

Abstract
We analyze mathematical models to examine how the genetic basis of fitness affects the persistence of a population suddenly encountering a harsh environment where it would go extinct without evolution. The results are relevant for novel introductions and for an established population whose existence is threatened by a sudden change in the environment. The models span a range of genetic assumptions, including identical loci that contribute to absolute fitness, a two-locus quantitative genetic model with nonidentical loci, and a model with major and minor genes affecting a quantitative trait. We find as a general (though not universal) pattern that prospects for persistence narrow as more loci contribute to fitness, in effect because selection per locus is increasingly weakened with more loci, which can even overwhelm any initial enhancement of fitness that adding loci might provide. When loci contribute unequally to fitness, genes of small effect can significantly reduce extinction risk. Indeed, major and minor genes can interact synergistically to reduce the time needed to evolve growth. Such interactions can also increase vulnerability to extinction, depending not just on how genes interact but also on the initial genetic structure of the introduced, or newly invaded, population.
Keywords: absolute mean fitness, extinction risk, major and minor genes, quantitative trait
The ‘holy grail’ of invasion biology has been the identification of both suites of traits of introduced species, and characteristics of the environment of invasion, that permit a reasonably accurate prediction of the success or failure of invasive species. Developing such a characterization of invasion success or failure has been a surprisingly difficult challenge (e.g., Williamson and Fitter 1996). There are many reasons for this. For instance, it is challenging to make reliable predictions in circumstances that are necessarily highly stochastic due, say, to the small population sizes of invasive propagules, or the haphazard and sporadic nature of inadvertent introductions. In addition, there is often only a very rough understanding of the ecological factors (e.g., local community structure) that permit population persistence in native ranges, an understanding that is needed for prediction of establishment success or failure to be effective in the arena of introduction (Peterson and Vieglais 2001).
In this paper, we explore another potentially important dimension of uncertainty in making predictions about invasion. In many situations, invasion may be facilitated, or may even absolutely require, adaptive evolution to novel environments, similar in some respects but differing in others from the environments found at the site of origin. One applied situation beyond the usual scenarios of invasion biology where this is particularly germane is the emergence of novel infectious diseases, where genetic adaptation to a novel host may be required for successful establishment and persistence (Holt and Hochberg 2002; Antia et al. 2003). To predict the success or failure of invasions, one thus must understand something about the genetics and evolutionary dynamics of the introduced species in a novel environment (Lee 2002; Lee and Gelembiuk 2008).
The mapping between genotype and phenotype is often highly complex, and any particular phenotype that might be exposed to selection could be produced by any of a wide range of genetic bases. Tolerance to an abiotic stressor, for instance, might be influenced by genetic variation at a single genetic locus with alleles of large effect on fitness or instead by the summation of effects across many loci, each with a small effect on fitness. When there are many loci involved, these could be either unlinked or linked to various degrees. Genetic loci could have additive effects upon the phenotype, or there could be nonadditive genetic interactions (epistasis). For all these reasons, among others, one expects substantial heterogeneity among species in the genetic capacity to adapt to novel environments. Superficially similar species might actually differ greatly in their capacity for evolutionary responses during adaptation to, and invasion of, novel environments. The dearth of knowledge about the genetic basis of traits relevant to adaptive evolution for most species (not just invasive species) suggests that this could be a significant reason (among others) for the difficulty in constructing a general, predictive theory of species invasions.
In this paper, we focus on a particular kind of novel environment, namely one where in the absence of evolution, an introduced species is expected to face extinction, because conditions there are outside its ecological niche (Holt et al. 2005). We define ‘adaptive colonization’ to be colonization that requires evolution of a colonizing population initially facing extinction, because its mean fitness (finite growth rate) is less than one. If the population mean fitness remains consistently less than unity, individuals are not being replaced across generations, on average, and the population will inexorably decline towards extinction in the absence of heritable genetic variation in fitness. If such genetic variation is present, and if selection can shift the mean fitness so that it exceeds one sufficiently rapidly, the population can begin to rebound and thus has a chance of persisting in the novel environment. The central issue we address in this article is how the genetic basis of fitness influences the likelihood of such adaptive colonization by an introduced species.
While invasive species must sometimes overcome inhospitable environmental conditions to become established, once a population is established, comparable issues arise when considering the potential impact of the invader upon resident species. Managers are not concerned with invasive species which, though present, are rare and unobtrusive, but rather with species such as Melaleuca quinquenervia in southern Florida, which has become sufficiently abundant and ecologically dominant to drastically alter the environment in unfavorable ways for a wide range of other species (Bodle et al. 1994). In some cases, the arrival of invaders may sharply degrade the environment for some resident species, and do so sufficiently to threaten their persistence. If the invader in effect pushes resident species out of their ancestral niches, adaptive evolution would be required to rescue these species from extinction in the new world created by the dominant invader.
As a possible example, on the island of Guam, the introduced brown treesnake (Boiga irregularis) severely impacted many native vertebrate species, including several species of geckos and skinks. Other gecko and skink species, however, have managed to coexist with the invasive snake (Fritts and Rodda 1998; Lockwood et al. 2007). These interspecific differences in responses among members of the native community could merely reflect differences in their degrees of exposure to the invasive species (e.g., microhabitat use), or instead could potentially reflect differences among taxa in the scope and rapidity of adaptive evolutionary responses to this novel threat in their environment.
In either scenario, successful establishment by invaders or resistance to invasion impacts by residents might depend on the genetic capacity species have to adapt rapidly enough to avoid extinction in novel environments—one component of their ‘evolvability’ (Houle 1992). Theory suggests that evolution is unlikely to rescue populations finding themselves suddenly exposed to severe conditions, such as those faced by a colonizing propagule entering habitats with conditions outside its fundamental niche, or by a resident species living in a world dominated by an aggressively successful invader (Gomulkiewicz and Holt 1995; Boulding and Hay 2001; Orr and Unckless 2008). However, little is known about how the detailed genetic basis of fitness might affect the prospects of successful adaptation in poor environments. Our contribution will be to investigate how the genetic complexity underlying absolute fitness impacts the capacity of invasive or invaded populations to adapt and persist when abruptly faced with harsh conditions. We use a theoretical approach to address this issue, comparing mathematical models that differ in their assumptions about the underlying genetics of fitness. We examine three deterministic models, which differ greatly in their assumptions about how genotypes map onto fitness, and then use simulations to examine the robustness of our conclusions to issues of demographic and genetic stochasticity, which are always present as a population declines toward extinction.
Invasion fitness determined by direct genetic effects
We begin with the biologically most abstract, but mathematically most tractable, case where postinvasion fitness is determined by summing over a set of loci, each of which impacts fitness. Assume a randomly mating diploid species with continuous, overlapping generations. Fitnesses are density-independent and each fitness-contributing locus has two alleles, A and a, with respective frequencies pt,i and qt,i = 1 −pt,i at locus i at time t. We assume that locus i has Malthusian fitness parameters mAA,i,, mAa,i, and maa,i for the three genotypes and that the loci contribute additively to total fitness. [Recall that Malthusian fitness may be thought of as the logarithm of fitness in discrete-time, nonoverlapping generation models (Crow and Kimura 1970, p. 7), so additive contributions across loci to Malthusian fitness correspond to multiplicative contributions to fitness in discrete generation models]. In addition, we assume Hardy-Weinberg proportions at each locus, a condition which is approached quickly for this continuous-time model if selection is weak (Nagylaki and Crow 1974; Nagylaki 1992). Given these conditions, the mean Malthusian fitness at time t is
| (1) |
where n is the number of loci contributing to fitness. Note that 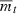 is also the intrinsic rate of increase: the population shrinks deterministically when it is negative and grows when it is positive.
is also the intrinsic rate of increase: the population shrinks deterministically when it is negative and grows when it is positive.
For further simplicity, we assume the parameterization 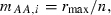
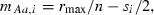 and
and 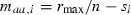 such that a population fixed for the A allele at all loci grows at intrinsic rate of increase rmax. These Malthusian fitnesses are called ‘additive’ (Crow and Kimura 1970) since each copy of A at locus i adds si/2 to individual fitness compared with that of an aa homozygote at that locus. We assume, without loss of generality, that the selection coefficient si > 0 so that a is the relatively deleterious allele at each locus. With this parameterization, the mean Malthusian fitness (1) simplifies to
such that a population fixed for the A allele at all loci grows at intrinsic rate of increase rmax. These Malthusian fitnesses are called ‘additive’ (Crow and Kimura 1970) since each copy of A at locus i adds si/2 to individual fitness compared with that of an aa homozygote at that locus. We assume, without loss of generality, that the selection coefficient si > 0 so that a is the relatively deleterious allele at each locus. With this parameterization, the mean Malthusian fitness (1) simplifies to
| (2) |
Since both si and qt.i are non-negative, growth can evolve only if rmax > 0. Given additive fitnesses, one can solve for qt,i as a function of the si, q0,i, and t (Crow and Kimura 1970, p. 193), 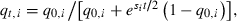 giving
giving
| (3) |
Our first objective is to determine how the time needed to evolve deterministically from a given initial intrinsic rate of decrease (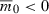 ) to a non-negative growth rate (
) to a non-negative growth rate (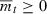 ) depends on n, the number of loci that determine fitness. If T is the first time the evolving population ceases to decline, then
) depends on n, the number of loci that determine fitness. If T is the first time the evolving population ceases to decline, then 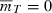 . Substituting this into (3) shows T is defined implicitly by
. Substituting this into (3) shows T is defined implicitly by
| (4) |
This equation can be solved numerically for the time T to evolve growth from 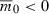 .
.
An explicit formula for T can be derived in the extreme case where loci have identical allele frequencies. Let q0,i = q0 and si = s for all i. Solving (4) for T gives
| (5) |
Suppose we hold constant the initial mean fitness 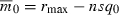 , the maximum possible growth rate rmax, and the initial frequency of the deleterious allele at each locus q0 as n varies. These conditions imply
, the maximum possible growth rate rmax, and the initial frequency of the deleterious allele at each locus q0 as n varies. These conditions imply
| (6) |
Substituting (6) into (5) shows
| (7) |
Expression (7) shows that the time to evolve growth increases linearly with the number of loci n. The cause of this is apparent in (6), which shows that the strength of selection per locus declines with the number of loci contributing to fitness.
Although our immediate interest is comparing invaders with the same initial rate of decline 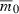 , it might make sense to keep quantities other than rmax or q0 constant for different numbers of loci. Indeed, suppose we keep
, it might make sense to keep quantities other than rmax or q0 constant for different numbers of loci. Indeed, suppose we keep 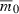 , rmax, and s—rather than q0—constant as n varies. Then
, rmax, and s—rather than q0—constant as n varies. Then 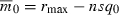 implies that the initial frequency of the deleterious allele must be inversely proportional to n:
implies that the initial frequency of the deleterious allele must be inversely proportional to n: 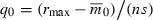 . Substituting this into (5) shows that T now decreases with the number of loci (Fig. 1A). Note that
. Substituting this into (5) shows that T now decreases with the number of loci (Fig. 1A). Note that 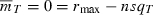 so that
so that 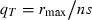 . Thus, the amount of allele frequency change required at each locus to achieve growth,
. Thus, the amount of allele frequency change required at each locus to achieve growth, 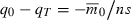 , declines with n while the strength of selection per locus remains constant by assumption. Genomes with more loci may experience the same selection per locus, but require less evolution at each locus to achieve growth than those with fewer loci.
, declines with n while the strength of selection per locus remains constant by assumption. Genomes with more loci may experience the same selection per locus, but require less evolution at each locus to achieve growth than those with fewer loci.
Figure 1.
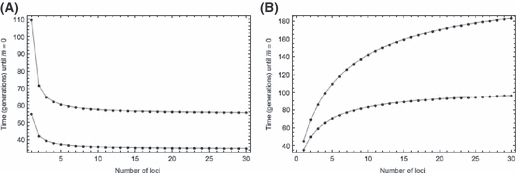
Time to evolve growth versus the number of identical, independent loci contributing to growth. Lower and upper sets of points correspond respectively to 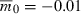 and
and 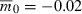 . (A) Holding per locus selection constant at s = 0.04 [equation 5 with
. (A) Holding per locus selection constant at s = 0.04 [equation 5 with 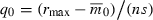 ] and rmax = 0.01. (B) Holding initial variance in fitness constant at V0 = 10−4 (equations 5 and 8) with rmax = 0.1.
] and rmax = 0.01. (B) Holding initial variance in fitness constant at V0 = 10−4 (equations 5 and 8) with rmax = 0.1.
Finally, consider how the number of loci affects T when assuming the total initial variance in fitness, 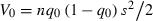 , along with
, along with 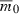 and rmax are held constant for different n. Keeping these quantities fixed implies
and rmax are held constant for different n. Keeping these quantities fixed implies
| (8a) |
and
| (8b) |
which can be substituted into (5) to express how the time to evolve growth T depends on the number of loci n (Fig. 1B) for invaders with the same 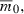 rmax, and V0. Since mean fitness evolves at a rate determined by the variance in fitness (Fisher 1930), fixing V0 in effect compares invaders with different n but the same initial per generation increase in mean fitness. Note that per locus selection is again weaker with larger n (equation 8a), as when q0 was held constant (equation 6), but this is to some extent countered by the lower initial frequency of deleterious alleles with more loci (equation 8b), similar to the case where s was assumed constant. Numerical evaluations suggest the net result is that T increases with n (Fig. 1B). Apparently, the reduction in total allele frequency change required for growth for genomes with larger n implied by (8b) fails to compensate for the slower evolution at individual loci due to the weaker selection per locus (equation 8a).
rmax, and V0. Since mean fitness evolves at a rate determined by the variance in fitness (Fisher 1930), fixing V0 in effect compares invaders with different n but the same initial per generation increase in mean fitness. Note that per locus selection is again weaker with larger n (equation 8a), as when q0 was held constant (equation 6), but this is to some extent countered by the lower initial frequency of deleterious alleles with more loci (equation 8b), similar to the case where s was assumed constant. Numerical evaluations suggest the net result is that T increases with n (Fig. 1B). Apparently, the reduction in total allele frequency change required for growth for genomes with larger n implied by (8b) fails to compensate for the slower evolution at individual loci due to the weaker selection per locus (equation 8a).
Clearly, then, conclusions about how the number of loci affects the time required by an invading population to evolve growth depend on what is held constant among populations. The contrast between our results when holding the fitness variance V0 (Fig. 1B) or allele frequency q0 (equation 7) constant versus holding the per locus selection coefficient s constant (Fig. 1A) could hardly be greater.
Our analyses assumed the growth rate at the start of invasion, 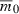 , was held constant regardless of how many or few loci contribute to fitness. In many cases, however, one expects that adding loci will directly alter fitness by, say, increasing metabolic capacity or changing the regulation of genetic factors. In the next two sections, we account for these new effects explicitly by assuming loci contribute to variation in a quantitative trait on which individual fitness and the mean fitness of the invading population depend. We also use this framework to relax the assumption that loci make identical contributions to individual fitness, and then examine the consequences of demographic stochasticity.
, was held constant regardless of how many or few loci contribute to fitness. In many cases, however, one expects that adding loci will directly alter fitness by, say, increasing metabolic capacity or changing the regulation of genetic factors. In the next two sections, we account for these new effects explicitly by assuming loci contribute to variation in a quantitative trait on which individual fitness and the mean fitness of the invading population depend. We also use this framework to relax the assumption that loci make identical contributions to individual fitness, and then examine the consequences of demographic stochasticity.
Invasion fitness determined by a quantitative trait
We assume that fitness depends on n loci via a quantitative trait z and model an invading population with random mating, discrete, nonoverlapping generations and density-independent growth. The finite growth rate at time t is 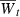 , the mean fitness. At the onset of invasion,
, the mean fitness. At the onset of invasion, 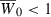 so that the population is initially in decline. Our goal is to determine how the period needed to evolve a mean fitness above one depends on the genetic assumptions. Unlike the previous section, we do not hold the initial growth rate constant as we alter the genetics. This could have consequences of its own on time to demographic recovery.
so that the population is initially in decline. Our goal is to determine how the period needed to evolve a mean fitness above one depends on the genetic assumptions. Unlike the previous section, we do not hold the initial growth rate constant as we alter the genetics. This could have consequences of its own on time to demographic recovery.
There are countless ways to map genotypes to phenotypes. We concentrate here on just two comparatively tractable biallelic diploid models—a two-locus model and the n-locus hypergeometic model—and leave a more general treatment for future investigation. For both models, we assume alleles combine additively to determine phenotypes z and employ the ‘quasi-linkage equilibrium’ (QLE) approximation. The QLE approximation presumes epistasis and selection are weak compared with recombination, such that the statistical associations among loci are small (on the same order as selection coefficients) and evolve more rapidly towards equilibrium than do allele frequencies (Barton and Turelli 1991).
The hypergeometric model (Barton 1992; Doebeli 1996; Shpak and Kondrashov 1999) assumes n loci with identical effects and allele frequencies. The loci are unlinked but can be statistically associated. Since all loci are identical, we need only follow the evolution of the shared allele frequency. By comparison, the two-locus model allows loci to differ and be linked. We use the QLE approximation to track allele frequencies at each locus separately.
We develop recursions assuming the trait z on which individual fitness depends (e.g., z might be body size or metabolic rate) is determined by adding allelic effects within and across loci. Let
| (9) |
where the genotype 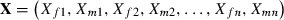 is a string of 2n indicator variables, each representing the presence (X = 1) or absence (X = 0) of a maternally (subscript mi) or paternally (subscript fi) derived allele with effect bi at locus i. An individual with all ‘0’ alleles has phenotype 0, which implies all phenotypic values z are scaled relative to that of this genotype. For simplicity, we ignore environmental effects meaning all individuals with a given genotypic value have the same phenotype. Assuming no sex differences
is a string of 2n indicator variables, each representing the presence (X = 1) or absence (X = 0) of a maternally (subscript mi) or paternally (subscript fi) derived allele with effect bi at locus i. An individual with all ‘0’ alleles has phenotype 0, which implies all phenotypic values z are scaled relative to that of this genotype. For simplicity, we ignore environmental effects meaning all individuals with a given genotypic value have the same phenotype. Assuming no sex differences 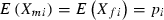 , the frequency of the ‘1’ allele at locus i, so the mean phenotype is
, the frequency of the ‘1’ allele at locus i, so the mean phenotype is
| (10) |
We model absolute fitness as a quadratic function of z, 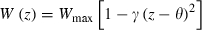 , where θ is the optimal phenotype, γ is a positive constant that governs effects of deviations from θ on fitness, and Wmax is the absolute fitness of an individual with the optimal phenotype. To ensure plausibility (i.e., absolute fitness must be non-negative), we assume the quadratic term is less than one. This quadratic fitness model is flexible enough to represent both primarily directional selection (when
, where θ is the optimal phenotype, γ is a positive constant that governs effects of deviations from θ on fitness, and Wmax is the absolute fitness of an individual with the optimal phenotype. To ensure plausibility (i.e., absolute fitness must be non-negative), we assume the quadratic term is less than one. This quadratic fitness model is flexible enough to represent both primarily directional selection (when 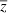 is far from θ) and primarily stabilizing selection (when
is far from θ) and primarily stabilizing selection (when 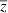 is near θ). The finite growth rate (mean fitness) is
is near θ). The finite growth rate (mean fitness) is
| (11) |
where  is given by (10) and
is given by (10) and
 |
(12) |
is the phenotypic variance given linkage disequilibrium coefficients Dij between loci i and j with qi = 1 −pi.
Assuming γ << 1 and that the conditions for the quasi-linkage equilibrium approximation hold, the linkage disequilibria converge quickly to quasi-equilibrium values  that depend only on the far more slowly evolving allele frequencies (Barton and Turelli 1991). For our model, it can be shown that these disequilibria are
that depend only on the far more slowly evolving allele frequencies (Barton and Turelli 1991). For our model, it can be shown that these disequilibria are
| (13) |
where rij is the recombination rate between loci i and j, and that the between-generation change in the pi is
| (14) |
to first order in γ. (A Mathematica notebook with the detailed derivation of these equations is available from the lead author.) Substituting (13) in (12) shows that, at QLE, the mean fitness (11) is a function solely of the slow changing allele frequencies.
For the two-locus QLE model there are two recursions (14), one each for p1 and p2. The mean phenotype in this case is  and the phenotypic variance is
and the phenotypic variance is
| (15) |
where r = r12 is the recombination rate between the two loci. For the n-locus hypergeometric model, we set pi = p, qi = q, bi = b, and rij = ½ for all i and j in equations 10–14. The mean (10) and variance (12) then simplify, respectively, to 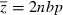 and
and
| (16) |
and the recursion for the shared allele frequency p is
| (17) |
Note that the phenotypic variance (16) in the hypergeometric case decreases with increasing n. Since absolute mean fitness increases as σ2 decreases, then for a fixed initial allele frequency p0, the initial growth rate 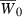 will be larger for invaders with more loci contributing to variation in the quantitative trait (Fig. 2). Could this initial advantage translate into a shorter period of decline, and less extinction risk, for invaders with larger n?
will be larger for invaders with more loci contributing to variation in the quantitative trait (Fig. 2). Could this initial advantage translate into a shorter period of decline, and less extinction risk, for invaders with larger n?
Figure 2.
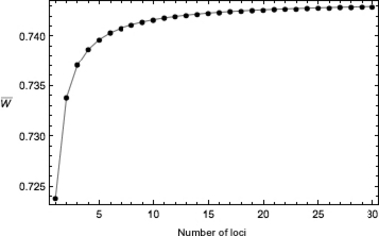
Finite growth rate 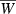 versus the number quantitative trait loci for the hypergeometric model (equations 10, 11, and 16) with Wmax = 1.1, θ = 2, γ = 0.1, p = 0.1, and b = 1/n.
versus the number quantitative trait loci for the hypergeometric model (equations 10, 11, and 16) with Wmax = 1.1, θ = 2, γ = 0.1, p = 0.1, and b = 1/n.
To address this conjecture, we numerically iterated the hypergeometric model recursions (17) using definitions (10), (11), and (16) in order to compute the time T needed to adapt from 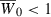 to
to 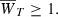 The results show that, while having more loci can indeed increase the initial growth rate (Fig. 2), the increase is generally not enough to overcome the concomitantly slower rate of adaptation that results presumably from the effect of spreading selection across loci, thus weakening selection at each locus (Fig. 3).
The results show that, while having more loci can indeed increase the initial growth rate (Fig. 2), the increase is generally not enough to overcome the concomitantly slower rate of adaptation that results presumably from the effect of spreading selection across loci, thus weakening selection at each locus (Fig. 3).
Figure 3.
Evolution of the finite growth rate 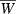 for the hypergeometric model (equations 10, 11, 16, and 17) with Wmax = 1.1, θ = 2, γ = 0.1, and b = 1/n, where n is the number of loci. Lines: red (n = 1), blue (n = 2), green (n = 4), purple (n = 8), orange (n = 16). Initial allele frequency for all is p0 = 0.1.
for the hypergeometric model (equations 10, 11, 16, and 17) with Wmax = 1.1, θ = 2, γ = 0.1, and b = 1/n, where n is the number of loci. Lines: red (n = 1), blue (n = 2), green (n = 4), purple (n = 8), orange (n = 16). Initial allele frequency for all is p0 = 0.1.
The hypergeometric model makes the obviously extreme assumption that frequencies and phenotypic effects of alleles are exactly the same at all loci. We used the two-locus QLE approximation (14) with (15) to explore how differences among loci might affect the capacity of an invader to evolve growth. We found that differences in initial allele frequencies and in phenotypic effects produce opposite results (Fig. 4). Indeed, differences in initial allele frequencies between the two loci lengthen the time needed to evolve growth relative to the hypergeometric model (with the same average initial allele frequency), and larger differences result in longer delays (Fig. 4A). In contrast, differences in phenotypic effects among loci shrink the time required for growth to evolve, compared with the hypergeometric model with the same average effect, and, moreover, larger differences produce steeper increases in mean fitness (Fig. 4B).
Figure 4.
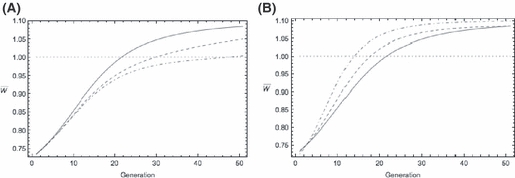
Evolution of the finite growth rate  for the two-locus QLE model (equations 10, 11, 14, and 15) with Wmax = 1.1, θ = 2, γ = 0.1, r12 = 0.5 (unlinked loci) and initial disequilibrium coefficient D12 = 0. Solid curve in both panels is the two-locus hypergeometric model with b = 0.5 and p0 = 0.1. (A) Different, but same average, initial allele frequencies at the two loci. Effect sizes are the same at both loci, b = 0.5. Dashed trajectory: p10 = 0.01, p20 = 0.19. Dot-dashed trajectory: p10 = 0.001, p20 = 0.199. (B) Different, but same average, effect sizes at the two loci. Initial frequencies are the same at both loci, p0 = 0.1. Dashed trajectory: b1 = 0.25, b2 = 0.75. Dot-dashed trajectory: b1 = 0.01, b2 = 0.99.
for the two-locus QLE model (equations 10, 11, 14, and 15) with Wmax = 1.1, θ = 2, γ = 0.1, r12 = 0.5 (unlinked loci) and initial disequilibrium coefficient D12 = 0. Solid curve in both panels is the two-locus hypergeometric model with b = 0.5 and p0 = 0.1. (A) Different, but same average, initial allele frequencies at the two loci. Effect sizes are the same at both loci, b = 0.5. Dashed trajectory: p10 = 0.01, p20 = 0.19. Dot-dashed trajectory: p10 = 0.001, p20 = 0.199. (B) Different, but same average, effect sizes at the two loci. Initial frequencies are the same at both loci, p0 = 0.1. Dashed trajectory: b1 = 0.25, b2 = 0.75. Dot-dashed trajectory: b1 = 0.01, b2 = 0.99.
When the two loci differ in both their allele frequencies and phenotypic effects, the consequences for the evolution of invasion fitness are, not surprisingly, more complicated though our results do suggest some general features. First, if initial allele frequencies and phenotypic effects are positively associated (i.e., the locus with larger allelic effect bi also has the higher initial frequency pi0), then growth evolves faster compared with a model in which only phenotypic effects differ among loci (Fig. 5A) whereas if the association is negative, then mean fitness evolves slowly compared to even the hypergeometric model (Fig. 5B). A positive association between bi and pi0 can overcome the reduced rate of evolution caused by variable allele frequencies (Fig. 5C). While a negative association can slow the rate of adaptation compared with allele frequency differences alone, it need not do so (Fig. 5D). Taken together, these results show that the time required for an invading or invaded population to evolve so that it can grow depends on not just the genetic basis of fitness (via the phenotypic effects at each locus) but also on the initial genetic structure of the population, which in turn reflects its history.
Figure 5.
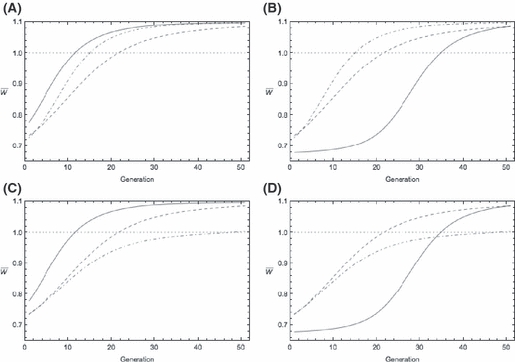
Impacts of jointly variable initial allele frequencies and effect sizes on evolution of the finite growth rate  (solid curves). Parameters as for Fig. 4 except where noted. Solid curve b1 = 0.1, b2 = 0.9, p10 = 0.001, p20 = 0.199 in both left-hand panels (i.e., initial frequencies and effects positively associated) and solid curve b1 = 0.1, b2 = 0.9, p10 = 0.199, p20 = 0.001 in both right-hand panels (initial frequencies and effects negatively associated). Dot-dashed curves, upper panels: effect sizes as for the solid curve but with initial allele frequency p0 = 0.1 at both loci. Dot-dashed curves, lower panels: initial allele frequencies as for the solid curve but with effect size b = 0.5 at both loci. The dashed curve in all four panels is the corresponding hypergeometric model (equations 16 and 17) with p0 = 0.1 and effect size b = 0.5.
(solid curves). Parameters as for Fig. 4 except where noted. Solid curve b1 = 0.1, b2 = 0.9, p10 = 0.001, p20 = 0.199 in both left-hand panels (i.e., initial frequencies and effects positively associated) and solid curve b1 = 0.1, b2 = 0.9, p10 = 0.199, p20 = 0.001 in both right-hand panels (initial frequencies and effects negatively associated). Dot-dashed curves, upper panels: effect sizes as for the solid curve but with initial allele frequency p0 = 0.1 at both loci. Dot-dashed curves, lower panels: initial allele frequencies as for the solid curve but with effect size b = 0.5 at both loci. The dashed curve in all four panels is the corresponding hypergeometric model (equations 16 and 17) with p0 = 0.1 and effect size b = 0.5.
Fitness determined by major and minor genes
The two-locus model results suggest that the evolution of population growth can be accelerated when fitness depends on a combination of mutations at different loci with major and minor effects (Figs 4B and 5A). However, it is often the case that numerous mutations of small effect contribute to fitness. To evaluate whether the two-locus findings generalize to genomes with a major gene at one locus that determines fitness along with many genes of minor effect, we utilized a model of major and minor genes proposed by Lande (1983).
Lande's model considers a quantitative character z influenced by a biallelic locus of major effect and by a genetic background determined by a large, unspecified number of loci of small effect. The major and minor loci are assumed in linkage equilibrium. The background variation is normally distributed with mean 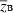 , fixed phenotypic variance σ2, and constant heritability h2. Lande's recursions are
, fixed phenotypic variance σ2, and constant heritability h2. Lande's recursions are
| (18a) |
| (18b) |
where 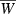 is mean fitness and p is the frequency of the ‘1’ allele at the major locus. We assume, as in the previous section, that the effect of trait z on fitness is quadratic with optimum phenotype θ, strength γ > 0, and maximum fitness Wmax > 1. We also assume, for simplicity, that alleles at the major locus do not have pleiotropic effects on fitness and that they act additively on the trait z, with each ‘1’ allele adding an amount b to the quantitative character. Then
is mean fitness and p is the frequency of the ‘1’ allele at the major locus. We assume, as in the previous section, that the effect of trait z on fitness is quadratic with optimum phenotype θ, strength γ > 0, and maximum fitness Wmax > 1. We also assume, for simplicity, that alleles at the major locus do not have pleiotropic effects on fitness and that they act additively on the trait z, with each ‘1’ allele adding an amount b to the quantitative character. Then
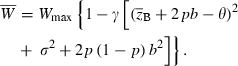 |
(18c) |
Equations (18) can be used to project evolution of the major locus, the background genetic variation, and the population's finite growth rate. Note that this approach does not explicitly consider allele frequencies or evolutionary dynamics of the individual minor loci.
Iteration of (18) shows that joint variation at major and minor genes can produce more substantial increases in mean fitness than can be achieved via evolution with either variation at the major locus alone (i.e., when h2 = 0 but p > 0) or minor gene variation alone (p = 0 but h2 > 0). For the example of dynamics shown in Fig. 6, the parameter values are such that it is impossible to evolve 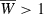 with variation at the major locus only (red symbols). With minor gene variation alone (blue symbols), the population can evolve
with variation at the major locus only (red symbols). With minor gene variation alone (blue symbols), the population can evolve 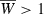 given sufficient time, but in Fig. 6 it is still well below this level after the 40 generations shown. Yet given both background and major locus genetic variation (purple symbols),
given sufficient time, but in Fig. 6 it is still well below this level after the 40 generations shown. Yet given both background and major locus genetic variation (purple symbols), 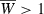 is achieved within the forty generations of adaptation. This demonstrates that major and minor genes can work synergistically to enhance the prospects of growth for an invader.
is achieved within the forty generations of adaptation. This demonstrates that major and minor genes can work synergistically to enhance the prospects of growth for an invader.
Figure 6.
Evolution of the finite growth rate 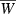 for Lande's major-minor locus model (equations 18) assuming Wmax = 1.1, θ = 2, γ = 0.1, b = 0.5, and σ2 = 0.64. Forty generations of joint dynamics of the major locus and background variation (initial value of
for Lande's major-minor locus model (equations 18) assuming Wmax = 1.1, θ = 2, γ = 0.1, b = 0.5, and σ2 = 0.64. Forty generations of joint dynamics of the major locus and background variation (initial value of 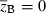 ) are plotted on the mean fitness contour surface. The straight dashed line corresponds to the phenotypic optimum,
) are plotted on the mean fitness contour surface. The straight dashed line corresponds to the phenotypic optimum, 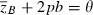 . Red symbols: variation at major locus only (p0 = 0.02, h2 = 0). Blue: background genetic variation only (p0 = 0, h2 = 0.1). Purple: background and major locus genetic variation (p0 = 0.02, h2 = 0.1).
. Red symbols: variation at major locus only (p0 = 0.02, h2 = 0). Blue: background genetic variation only (p0 = 0, h2 = 0.1). Purple: background and major locus genetic variation (p0 = 0.02, h2 = 0.1).
The apparent synergy between major and minor genes can be traced to conflicting effects of the major locus on mean fitness. As the favored ‘1’ allele spreads from low frequency, it quickly moves the population mean phenotypes towards the optimum θ (indicated by the straight dashed line, 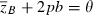 , in Fig. 6), which enhances mean fitness. But this progress is countered by the (transiently) increasing phenotypic variance contributed by the major locus, which increasingly slows the rise of mean fitness (see the last term in equation 18c). The comparable contribution to phenotypic variance from minor gene variation, σ2, is constant by assumption so the resistance to improving mean fitness of this variance is relatively constant. In Fig. 6 (purple symbols), spread of the favored allele at the major locus boosts the steady adaptation due to the minor genes before the increasing phenotypic variance can significantly decelerate the progress of mean fitness. While we did not find such synergy in all cases we examined, our results clearly demonstrate the potential for major-minor gene interactions to enhance the evolution of growth in invading or invaded populations experiencing harsh novel environments.
, in Fig. 6), which enhances mean fitness. But this progress is countered by the (transiently) increasing phenotypic variance contributed by the major locus, which increasingly slows the rise of mean fitness (see the last term in equation 18c). The comparable contribution to phenotypic variance from minor gene variation, σ2, is constant by assumption so the resistance to improving mean fitness of this variance is relatively constant. In Fig. 6 (purple symbols), spread of the favored allele at the major locus boosts the steady adaptation due to the minor genes before the increasing phenotypic variance can significantly decelerate the progress of mean fitness. While we did not find such synergy in all cases we examined, our results clearly demonstrate the potential for major-minor gene interactions to enhance the evolution of growth in invading or invaded populations experiencing harsh novel environments.
Simulations
Realistic population and evolutionary dynamics contain a number of stochastic elements, which are expected to be particularly important in small, extinction-prone populations such as colonizing groups in harsh environments. The models and analyses above completely ignore stochasticity and besides use a number of other approximations. Moreover, we measured extinction risk in terms of the evolution of mean fitness. This may not always be the best measure of the potential for evolutionary rescue. For instance, if one were dealing with a genetically heterogeneous clonal species, this measure clearly could be misleading; for population persistence, deterministically it suffices that a single clone be present with a positive growth rate, regardless of mean fitness across all clones. Despite this caveat, we have found in the past that analyzing how evolution changes mean fitness in deterministic models yields useful insights into adaptation into harsh environments using more complex models that take proper account of stochasticity (e.g., Holt and Gomulkiewicz 1997; Holt et al. 2003; Holt et al. 2005). In order to assess the heuristic utility of the deterministic approaches for gauging extinction risk utilized in the above models, we performed individual-based stochastic simulations to assess directly whether our conclusions are qualitatively robust regarding how genetics impacts the extinction risk of invaders in harsh environments.
Specifically, we based our simulations on the quantitative trait model corresponding to recursion (14). For each set of genetic assumptions and parameter values, we simulated 10 000 replicate populations and estimated the associated probability of extinction as the fraction of these that went extinct within 200 generations.
Details of the simulations are as follows. Each replicate started with N0 diploid hermaphroditic adult individuals. Each position at locus i within an individual was initially assigned the ‘1’ allele with probability p0i and the ‘0’ allele otherwise. Each adult could mate as a female, male, or both. If there were 500 or fewer adults, then all mated as females. Otherwise 500 adults were chosen randomly without replacement to be female parents. For each mating female, an individual was chosen at random from all adults (with replacement) to act as the male parent; self-fertilization was allowed. Each mated pair produced a binomially distributed number of offspring with expectation Wmax and probability of success Wmax/2. The parent generation died after all matings were completed. There was no mutation, and each parent contributed a haploid gamete with free recombination to each of its offspring. An offspring survived to adulthood with probability 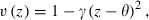 where z is its phenotype (determined via equation 9), θ is the optimum phenotype, and γ is the strength of stabilizing selection. Extinction occurred if no offspring survived to adulthood.
where z is its phenotype (determined via equation 9), θ is the optimum phenotype, and γ is the strength of stabilizing selection. Extinction occurred if no offspring survived to adulthood.
Results from these stochastic simulations are, on average, in quantitative agreement with the predictions of our deterministic analyses, even for initial population sizes as small as four (see Fig. S1). The simulations also confirm that the time needed to evolve growth is positively related to the probability of extinction. In particular, Fig. 7 confirms that genomes with more loci have higher probabilities of extinction, as suggested by our deterministic analyses of the hypergeometric model (cf., Fig. 3). Our two-locus simulations likewise show that differences among loci in phenotypic effects reduce extinction risk (compare Figs 8A and 4B), that differences in initial allele frequencies increase vulnerability to extinction (Fig. 8A versus Fig. 4A), and that positive/negative association between phenotypic effects and initial allele frequencies further reduce/enhance extinction hazard (Fig. 8B versus Fig. 5). In sum, the simulation results suggest strongly that our relatively tractable deterministic approach focused on the dynamics of mean fitness produces qualitative conclusions about population survival emerging from evolutionary rescue consistent with the far more analytically complex stochastic realities of extinction and evolution.
Figure 7.
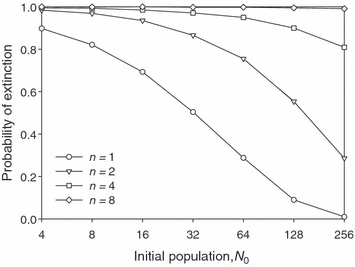
Estimated probability of extinction (fraction of 10 000 replicates extinct by generation 200) versus initial population size assuming different numbers of identical quantitative trait loci for the individual-based simulation model. Parameter values as in Fig. 3.
Figure 8.
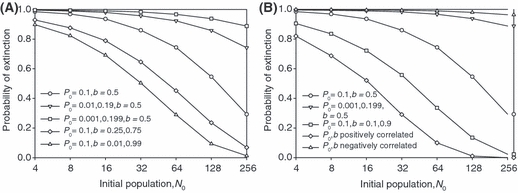
Probability of extinction (fraction of 10 000 replicates extinct by generation 200) versus initial population size for individual-based simulations with two loci. (A) Influences of between locus differences in initial allele frequencies or phenotypic effects. (B) Influences of simultaneous differences in allele frequencies and phenotypic effects. Parameters as in Figs 4 and 5.
Discussion
We have used a series of mathematical models to examine the impact of genetics on a new population invading a harsh environment in which it would go extinct without evolutionary change. We applied the term ‘adaptive colonization’ to an invading population, initially declining in size, which evolves so that it can eventually grow and become self-sustaining. Note that our findings are relevant to any population that finds itself in decline regardless of cause, including a resident population whose existence is suddenly threatened by an exotic invasion (or for that matter any other source of degradation in its environment, such as abrupt climate change or a surge in the concentration of a toxic pollutant).
Our results show that the prospects of adaptive colonization narrow considerably with an increase in the number of genetic loci on which adaptation depends, given a fixed initial variance in fitness. This is because per locus selection must be adjusted downward as more loci are added in the model to keep the fitness variance constant. The effect is to ultimately slow the pace of adaptation and extend the time before growth evolves, increasing the risk of extinction. Interestingly, just the opposite holds in model (1) if, instead of initial variance in fitness, the per-locus strength of selection is fixed regardless of the number of loci. This is because, with more loci, the amount of allele frequency change per locus required to achieve a mean fitness consistent with growth is smaller while the strength of selection at each locus is assumed to be the same. Invading populations in nature will undoubtedly differ in genetic architecture, variability in fitness, and how they experience selection. Our results emphasize that predicting when invasion will be successful will require understanding how those factors affect the strength of selection on, and the amount of genetic change at, individual loci ultimately required for overall positive growth.
Having more loci contribute to fitness can sometimes enhance the mean fitness and growth rate of an invading population. Our findings show, however, that this initial enhancement tends to be far too small to compensate for weakened per locus strength of selection if the effects of fitness are divided among more and more loci. Our results also show, intriguingly, that loci of small effect can make significant contributions to rapid increases in mean fitness. Indeed, we found that adaptive colonization was more likely when major genes interact with variation at minor loci compared with the progress achievable given their separate effects. However, these interactions can also greatly impede adaptive colonization, depending not on how genes interact, but rather the initial genetic structure—the history—of the invading population.
Our conclusions regarding the importance of major and minor genes in a population faced with a considerable adaptive challenge, such as an new invader, differ from those of Lande (1983) and Macnair (1991). Lande showed that rapid evolution toward a new distant phenotypic optimum can be accomplished just as easily with polygenes as with a major mutation and, indeed, is more likely if the major mutation is initially rare or initially common but doomed to be lost. Macnair countered that if a large phenotypic response is required for population persistence, it can only be achieved via spread of rare mutations of large effect.
Lande's analyses focused on how major and minor genes contribute to the attainment of adaptive equilibrium given the new phenotypic optimum (Lande 1983). Our analyses, instead, concentrated on how major and minor genes affect the time needed to attain a mean fitness that allows population growth, which can occur when the population is still far from its evolutionary equilibrium. In fact, we found cases in which a mutation at the major locus is ultimately lost but nevertheless helps reduce the time needed to evolve positive growth, compared with a population whose adaptation relies strictly on background genetic variation (results not shown). Macnair emphasized, as we have here, the importance of evolving far enough to ensure persistence (Macnair 1991, p. 214), but his treatment did not explicitly quantify the phenotypic distance required for growth. By comparison, this distance is explicit in our models.
The contrast between the conclusions of Lande and Macnair versus our finding that major and minor genes can lead to the evolution of growth most rapidly together can thus be traced to our focus on the explicit, transient evolutionary dynamics of the population mean fitness. We believe this focus is particularly relevant for an invading population whose successful establishment depends on it evolving such that it can increase its numbers. More generally, this comparison highlights the difference between adaptation and adaptive colonization, as we have defined it here. The latter accounts for the demographic fate of a population and focuses on transient dynamics, whereas the former term usually refers to long-term evolutionary outcomes, often regardless of their connection to population dynamics.
The models and analyses considered in the paper expand our conceptual understanding of how evolution contributes to biological invasions. They also serve as important building blocks for developing models tailored to particular empirical systems that could be used to predict the prospects and risks of invasion. Moreover, our results have ramifications for applications beyond invasion biology. For example, our results may help to explain the evolution of emerging infectious diseases. Specifically, our findings suggest that the genetic underpinnings of pathogen-host adaptation may explain why some pathogens successfully shift onto novel hosts whereas others do not. Any environmental change that is experienced abruptly by a species, and in which adaptation by natural selection is required for it to persist, can in principle be addressed with models along the lines of those we have explored here. For instance, a sudden increase in harvesting pressure might threaten overexploited species, which could potentially survive by altering body size to escape a particular mesh of net. A pesticide might incidentally and quickly reduce the abundance of a nontarget insect species, which to persist must evolve adaptation to the novel toxin. In all such cases, heterogeneity among species in their observed abilities to persist in the novel environments might reflect hidden variation in the genetic architecture underlying the traits determining fitness in these environments.
This study relied, for the most part, on biologically simplistic, deterministic models, and a relatively simple surrogate for extinction risk—the time a population spends declining in size—allowed for clear comparison of specific genetic effects on invasion success or failure. Similar to an empirical experiment, our theoretical approach provides a highly controlled assessment of precisely manipulated features, which in our case are the number of loci that contribute to fitness and the variability of their effects. But like experimental controls, our simplifying assumptions are simultaneously limiting in that they preclude insight into the potential impacts of numerous demographic and genetic features that were ignored—small population size, demographic stochasticity and heterogeneity, linkage, random genetic drift, epistasis, and dominance to name just a few. Our simulations showed that the qualitative results of our deterministic analyses are robust to violations of several of these assumptions and thus suggest that the deterministic models may provide a reasonable starting point for development of more complex models of adaptive colonization, tailored to the biological details of particular invasive species.
Acknowledgments
We are grateful to Carol Lee for inviting us to contribute to the Special Workshop: Synthesizing Ecology and Evolution for the Study of Invasive Species working group. We thank the Associate Editor and two anonymous reviewers for their helpful comments. This research was supported by NSF grants DEB 0613357 and DMS 0540524 to RG, DMS 0540392 to SLN, and EID 0525751 to RDH, who also thanks the University of Florida Foundation for funding. RG thanks the Section of Evolution and Ecology at UC Davis for providing access to their facilities during preparation of this manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Average trajectories of population growth rate (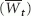 ) for simulations (based on initial population sizes ofN = 4, 16, and 64) compared with analytic results obtained by iterating (17) for one-locus (A) and two-locus (B) hypergeometric model assumptions. Parameter values are as in Fig. 3 of the main text. Simulation results at each generation were averaged over all populations that survived to that time.
) for simulations (based on initial population sizes ofN = 4, 16, and 64) compared with analytic results obtained by iterating (17) for one-locus (A) and two-locus (B) hypergeometric model assumptions. Parameter values are as in Fig. 3 of the main text. Simulation results at each generation were averaged over all populations that survived to that time.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Antia R, Regoes RR, Koella JC, Bergstrom CT. The role of evolution in the emergence of infectious diseases. Nature. 2003;426:658–661. doi: 10.1038/nature02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. The divergence of a polygenic system subject to stabilizing selection, mutation, and drift. Genetical Research of Cambridge. 1992;54:59–77. doi: 10.1017/s0016672300028378. [DOI] [PubMed] [Google Scholar]
- Barton NH, Turelli M. Natural and sexual selection on many loci. Genetics. 1991;127:229–255. doi: 10.1093/genetics/127.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodle MJ, Ferriter AP, Thayer DD. The biology, distribution, and ecological consequences of Maleleuca quinquenervia in the Everglades. In: Davis SM, Ogden JC, editors. Everglades: The Ecosystem and Its Restoration. Boca Raton, Florida: St. Lucie Press; 1994. pp. 341–355. [Google Scholar]
- Boulding EG, Hay T. Genetic and demographic parameters determining population persistence after a discrete change in the environment. Heredity. 2001;86:313–324. doi: 10.1046/j.1365-2540.2001.00829.x. [DOI] [PubMed] [Google Scholar]
- Crow JF, Kimura M. Introduction to Population Genetics Theory. Minneapolis, MN: Burgess Publishing Company; 1970. [Google Scholar]
- Doebeli M. Quantitative genetics and population dynamics. Evolution. 1996;50:532–546. doi: 10.1111/j.1558-5646.1996.tb03866.x. [DOI] [PubMed] [Google Scholar]
- Fisher RA. The Genetical Theory of Natural Selection. Oxford: Clarendon Press; 1930. [Google Scholar]
- Fritts TH, Rodda GH. The role of introduced species in the degradation of island ecosystems: a case history of Guam. Annual Review of Ecology and Systematics. 1998;29:113–140. [Google Scholar]
- Gomulkiewicz R, Holt RD. When does evolution by natural selection prevent extinction? Evolution. 1995;49:201–207. doi: 10.1111/j.1558-5646.1995.tb05971.x. [DOI] [PubMed] [Google Scholar]
- Holt RD, Gomulkiewicz R. The evolution of species’ niches: a population dynamic perspective. In: Othmer HG, Adler FR, Lewis MA, editors. Case Studies in Mathematical Modeling—Ecology, Physiology, and Cell Biology. New York: Prentice-Hall; 1997. pp. 25–50. [Google Scholar]
- Holt RD, Hochberg ME. Virulence on the edge: a source-sink perspective. In: Dieckmann U, Metz JAJ, Sabelis MW, Sigmund K, editors. Adaptive Dynamics of Infectious Diseases: In Pursuit of Virulence Management. Cambridge, UK: Cambridge University Press; 2002. pp. 104–120. [Google Scholar]
- Holt RD, Gomulkiewicz R, Barfield M. The phenomology of niche evolution via quantitive traits in a ‘black-hole’ sink. Proceedings of the Royal Society of London Series B-Biological Sciences. 2003;270:215–224. doi: 10.1098/rspb.2002.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RD, Barfield M, Gomulkiewicz R. Theories of niche conservatism and evolution: could exotic species be potential tests? In: Sax DF, Stachowicz JJ, Gaines SD, editors. Species Invasions: Insights into Ecology, Evolution, and Biogeography. Sunderland, MA: Sinauer Associates Inc; 2005. pp. 259–290. [Google Scholar]
- Houle D. Comparing evolvability and variability of quantitative traits. Genetics. 1992;130:195–204. doi: 10.1093/genetics/130.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R. The response to selection on major and minor mutations affecting a metrical trait. Heredity. 1983;50:47–65. [Google Scholar]
- Lee CE. Evolutionary genetics of invasive species. Trends in Ecology and Evolution. 2002;17:386–391. [Google Scholar]
- Lee CE, Gelembiuk GW. Evolutionary origins of invasive populations. Evolutionary Applications. 2008;1:427–448. doi: 10.1111/j.1752-4571.2008.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood JL, Hoopes MF, Marchetti MP. Invasion Ecology. Malden, MA: Blackwell Publishing; 2007. [Google Scholar]
- Macnair MR. Why the evolution of resistance to anthropogenic toxins normally involves major gene changes: the limits to natural selection. Genetica. 1991;84:213–219. [Google Scholar]
- Nagylaki T. Introduction to theoretical population genetics. In: Levin S, editor. Biomathematics. Vol. 21. Berlin: Springer-Verlag; 1992. pp. 79–91. [Google Scholar]
- Nagylaki T, Crow JF. Continuous selective models. Theoretical Population Biology. 1974;5:257–283. doi: 10.1016/0040-5809(74)90045-8. [DOI] [PubMed] [Google Scholar]
- Orr HA, Unckless RL. Population extinction and the genetics of adaptation. The American Naturalist. 2008;172:160–169. doi: 10.1086/589460. [DOI] [PubMed] [Google Scholar]
- Peterson AT, Vieglais DA. Predicting species invasions using ecological niche modeling. BioScience. 2001;51:363–371. [Google Scholar]
- Shpak M, Kondrashov AS. Applicability of the hypergeometric phenotypic model to haploid and diploid populations. Evolution. 1999;53:600–604. doi: 10.1111/j.1558-5646.1999.tb03794.x. [DOI] [PubMed] [Google Scholar]
- Williamson M, Fitter A. The varying success of invaders. Ecology. 1996;77:1661–1666. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.