across all tick races (constant) or as differing for each seabird-specific tick race (tick race). We modeled the detection probability (p) as a constant (constant), as a function of tick race (tick race), and as a function of the bacteria quantity index using both linear (quantity) and quadratic (quantity2) relationships. We also included an effect of engorgement status (engorge) for detection in each tick. Ixodes uriae feed only once per life stage, thus engorgement status was considered as a binomial variable designating whether the tick had had a blood meal when collected (0: nonengorged, 1: engorged). We expected that detection would be higher in engorged ticks as bacteria start to multiply in the midgut of the tick vector once the blood meal is engaged, thus increasing the overall spirochete density in the tick (Piesman et al. 1990). We ran all combinations of additive effects and constructed a total of 24 alternative models.
Model selection and parameter estimation were performed using Program mark (White and Burnham 1999; http://welcome.warnercnr.colostate.edu/~gwhite/mark/mark.htm). We calculated a 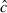 value (i.e. overdispersion factor) using the median
value (i.e. overdispersion factor) using the median 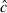 procedure available in Program mark. The
procedure available in Program mark. The 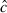 can help correct for overdispersion in the data and improve model goodness-of-fit. We used Akaike's Information Criterion, adjusted for
can help correct for overdispersion in the data and improve model goodness-of-fit. We used Akaike's Information Criterion, adjusted for 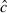 and with a small sample size correction (QAICc) to rank models (Burnham and Anderson 2002). We used QAICc weights to help interpret the strength of evidence for each model. These model weights sum to 1. We also computed cumulative QAICc weights (0 ≤ ∑ wi ≤ 1), or importance values, for each variable. Cumulative weights evaluate the strength of evidence for each variable and are a sum of the model weights associated with each model in which the variable appears. We also present model-averaged prevalence estimates across the entire model set to include model uncertainty in our estimates (Burnham and Anderson 2002; Anderson 2008b).
and with a small sample size correction (QAICc) to rank models (Burnham and Anderson 2002). We used QAICc weights to help interpret the strength of evidence for each model. These model weights sum to 1. We also computed cumulative QAICc weights (0 ≤ ∑ wi ≤ 1), or importance values, for each variable. Cumulative weights evaluate the strength of evidence for each variable and are a sum of the model weights associated with each model in which the variable appears. We also present model-averaged prevalence estimates across the entire model set to include model uncertainty in our estimates (Burnham and Anderson 2002; Anderson 2008b).
Results
Borrelia detection and infection intensity
Of the 95 I. uriae ticks (both of known and unknown infection status) subjected to single-nested PCR and qPCR analyses, 29 (30.5%) ticks were positive and 39 (41.1%) negative by both methods. The remaining 27 (28.4%) ticks were positive only by qPCR. The sequence analysis of Borrelia isolates revealed the majority of strains corresponded to the B. garinii genospecies. However, some atypical strains were detected and grouped most closely with B. lusitaniae in phylogenetic analyses (Duneau et al. 2008).
The index of the number of spirochetes in the tick DNA extracts used in PCR varied from 0 to a maximum value of 1957. Among infected ticks, this index showed a median of 2.97 and a geometric mean of 5.01 ± 0.53 (no. bacteria ± SE). These are relative index values of the total number of spirochetes per tick because quantitative analyses were performed on standardized tick DNA concentrations (40–50 ng of DNA). These values cannot be easily extrapolated to the entire tick because spirochetes are not homogeneously distributed in the tick's body and tick DNA concentrations in the extracts change with the engorgement status. However, as there was no strong association between host type and tick engorgement status (Spearman's rho = 0.02, N = 91, P = 0.82), we assume this underestimation would not affect our overall results (i.e. testing differences among host-associated tick races in infection prevalence and intensity). Overall, the relative infection intensity was greater in puffin ticks (148.89 ± 90.57) than in murre (52.47 ± 45.74) and kittiwake ticks (13.50 ± 6.83) (mean no. bacteria ± SE) (Fig. 1).
Figure 1.
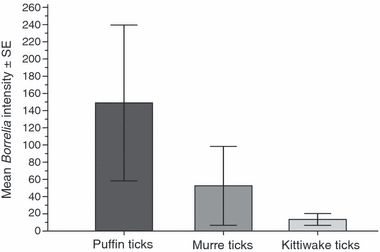
Intensity of infection of Lyme borreliosis bacteria (mean number of spirochetes per infected sample) in three tick races associated with puffins, murres, and kittiwakes, respectively. The error bars are standard errors (SE).
Borrelia detection probabilities
We estimated 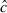 to be 1.7 and adjusted our model set accordingly. Model-selection results (Table 1) showed that Borrelia detection was primarily a function of tick race (cumulative QAICc wt = 1.0 (βmurre = −2.44, 95% CI: −3.71,−1.16; βkittiwake = −1.56, 95% CI: 0.01, 0.41) and bacteria quantity (quantity2; cumulative QAICc weight = 1.0, Fig. 2) and, to a lesser extent, engorgement status (cumulative QAICc weight = 0.31). The best model that contained an engorgement effect, predicted a positive relationship (β = 0.46, 95% CI: −0.53, 1.46), but the 95% confidence interval widely overlapped zero and was probably a ‘pretending variable’ (Anderson 2008a). Such variables rank highly because they are only one parameter different from other high-ranking models and by definition AIC will only drop by a maximum of 2 with an addition of a single parameter. The bacteria quantity index was also positive (
to be 1.7 and adjusted our model set accordingly. Model-selection results (Table 1) showed that Borrelia detection was primarily a function of tick race (cumulative QAICc wt = 1.0 (βmurre = −2.44, 95% CI: −3.71,−1.16; βkittiwake = −1.56, 95% CI: 0.01, 0.41) and bacteria quantity (quantity2; cumulative QAICc weight = 1.0, Fig. 2) and, to a lesser extent, engorgement status (cumulative QAICc weight = 0.31). The best model that contained an engorgement effect, predicted a positive relationship (β = 0.46, 95% CI: −0.53, 1.46), but the 95% confidence interval widely overlapped zero and was probably a ‘pretending variable’ (Anderson 2008a). Such variables rank highly because they are only one parameter different from other high-ranking models and by definition AIC will only drop by a maximum of 2 with an addition of a single parameter. The bacteria quantity index was also positive (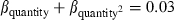 , 95% CI: 0.01, 0.04) and differed by tick race (Fig. 2). The puffin tick race had the highest detection probability and the strongest relationship with bacteria quantity (Fig. 2A), whereas the murre tick race had the lowest (Fig. 2C).
, 95% CI: 0.01, 0.04) and differed by tick race (Fig. 2). The puffin tick race had the highest detection probability and the strongest relationship with bacteria quantity (Fig. 2A), whereas the murre tick race had the lowest (Fig. 2C).
Table 1.
Model selection results for models estimating prevalence (ψ) of Borrelia spp. in Ixodes uriae host races
| Model | ΔQAICc | QAICc weights | Model likelihood | Number of parameters | Deviance |
|---|---|---|---|---|---|
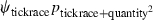 |
0.00 | 0.48 | 1.00 | 8 | 168.68 |
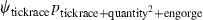 |
1.59 | 0.21 | 0.45 | 9 | 167.83 |
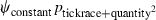 |
1.65 | 0.21 | 0.44 | 6 | 175.05 |
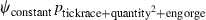 |
3.14 | 0.10 | 0.21 | 7 | 174.21 |
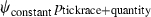 |
13.16 | 0.00 | 0.00 | 5 | 188.84 |
We modeled Borrelia occupancy of ticks as a constant regardless of which seabird species they fed on (ψconstant) and as a function of tick race (ψtickrace). We modeled detection as a constant (pconstant), a function of tick race (ptickrace), a function of the relative quantity of bacteria in a linear (pquantity), and quadratic (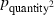 ) fashion, as well as a function of engorgement status (pengorge). We ran all combinations of additive effects. Only the top five models are presented. ΔQAICc is the QAICc scaled so the lowest value is zero (for ease of interpretation). QAICc weights are the model weights. The model likelihood is the QAICc weight of the model of interest divided by the QAICc weight of the best model. This value is the strength of evidence of one model relative to other models in the set. Number of parameters and deviance [difference in the −2log(likelihood) of the model of interest and the −2log(likelihood) of the saturated model] are also given.
) fashion, as well as a function of engorgement status (pengorge). We ran all combinations of additive effects. Only the top five models are presented. ΔQAICc is the QAICc scaled so the lowest value is zero (for ease of interpretation). QAICc weights are the model weights. The model likelihood is the QAICc weight of the model of interest divided by the QAICc weight of the best model. This value is the strength of evidence of one model relative to other models in the set. Number of parameters and deviance [difference in the −2log(likelihood) of the model of interest and the −2log(likelihood) of the saturated model] are also given.
Figure 2.
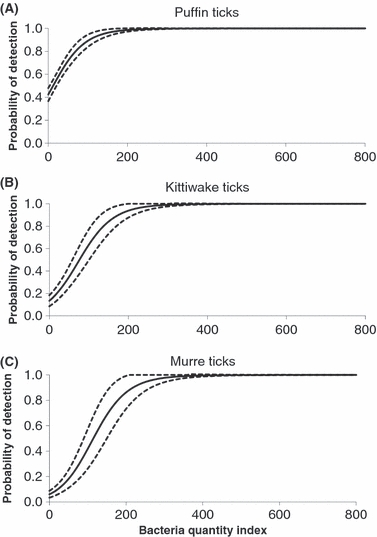
Detection probability of Borrelia spp. as a function of the quantity of bacteria in tick races associated with three seabird species (A: puffin; B: kittiwake; C: murre). The dotted lines are standard errors (SE).
Modeled Borrelia prevalence
Our model-selection analysis suggested a difference in prevalence (corrected for detection) according to tick race (Table 1), as indicated by our top two models (cumulative QAICc weight = 0.69). However, the third ranked model provided some support for no difference among tick races (QAICc weight = 0.21). We therefore calculated tick race-specific prevalence estimates by model-averaging estimates over our entire model set (Table 2). This accounted for model-selection uncertainty (Burnham and Anderson 2002). Average prevalence across all tick races was 0.40 ± 0.14, with the highest prevalence in the puffin tick race and lowest in the kittiwake tick race (Table 2).
Table 2.
Comparisons of naïve and corrected prevalence for three host races of Ixodes uriae (±1 SE)
| Tick race | Naïve prevalence | Corrected prevalence (Ψ) | Bias |
|---|---|---|---|
| Puffin | 0.53 (0.13) | 0.57 (0.15) | 0.04 |
| Kittiwake | 0.12 (0.07) | 0.22 (0.14) | 0.10 |
| Murre | 0.11 (0.08) | 0.41 (0.32) | 0.30 |
| All races | 0.22 (0.06) | 0.40 (0.14) | 0.18 |
The naïve prevalence was calculated from the raw data as the number of ticks observed with Borrelia spp. divided by the total number of ticks tested and the SE was based on a binomial distribution. The corrected prevalence takes into account the detection probability and is the model-averaged estimate across the entire model set, and thus accounts for both uncertainty in the estimation process, the detection process, and the model set. The corrected estimate for all tick races combined was from the highest ranking model in which prevalence was considered constant across tick races. Bias was calculated as the corrected prevalence minus the naïve prevalence. Naïve and corrected prevalence are based on the unknown tick sample only (see Materials and methods section).
Accounting for detection was important in this dataset as a naive overall estimate would have been 0.22 if all five PCR tests were considered (Table 2) and would have ranged from 0.10 to 0.16 if only a single-nested PCR test was considered. Race-specific prevalence estimates changed rank order of importance after accounting for detection (Table 2), further underlining the importance of accounting for nondetection in parameter estimates.
Discussion
Imperfect pathogen detection
Site-occupancy analyses showed that conventional PCR methods can strongly underestimate the presence of LB Borrelia in vector ticks and that this underestimation varies among host-specific tick groups. Several factors could contribute to the low performance, or sensitivity, of the diagnostic test (Banoo et al. 2008). For example, the intrinsic variability of the nested PCR approach, i.e. reproducibility, may lead to the imperfect detection of LB spirochetes (Altwegg 1995; Valkinas et al. 2006). However, in the present study, experimental conditions were carefully standardized to minimize this effect and such variability should not lead to race-associated differences in detection. More likely, the sensitivity of the detection technique to the relative concentrations of bacterial and vector DNA within the extract played a role (Cogswell et al. 1996; Banoo et al. 2008). Indeed, imperfect detection is often associated with poorly adapted pathogen strains or species as they tend to be present in low numbers within a given host (McKenzie et al. 2003). The qPCR approach we applied here showed that LB detection was related to the quantity of bacteria within an individual vector. The effect of infection intensity on amplification success was also supported by experimental tests using variable combinations of pure bacterial and host DNA; as host DNA concentrations increase relative to that of the bacteria, pathogen detection is considerably reduced (results not shown). However, despite the importance of infection intensity for detection, this factor could not entirely explain vector-specific differences. In particular, detection probability was the lowest in murre ticks, even though these tick vectors did not show the lowest average infection intensity. Detection probabilities could therefore also be linked to the presence of inhibitors that originate from the tick itself and that vary among races (Cogswell et al. 1996; Schwartz et al. 1997). Likewise, there may be differences among Borrelia strains found in the different races that alter detection probabilities. In the terrestrial cycle, previous evidence shows differences in host specificity and selectivity of Borrelia genospecies (Kurtenbach et al. 2002). In our system, a few atypical strains (non-B. garinii) have been previously isolated from I. uriae ticks (B. lusitaniae and B. burgdorferii-like isolates). These atypical strains tend to show lower detection probabilities compared with B. garinii, but the prevalence is too low to make inferences regarding patterns of infection among vector groups. In addition, no support for vector-related or host-related strain specificity in this system has yet been found (Duneau et al. 2008).
In most current studies, researchers employ PCR procedures (simple or nested) to determine the infective status of LB vector ticks (e.g. Ferquel et al. 2006; Halos et al. 2006; Kipp et al. 2006; Brisson et al. 2008). Despite the improvement that these techniques represent compared with immunofluoresence methods (Kahl et al. 1998), our results show that they can still have low sensitivity and result in a significant underestimate of pathogen presence. qPCR procedures are becoming more common and can improve detection sensitivity (as seen here). However, given the restricted conditions for the use of such methods, the targeted pathogen range can be large and the amplified sequence cannot always be used for isolate identification. Therefore, the method of choice will depend on the questions to be addressed and the system under consideration. Regardless of the chosen detection method, no method is likely to be perfect. In this way, the inclusion of site-occupancy modeling can be an efficient means to examine the accuracy of naïve estimates of pathogen prevalence and for determining potential sources of detection bias. We therefore advocate the inclusion of such models for all pathogen survey techniques.
Vector-specific patterns of infection
Model estimates indicated that, after taking into account vector-specific detection probabilities, Borrelia prevalence varied among the different host-associated tick races. Puffin ticks showed the highest prevalence and kittiwake ticks the lowest. As the current study was specifically designed to address the implications of cryptic vector structure on pathogen infection, we were not able to include spatial structure in our models. However, infected ticks of different host species came from all four geographic locations and preliminary analyses indicated no obvious effect of geography on infection intensity (results not shown). Future studies, with a more complete geographical coverage, will be designed to address spatial variation in detection and prevalence in this system.
Several alternative hypotheses could explain vector-specific patterns of infection. For example, differences in prevalence among the different tick groups could be due to differences in the life stage of the sampled ticks (Wang et al. 2003) or to their engorgement state (i.e. engorged or unengorged; Piesman et al. 1990). In the present study, all ticks were adults that were directly sampled from their specific hosts. Ixodes uriae host races are very faithful to their seabird host and therefore blood meals at the larval and nymphal stages have most likely been collected from the same host type (McCoy et al. 2001, 2005; Kempf et al. 2009a). Similarly, there were no statistical differences among seabird-tick races in their engorgement status (see Materials and methods section). Thus, inter-vectorial differences in infection are more likely associated with intrinsic differences in either the reservoir capacity of the different seabird host species or the relative competence of the different vector groups.
Successful transmission depends on the capacity of the vector to acquire, maintain, and transmit the pathogen (i.e. vector competence; Lane 1994; Edman 2002). Differences among tick races in Borrelia infection intensity and prevalence may therefore indicate differences in the ability of the bacteria to survive and replicate in the three vector groups. Similar patterns among different vector species have been found for a variety of zoonoses, including for example mosquito vectors of Plasmodium vivax (Adak et al. 1999), fleas transmitting Yersinia pestis (Lorange et al. 2005), or tick vectors of Anaplasma phagocytophilum (Teglas and Foley 2006). However, differences in infection among vector groups could also be linked to the seabird host species, rather than to the vector itself. Other studies support the idea that host susceptibility to infection and reservoir potential may be more important than variability in vector competence for the epidemiology of the pathogenic agent (Teglas and Foley 2006; Eisen et al. 2008). For example, under the ‘dilution effect’ model, the diversity and composition of the host community can have profound implications for disease epidemiology (LoGiudice et al. 2003). Based on this model, different host species show different reservoir capacities and vary in their level of infestation by the vector such that the presence of certain host species in the community limits the transmission potential of the pathogen and thus lowers disease risk. The differences that we find in Borrelia infection may thus be related to differences in the reservoir abilities of the various sympatric seabird host species (Ostfeld et al. 2006; Brunner et al. 2008). For instance, higher bacterial intensities in the tick could result from a higher infection dose received from the infecting host. Alternatively, host-associated blood factors could differentially favor the maintenance and replication of Lyme disease bacteria within the tick (Eisen et al. 2008). In particular, seabird-specific immune factors (i.e. bacteriolytic or inhibitory factors) or the seabird blood itself may affect bacterial fitness in the vector. Significant inter-host heterogeneity in infection can also occur within host populations due to individual differences in pathogen exposure and susceptibility (e.g. Staszewski et al. 2007) and is thus an important factor to take into account in sampling designs. Here, although adult ticks were sampled from numerous host individuals within each population, our dataset remains limited to fully explore this type of heterogeneity.
Vector-specific infection and disease dynamics
Regardless of whether vector or host-associated factors are responsible for the race-specific patterns we find in detection and infection, cryptic vector population structure can have important epidemiological implications. In particular, certain vector–host combinations are going to be more important than others in pathogen transmission. For example, among the three host–vector combinations considered here, our results indicate that a particular effort should be made to survey the presence and structure of LB spirochetes associated with Atlantic puffins. The potential role of this species in Lyme disease epidemiology is further amplified by the fact that puffins seem to move at larger spatial scales than either kittiwakes or common murres, and therefore may disperse vectors and disease agents more widely (McCoy et al. 2003). Pathogen strains carried by this host–vector combination should therefore be distributed at larger spatial scales, should be less impacted by the effects of local drift, and may be exchanged more readily with strains from the terrestrial cycle. Large-scale dispersal may also favor pathogen emergence by generating novel diversity via the horizontal transfer of plasmid-borne genes (Qiu et al. 2004). These factors, in combination with other important variables that may affect pathogen transmission rates such as host community composition, host density, and vector basic reproductive number (R0) (LoGiudice et al. 2003, 2008; Kurtenbach et al. 2006; Ostfeld et al. 2006; Hartemink et al. 2008), will likely have a significant impact on LB dynamics over space and time and should therefore be considered more generally in epidemiological models of this disease.
Concluding remarks
The differences we find in pathogen infection and detection among different vector groups are relevant to many vector-borne zoonoses and should be considered more explicitly in other systems of medical and economic importance. Such differences can lead to vector-specific patterns of transmission which, in turn, can affect the ecological and evolutionary dynamics of the system, e.g. selection on the pathogen, and ultimately, the epidemiological patterns of the associated disease. Likewise, prevalence underestimation and biases in detection may be common in many parasitological and epidemiological studies. Efforts to standardize pathogen detection methods are difficult under certain sampling conditions (e.g. Banoo et al. 2008), and thus estimating detection directly is advocated. The site-occupancy, and related, modeling methods continue to be developed and can be applied in many novel ways to host–parasite studies and disease epidemiology (Mackenzie et al. 2006, 2009; Kendall 2008; Conn and Cooch 2009). Such analyses can provide more robust parameter estimates and can reveal new avenues to explore for our understanding of host–pathogen systems.
Acknowledgments
We thank T. Boulinier, R.T. Barrett, T. Tverra, A. Petersen, and members of the Working group ‘Tique et Maladies à Tiques’ of the ‘Réseau Ecologique des Interactions Durables’ for help and advice at different stages of this work. We thank comments and discussions provided by three anonymous referees on previous versions of the manuscript. Financial support was provided by the ‘Institut Polaire-Paul Emile Victor’ (IPEV, Program No. 333), the Centre National de la Recherche Scientifique (CNRS), and the Agence National de la Recherche (ANR-06-JCJC-0095-01). E.G.-D. was supported by a Marie Curie Fellowship No. PIEF-GA-2008-221243. Borrelia reference strains were kindly provided by the Pasteur Institute (Paris, France), and we thank R. Wallich (Heidelberg, Germany) for the plasmid pB31/41-9.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Number of ticks analyzed pergeographic site (N) and seabird host species and number ofdetections of Borrelia spirochetes by two different methods (quantitative and nested PCR). Seabird host species include the Black-legged Kittiwake (KT), the Common Murre (CM) and the Atlantic Puffin (PF).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Literature cited
- Adak T, Sarbjit K, Singh OP. Comparative susceptibility of different members of the Anopheles culicifacies complex to Plasmodium vivax. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:573. doi: 10.1016/s0035-9203(99)90052-4. [DOI] [PubMed] [Google Scholar]
- Altwegg M. General problems associated with diagnostic applications of amplification methods. Journal of Microbiological Methods. 1995;23:21. [Google Scholar]
- Altwegg R, Wheeler M, Erni B. Climate and the range dynamics of species with imperfect detection. Biology Letters. 2008;4:581. doi: 10.1098/rsbl.2008.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DR. Model Based Inference in the Life Sciences: A Primer on Evidence. New York, NY: Springer-Verlag; 2008a. [Google Scholar]
- Anderson DR. Model Based Inference in the Life Sciences – A Primer on Evidence. New York, NY: Springer; 2008b. [Google Scholar]
- Bailey LL, Simons TR, Pollock KH. Estimating site occupancy and species detection probability parameters for terrestrial salamanders. Ecological Applications. 2004;14:692–702. [Google Scholar]
- Banoo S, Bell D, Bossuyt P, Herring A, Mabey D, Poole F, Smith PG, et al. Evaluation of diagnostic tests for infectious diseases: general principles. Nature Reviews in Microbiology. 2008;5:S16–S28. [PubMed] [Google Scholar]
- Brisson D, Dykhuizen DE, Ostfeld RS. Conspicuous impacts of inconspicuous hosts on the Lyme disease epidemic. Proceedings of the Royal Society of London, Series B: Biological Sciences. 2008;275:227. doi: 10.1098/rspb.2007.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner JL, LoGiudice K, Ostfeld RS. Estimating reservoir competence of Borrelia burgdorferi hosts: prevalence and infectivity, sensitivity, and specificity. Journal of Medical Entomology. 2008;45:139–147. doi: 10.1603/0022-2585(2008)45[139:ercobb]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Burnham K, Anderson D. Model Selection and Multimodel Inference. Practical Information-Theoretic Approach. New York, NY: Springer; 2002. [Google Scholar]
- Clark K. Borrelia species in host-seeking ticks and small mammals in northern Florida. Journal of Clinical Microbiology. 2004;42:5076–5086. doi: 10.1128/JCM.42.11.5076-5086.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogswell FB, Bantar CE, Hughes TG, Gu Y, Philipp M. Host DNA can interfere with detection of Borrelia burgdorferi in skin biopsy specimens by PCR. Journal of Clinical Microbiology. 1996;34:980–982. doi: 10.1128/jcm.34.4.980-982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PB, Cooch EG. Multistate capture-recapture analysis under imperfect state observation: an application to disease models. Journal of Applied Ecology. 2009;46:486–492. [Google Scholar]
- De Meeûs T, Lorimier Y, Renaud F. Lyme borreliosis agents and the genetics and sex of their vector, Ixodes ricinus. Microbes and Infection. 2004;6:299–304. doi: 10.1016/j.micinf.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Duneau D, Boulinier T, Gómez-Díaz E, Petersen A, Tveraa T, Barrett RT, McCoy KD. Prevalence and diversity of Lyme borreliosis bacteria in marine birds. Infection, Genetics and Evolution. 2008;8:352. doi: 10.1016/j.meegid.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Edman JD. Arthropod transmission of vertebrate parasites. In: Eldridge BF, Edman JD, editors. Medical Entomology. Boston, MA: Kluwer Academic Publications; 2002. pp. 151–163. [Google Scholar]
- Eisen RJ, Vetter SM, Holmes JL, Bearden SW, Montenieri JA, Gage KL. Source of host blood affects prevalence of infection and bacterial loads of Yersinia pestis in fleas. Journal of Medical Entomology. 2008;45:933. doi: 10.1603/0022-2585(2008)45[933:sohbap]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Ferquel E, Garnier M, Marie J, Bernede-Bauduin C, Baranton G, Perez-Eid C, Postic D. Prevalence of Borrelia burgdorferi Sensu Lato and Anaplasmataceae members in Ixodes ricinus ticks in Alsace, a focus of Lyme borreliosis endemicity in France. Applied and Environmental Microbiology. 2006;72:3074–3078. doi: 10.1128/AEM.72.4.3074-3078.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Guiguen C. Marseille: Faculté de Médecine de Marseille; 1988. Anthropozoonoses et oiseaux marins: contribution à l’étude des ectoparasites hématophages des espèces nicheuses sur les côtes françaises continentales et insulaires. Doctoral dissertation. [Google Scholar]
- Halos LG, Mavris M, Vourc'h G, Maillard R, Barnouin J, Boulouis HJ, Vayssier-Taussat M. Broad-range PCR-TTGE for the first-line detection of bacterial pathogen DNA in ticks. Veterinary Research. 2006;37:245–253. doi: 10.1051/vetres:2005055. [DOI] [PubMed] [Google Scholar]
- Hartemink NA, Randolph SE, Davis SA, Heesterbeek JAP. The basic reproduction number for complex disease systems: defining R0 for Tick-Borne infections. The American Naturalist. 2008;171:743–754. doi: 10.1086/587530. [DOI] [PubMed] [Google Scholar]
- Jennelle CS, Cooch EG, Conroy MJ, Senar JC. State-specific detection probabilities and disease prevalence. Ecological Applications. 2007;17:154–167. doi: 10.1890/1051-0761(2007)017[0154:sdpadp]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Johannesen J, Lux B, Michel K, Seitz A, Maixner M. Invasion biology and host specificity of the grapevine yellows disease vector Hyalesthes obsoletus in Europe. Entomologia Experimentalis et Applicata. 2008;126:217–227. [Google Scholar]
- Johnson B, Happ C, Mayer L, Piesman J. Detection of Borrelia burgdorferi in ticks by species-specific amplification of the flagellin gene. American Journal of Tropical Medicine and Hygiene. 1992;47:730–741. doi: 10.4269/ajtmh.1992.47.730. [DOI] [PubMed] [Google Scholar]
- Joy DA, Gonzalez-Ceron L, Carlton JM, Gueye A, Fay M, McCutchan TF, Su X. Local Adaptation and Vector-Mediated Population Structure in Plasmodium vivax Malaria. Molecular Biology and Evolution. 2008;25:1245–1252. doi: 10.1093/molbev/msn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahl O, Gern L, Gray J, Guy E, Jongejan F, Kirstein F, Kurtenbach K, et al. Detection of Borrelia burgdorferi sensu lato in ticks: immunofluorescence assay versus polymerase chain reaction. Zentralblatt für Bakteriologie. 1998;287:205–210. doi: 10.1016/s0934-8840(98)80122-9. [DOI] [PubMed] [Google Scholar]
- Kempf F, Boulinier T, De Meeûs T, Arnathau C, McCoy KD. Recent evolution of host-associated specialization in the seabird tick Ixodes uriaeMolecular Ecology. 2009a;18:4450–4462. doi: 10.1111/j.1365-294X.2009.04356.x. [DOI] [PubMed] [Google Scholar]
- Kempf F, De Meeüs T, Arnathau C, Degeilh B, McCoy KD. Assortative Pairing in Ixodes ricinus L. (Acari: Ixodidae), the European Vector of Lyme Borreliosis. Journal of Medical Entomology. 2009b;46:471–474. doi: 10.1603/033.046.0309. [DOI] [PubMed] [Google Scholar]
- Kendall WL. One size does not fit all: adapting mark-recapture and occupancy models for state uncertainty. Environmental and Ecological Statistics. 2008;3:765–780. [Google Scholar]
- Kipp S, Goedecke A, Dorn W, Wilske B, Fingerle V. Role of birds in Thuringia, Germany, in the natural cycle of Borrelia burgdorferi sensu lato, the Lyme disease spirochaete. International Journal of Medical Microbiology. 2006;296:125. doi: 10.1016/j.ijmm.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Kurtenbach K, DeMichelis S, Etti S, Schäfer SM, Sewell H-S, Brade V, Kraiczy P. Host association of Borrelia burgdorferi sensu lato - the key role of host complement. Trends in Microbiology. 2002;10:74–79. doi: 10.1016/s0966-842x(01)02298-3. [DOI] [PubMed] [Google Scholar]
- Kurtenbach K, Hanincova K, Tsao JI, Margos G, Fish D, Ogden NH. Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nature Reviews in Microbiology. 2006;4:660–669. doi: 10.1038/nrmicro1475. [DOI] [PubMed] [Google Scholar]
- Lambrechts L, Halbert J, Durand P, Gouagna L, Koella J. Host genotype by parasite genotype interactions underlying the resistance of anopheline mosquitoes to Plasmodium falciparum. Malaria Journal. 2005;4:3. doi: 10.1186/1475-2875-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RS. Competence of ticks as vectors of microbial agents with an emphasis on Borrelia burgdorferi. In: Sonenshine D, Mather T, editors. Ecological Dynamics of Tick-Borne Zoonoses. New York, NY: Oxford University Press; 1994. pp. 45–67. [Google Scholar]
- LoGiudice K, Ostfeld RS, Schmidt KA, Keesing F. The ecology of infectious disease: effects of host diversity and community composition on Lyme disease risk. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:567–571. doi: 10.1073/pnas.0233733100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoGiudice K, Duerr STK, Newhouse MJ, Schmidt KA, Killilea ME, Ostfeld RS. Impact of host community composition on lyme disease risk. Ecology. 2008;89:2841–2849. doi: 10.1890/07-1047.1. [DOI] [PubMed] [Google Scholar]
- Lorange EA, Race BL, Sebbane F, Joseph Hinnebusch B. Poor vector competence of fleas and the evolution of hypervirulence in Yersinia pestis. The Journal of Infectious Diseases. 2005;191:1907–1912. doi: 10.1086/429931. [DOI] [PubMed] [Google Scholar]
- Mackenzie DI, Nichols JD, Lachman GB, Droege S, Andrew Royle J, Langtimm CA. Estimating site occupancy rates when detection probabilities are less than one. Ecology. 2002;83:2248–2255. [Google Scholar]
- Mackenzie DI, Nichols JD, Royle JA, Pollock KH, Bailey LL, Hines JE. Occupancy Estimation and Modeling: Inferring Patterns and Dynamics of Species Occurence. Amsterdam: Elsevier; 2006. [Google Scholar]
- Mackenzie DI, Nichols JD, Seamans ME, Gutierrez R. Modeling species occurrence dynamics with multiple states and imperfect detection. Ecology. 2009;90:823–835. doi: 10.1890/08-0141.1. [DOI] [PubMed] [Google Scholar]
- Margolis L, Esch GW, Holmes JC, Kuris AM, Schad GA. The use of ecological terms in parasitology. Journal of Parasitology. 1982;68:131–133. [Google Scholar]
- McCoy KD. The population genetic structure of vectors and our understanding of disease epidemiology. Parasite-Journal De La Societe Francaise De Parasitologie. 2008;15:444–448. doi: 10.1051/parasite/2008153444. [DOI] [PubMed] [Google Scholar]
- McCoy KD, Boulinier T, Tirard C, Michalakis Y. Host specificity of a generalist parasite: genetic evidence of sympatric host races in the seabird tick Ixodes uriae. Journal of Evolutionary Biology. 2001;14:395. [Google Scholar]
- McCoy KD, Boulinier T, Tirard C, Michalakis Y. Host-dependent genetic structure of parasite populations: differential dispersal of seabird tick host races. Evolution. 2003;57:288–296. doi: 10.1111/j.0014-3820.2003.tb00263.x. [DOI] [PubMed] [Google Scholar]
- McCoy KD, Chapuis E, Tirard C, Boulinier T, Michalakis Y, Le Bohec C, Le Maho Y, et al. Recurrent evolution of host-specialized races in a globally distributed parasite. Proceedings of the Royal Society of London, Series B: Biological Sciences. 2005;272:2389. doi: 10.1098/rspb.2005.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie FE, Sirichaisinthop J, Miller RS, Gasser RAJ, Wongsrichanalai C. Dependence of malaria detection and species diagnosis by microscopy on parasite density. American Journal of Tropical Medicine and Hygiene. 2003;69:372–376. [PMC free article] [PubMed] [Google Scholar]
- Olsen B. The role of birds in the ecology and epidemiology of Lyme borreliosis and ehrlichiosis. Infectious Diseases. 2003;2/2003:4–7. [Google Scholar]
- Olsen B. Borrelia. In: Thomas NJ, Hunter DB, Atkinson CT, editors. Infectious Diseases of Wild Birds. Oxford, UK: Blackwell Publishing; 2007. pp. 341–351. [Google Scholar]
- Olsen B, Jaenson TGT, Noppa L, Bunikis J, Bergström S. A Lyme borreliosis cycle in seabirds and Ixodes uriae ticks. Nature. 1993;362:340. doi: 10.1038/362340a0. [DOI] [PubMed] [Google Scholar]
- Olsen B, Duffy DC, Jaenson TGT, Gylfe A, Bonnedahl J, Bergström S. Transhemispheric exchange of Lyme disease spirochetes by seabirds. Journal of Clinical Microbiology. 1995;33:3270–3274. doi: 10.1128/jcm.33.12.3270-3274.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostfeld RS, Keesing F, LoGiudice K. Community ecology meets epidemiology: the case of Lyme disease. In: Collinge SK, Ray C, editors. Disease Ecology: Community Structure and Pathogen Dynamics. New York, NY: Oxford University Press; 2006. pp. 28–40. [Google Scholar]
- Piesman J, Oliver JR, Sinsky RJ. Growth kinetics of the Lyme disease spirochete (Borrelia Burgdorferi) in vector ticks (Ixodes Dammini. American Journal of Tropical Medicine and Hygiene. 1990;42:352–357. doi: 10.4269/ajtmh.1990.42.352. [DOI] [PubMed] [Google Scholar]
- Qiu W-G. Geographic Uniformity of the Lyme Disease Spirochete (Borrelia burgdorferi) and its shared history with tick vector (Ixodes scapularis) in the Northeastern United States. Genetics. 2002;160:833–849. doi: 10.1093/genetics/160.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W-G, Schutzer SE, Bruno JF, Attie O, Xu Y, Dunn JJ, Fraser CM, Casjens SR, Luft BJ. Genetic exchange and plasmid transfers in Borrelia burgdorferi sensu stricto revealed by three-way genome comparisons and multilocus sequence typing. Proceedings of the National Academy of Science of the United States of America. 2004;101:14150–14155. doi: 10.1073/pnas.0402745101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel S, Hervé J, Diarrassouba S, Kone A, Cuny G. Microsatellite markers for population genetic studies in Aedes aegypti (Diptera: Culicidae) from Côte d'Ivoire: evidence for a microgeographic genetic differentiation of mosquitoes from Bouaké. Acta Tropica. 2002;82:39. doi: 10.1016/s0001-706x(02)00028-1. [DOI] [PubMed] [Google Scholar]
- Ravel S, De Meeûs T, Dujardin JP, Zézé D, Gooding RH, Dusfour I, Sané B, et al. The tsetse fly Glossina palpalis palpalis is composed of several genetically differentiated small populations in the sleeping sickness focus of Bonon, Côte d'Ivoire. Infection, Genetics and Evolution. 2007;7:116–125. doi: 10.1016/j.meegid.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Rothschild M, Clay T. Fleas, Flukes and Cuckoos: A Study of Bird Parasites. London: Arrow Books Ltd; 1961. [Google Scholar]
- Schwartz I, Varde S, Nadelman RB, Wormser GP, Fish D. Inhibition of efficient polymerase chain reaction amplification of Borrelia burgdorferi DNA in blood-fed ticks. American Journal of Tropical Medicine and Hygiene. 1997;56:339–342. doi: 10.4269/ajtmh.1997.56.339. [DOI] [PubMed] [Google Scholar]
- Stanek G, Strle F. Lyme borreliosis. The Lancet. 2003;362:1639. doi: 10.1016/S0140-6736(03)14798-8. [DOI] [PubMed] [Google Scholar]
- Staszewski V, McCoy KD, Tveraa T, Boulinier T. Interannual dynamics of antibody levels in naturally infected long-lived colonial birds. Ecology. 2007;88:3183–3191. doi: 10.1890/07-0098.1. [DOI] [PubMed] [Google Scholar]
- Staszewski V, McCoy KD, Boulinier T. Variable exposure and immunological response to Lyme disease Borrelia among North Atlantic seabird species. Proceedings of the Royal Society of London, Series B: Biological Sciences. 2008;275:2101. doi: 10.1098/rspb.2008.0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. Journal of Clinical Investigations. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teglas M, Foley J. Differences in the transmissibility of two Anaplasma phagocytophilum strains by the North American tick vector species, Ixodes Pacificus and Ixodes Scapularis (Acari: Ixodidae) Experimental and Applied Acarology. 2006;38:47. doi: 10.1007/s10493-005-5293-5. [DOI] [PubMed] [Google Scholar]
- Thompson KG. Use of site occupancy models to estimate prevalence of Myxobolus cerebralis infection in trout. Journal of Aquatic Animal Health. 2007;19:8–13. doi: 10.1577/H06-016.1. [DOI] [PubMed] [Google Scholar]
- Valkinas G, Bensch S, Iezhova TA, Kri anauskien A, Hellgren O, Bolshakov CV. Nested Cytochrome B Polymerase Chain Reaction Diagnostics Underestimate Mixed Infections of Avian Blood Haemosporidian Parasites: microscopy is Still Essential. Journal of Parasitology. 2006;92:418–422. doi: 10.1645/GE-3547RN.1. [DOI] [PubMed] [Google Scholar]
- Wang G, Liveris D, Brei B, Wu H, Falco RC, Fish D, Schwartz I. Real-Time PCR for simultaneous detection and quantification of Borrelia burgdorferi in field-collected Ixodes scapularis ticks from the Northeastern United States. Applied and Environmental Microbiology. 2003;69:4561–4565. doi: 10.1128/AEM.69.8.4561-4565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller TJ. Using occupancy estimation to assess the effectiveness of a regional multiple-species conservation plan: bats in the Pacific Northwest. Biological Conservation. 2008;141:2279. [Google Scholar]
- White GC, Burnham KP. Program MARK: survival estimation from populations of marked animals. Bird Study. 1999;46:120–139. [Google Scholar]
- Whiteman NK, Matson KD, Bollmer JL, Parker PG. Disease ecology in the Galapagos Hawk (Buteo galapagoensis): host genetic diversity, parasite load and natural antibodies. Proceedings of the Royal Society of London, Series B: Biological Sciences. 2006;273:797. doi: 10.1098/rspb.2005.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson N. Acarina: Metastigmata: Ixodidae of South Georgia, Heard and Kerguelen. Pacific Insects Monographs. 1970;23:78–88. [Google Scholar]
- Winchell CS, Doherty PF. Using California gnatcatcher to test underlying models in habitat conservation plans. Journal of Wildlife Management. 2008;72:1322–1327. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.