

Abstract
Populations that are small and isolated can be threatened through loss of fitness due to inbreeding. Nevertheless, an increased frequency of recessive homozygotes could increase the efficiency of selection against deleterious mutants, thus reducing inbreeding depression. In wild populations, observations of evolutionary changes determined by selection against inbreeding are few. We used microsatellite DNA markers to compare the genetic features of tadpoles immediately after hatch with those of metamorphosing froglets belonging to the same cohort in a small, isolated population of the threatened frog Rana latastei. Within a generation, the inbreeding coefficient (FIS) decreased: at hatch, FIS was significantly >0, whereas FIS was <0 after metamorphosis. Furthermore, heterozygosity increased and allelic frequencies changed over time, resulting in the loss of genotypes at metamorphosis that were present in hatchlings. One microsatellite locus exhibited atypically large FST values, suggesting it might be linked to a locus under selection. These results support the hypothesis that strong selection against the most inbred genotypes occurred among early life-history stages in our population. Selective forces can promote changes that can affect population dynamics and should be considered in conservation planning.
Keywords: genetic purging, heterozygosity, microsatellites, Rana latastei, temporal variation
Introduction
Small, isolated populations are particularly subject to inbreeding, and are thus intrinsically vulnerable to extirpation (Keller and Waller 2002). Both laboratory and field studies have shown that increased inbreeding associated with small and isolated populations can strongly reduce both the variance and mean of traits that are important components of fitness, including survival, fertility and individual growth rate (Saccheri et al. 1998; Keller and Waller 2002; Rowe and Beebee 2003; Frankham 2005; Wright et al. 2008). The reduced survival of inbred individuals can further reduce population size and increase susceptibility to stochastic processes, such that mutual reinforcement among environmental, demographic and genetic stochasticity creates an ‘extinction vortex’ (Gilpin and Soulé 1986). However, if enough genetic variability is retained by the population, less inbred individuals, or individuals not carrying deleterious alleles can be favoured through selection. If inbreeding depression is mainly caused by the effect of strongly deleterious, recessive alleles, natural selection may be particularly effective against them when they occur in the homozygous state (Wang 2000). Two processes can thus occur in small populations. First, the loss of inbred individuals can cause a further reduction of population size, leading to cycles of exacerbated inbreeding and drift in subsequent generations (referred to as ‘mutational meltdown’; Lynch et al. 1995). On the other hand, selection against inbred genotypes can purge inbreeding depression (Hedrick 2001). In this case, populations may be able to recover demographically despite initial demographic losses by removing inbred individuals and reducing the frequency of deleterious alleles. Selection can be most effective at countering inbreeding depression under particular conditions, such as when the fitness disadvantages of deleterious mutations are particularly strong, inbreeding does not occur randomly, and migrants are not (re)introducing deleterious alleles (Glemin 2003; Edmands 2007).
Detecting the ongoing population genetic processes that lead to mutational meltdown or recovery can provide key insights for the management of threatened species at greater risk of inbreeding. However, detecting either of these patterns in wild animal populations can be challenging. Under natural settings, measuring the relative performance of individuals with respect to their inbreeding status usually requires long-term capture/mark/recapture analyses over multiple generations and the reconstruction of pedigrees. For this and other reasons, the measurement of inbreeding depression in the wild has been done primarily in larger vertebrates where parentage is easier to determine and the number of offspring per female is small (Keller and Waller 2002; Kruuk and Clutton-Brock 2008). For taxa where such parameters are not so amenable to measurement, two other approaches are used. Inbreeding depression can be measured under laboratory conditions, however inbreeding depression is frequently stronger under natural (stressful) conditions, and results obtained by laboratory studies may not reflect the complexity of natural conditions (Bijlsma et al. 1999; Joron and Brakefield 2003; Halverson et al. 2006). Alternatively, averaged population responses (e.g., FIS) can be used, by comparing populations with different inbreeding histories rather than measuring fitness of individuals (Saccheri et al. 1998).
Molecular genetics can be a powerful approach for the study of microevolutionary changes over a range of temporal scales, from a few generations to thousands years. In anuran amphibians, typically 80–95% of mortality occurs between hatch and metamorphosis (Vonesh and De la Cruz 2002) and inbreeding depression may be most readily detected through the measurement of relevant population genetics parameters during these periods of heavy mortality (Rowe and Beebee 2003; Halverson et al. 2006; Chapman et al. 2009). In this study, we used microsatellite DNA polymorphisms to study temporal variation of genetic features in small, isolated population of a threatened frog suffering strong inbreeding depression (Ficetola et al. 2007). We analyzed the genetic features of the same cohort at two subsequent life-history stages, immediately after hatch and at metamorphosis; our sampling protocol was therefore designed to compare the genetic features of a cohort before and after the effects of potentially strong natural selection. We hypothesized that strong selection against the most inbred genotypes might cause strong shifts in genetic features of the population. Evolutionary signatures of selection against inbreeding include an increase in heterozygosity, a decrease in the inbreeding coefficient FIS, and loci exhibiting outlying values of FST (Beaumont 2005). Moreover, the reduced frequency or even elimination of specific alleles all may occur if selection is removing deleterious alleles expressed in the homozygous condition.
Methods
Study species and area
The Italian agile frog, Rana latastei Boulenger, is endemic to Northern Italy and adjacent countries and is classified as vulnerable to extinction by the IUCN (Andreone et al. 2008). Habitat requirements are river washes, forest brooks and ponds associated with lowland forest cover, primarily along the Po River drainage in Italy. In this area, habitat has been subject to intense urbanization and agriculture. In particular, the western part of the distribution of R. latastei is broken into a limited number of small and isolated forest fragments. Many breeding sites are characterized by small census size and geographic isolation (Ficetola et al. 2007) and this combination of isolation and small population size has increased the risk of extinction of populations (Ficetola and De Bernardi 2004). Further, the part of the species range from which our study population comes from is characterized by a 3× reduction in genetic variability when compared to the eastern part of the range (Garner et al. 2004) and experiments have shown that reductions in genetic variability correlate well with reduced fitness (Pearman and Garner 2005; Ficetola et al. 2007). Breeding occurs in late winter; females lay a single egg mass per year (Barbieri and Mazzotti 2006), therefore the number of clutches represents both annual population reproductive output and the number of breeding females in a population. Multiple paternity has never been observed in R. latastei. Some studies have described multiple paternity in related species, but the offspring of polyandrous mating is a small proportion. For instance, in Rana temporaria, clutch piracy affected 84% of clutches, but pirate males fertilized only 26% of pirated clutches, and in these clutches 24% of embryos were fertilized by pirates, i.e., 5% of all eggs were sired by a secondary male (Vieites et al. 2004). Similarly, in Rana dalmatina only 4% of all eggs are sired by a secondary male (Lodé et al. 2005). Our results are robust even with the removal of 5% of larvae from the analyses, therefore the occurrence of multiple paternity at rates similar to the ones observed in related frogs would have minimal influence on the outcome of this study (see also Ficetola and De Bernardi 2009; Ficetola et al. 2010 for further details).
We studied one population located close to the Alserio Lake, in Northern Italy (population AL in the map in Ficetola et al. 2007). At this location, we detected a total of 43 clutches during the 2003 breeding season, most of which were found in a single pond (hereafter: study pond; 32 egg masses in 2003). The study pond is small (surface area 60 m2), shallow (maximum water depth: 50 cm) and with clear water; clutches are deposited onto submerged tree roots and sticks below the water surface, thus, they are easily detected. Clutch counts therefore provide a reliable standard census method for agile frogs (Salvidio 2009). A few clutches were found in a small satellite wetland within 50 m of the study pond; clutches from the satellite pond were not considered here. The study population is isolated: the next available breeding location was located 3 km away, and the intervening distance was composed of unsuitable habitat, lacking both suitable breeding locations and contiguous forested habitat; a motorway acts as a further barrier.
Sampling protocol and analyses
We subsampled all 32 viable clutches deposited in the study pond in March 2003, taking a single tadpole per egg mass for DNA extraction. This sample therefore represented the entire hatching cohort. In late June–early July, we returned to the study pond and we sampled the pond banks to collect metamorphic froglets (stage 39–42; Gosner 1960). We captured active individuals; we also used a net to collect leaf litter and cryptic individuals; overall, we obtained 29 froglets. We collected a small tissue sample (remnant tail tip) from each metamorph for DNA extraction. We restricted our sampling to these semiaquatic stages to prevent sampling any possible metamorphs that may have come from the adjacent woodland.
Overall, 78 microsatellite loci developed either for R. latastei or for other Rana species have been tested on R. latastei samples. Thirty-one of these loci were monomorphic, and 41 did not amplify or showed presence of null alleles, therefore only six polymorphic loci are actually available for this species, a proportion significantly lower than in other frog species (Garner and Tomio 2001; Primmer and Merilä 2002; T.W.J.G. unpublished data); this is probably caused by the colonization history of this small range species combined with the extreme landscape fragmentation (Garner et al. 2004; Ficetola et al. 2007). DNA was extracted using a Chelex-based protocol and amplified at variable microsatellites following Garner and Tomio (2001). [33P]-labeled fragments were electrophoresed through 6% polyacrylamide gels and fragments were detected using autoradiography. Alleles were visually scored against a pGem®-3zf+ (Promega, Madison, Wisconsin, USA) sequence reference marker. Further details on genetic analyses are provided in Ficetola et al. (2007).
We used Genepop 3.4 (Raymond and Rousset 1995) to test for linkage disequilibrium among the microsatellite loci that proved to be variable, and to test for Hardy–Weinberg equilibrium in larvae and metamorphs. To compare the genetic variability of hatchlings and of metamorphs, we estimated the percentage of polymorphic loci, allelic richness, expected and observed heterozygosity for both developmental stages. We also tested for genetic differences among developmental stages by calculating respective FST values, and permuting these values 10 000 times to test for significance.
We calculated FIS for hatchlings and for metamorphs as a measure of the level of inbreeding in a cohort (Wright 1969); we estimated 95% confidence intervals for each coefficient by bootstrapping 10 000 times, and tested for significant differences in the inbreeding coefficient among developmental stages. We estimated FIS separately for hatchlings and metamorphs, therefore differences in FIS might be in principle due to differences in the reference population considered for FIS estimation. To address this, we used simulations to evaluate whether random mortality or sampling, and changes in the reference population, may have caused the observed changes in FIS. We simulated 1000 individuals with the same FIS of hatchlings and the same allele frequencies detected in the hatchlings. Subsequently, we randomly selected 29 individuals from the simulated population to form the metamorphic sample, and we estimated FIS for these individuals. This process was repeated 1 000 000 times.
We used Fdist2 (Beaumont and Nichols 1996; Beaumont 2000) to identify loci showing the potential signature of selection. This approach is based on the principle that loci under selection or tightly linked to a locus under selection should exhibit outlying FST values (outliers) (Beaumont 2005). We generated a null distribution based on 30 000 simulated loci; the mean FST was calculated on the basis of empirical distribution and then iteratively modified as outliers were identified and removed (Beaumont and Nichols 1996). To evaluate the robustness of our results to different approaches in the estimation of expected FST, this analysis was repeated by calculating FST without the iterative removal of outliers, which yields more conservative results. This second analysis produced very similar results (not shown). Results from Fdist2 may be misleading when analyzing multiple populations grouped in phylogenetic hierarchies (Latta 2008). For analysis with Fdist2, we treated the larval cohort as the first ‘population’, and the metamorphic cohort as the second ‘population’. Therefore, phylogenetic hierarchies are absent from our data.
Finally, we used a Correspondence Analysis (CA) to detect differences in genotype distributions between tadpoles and metamorphs by graphically projecting all the individuals on the space defined by factors extracted on the basis of the similarity of their allelic states. This last analysis was completed using Genetix 4.05 (Belkir 2004).
Results
Three loci (RlatCa17, RlatCa21 and Rt2Ca9) were monomorphic across the entire sample: therefore all subsequent analyses were performed using the remaining three polymorphic loci (RlatCa9, RlatCa27 and RlatCa41). We did not detect evidence of linkage disequilibrium among any of these loci (all P > 0.08). In the earlier (larval) life-history stage, analysis of Hardy–Weinberg equilibrium showed a nonsignificant trend for heterozygote deficiency (P = 0.07). When loci were examined individually, only locus RlatCa27 exhibited a significant heterozygote deficit in larvae (P = 0.004). Conversely, in the later, postmetamorphic life-history stage we observed a significant global heterozygote excess (P = 0.013), despite the lack of any effect when any one locus was analyzed independently (all P > 0.1). Percentage of polymorphic loci, allelic richness and expected heterozygosity were similar in both stages. Observed heterozygosity of the cohort increased from 0.404 at hatch to 0.547 in metamorphs; nevertheless, standard errors were large (Table 1). At hatch, the inbreeding coefficient was significantly >0 (FIS = 0.183, 95% CI: 0.027 to 0.305). In contrast, the inbreeding coefficient of metamorphic individuals was negative (FIS = −0.169, 95% CI: −0.027 to −0.337, Fig. 1) and significantly different from the hatch value (P < 0.001). This decrease in the inbreeding coefficient over life-history stages was strongly influenced by one locus (RlatCa27) but was evident at all three of the variable loci (Fig. 1). Exclusion of locus RlatCa27 from the analysis resulted in a marginally nonsignificant trend (P = 0.057). Simulations showed that the observed changes in FIS could not be explained by random mortality/sampling, as the observed FIS of metamorphs was lower than the first percentile of the distribution of simulated data (Fig. 2).
Table 1.
Genetic features of tadpoles immediately after hatch and of metamorphosing froglets in a population of Rana latastei. Data derived from polymorphisms at microsatellite loci. Standard errors are in parentheses
| Hatchlings | Metamorphs | |
|---|---|---|
| N polymorphic loci | 3 | 3 |
| Number of alleles | 10 | 10 |
| Allelic richness | 3.31 | 3.33 |
| Expected heterozygosity | ||
| All loci | 0.484 (0.108) | 0.461 (0.099) |
| RlatCa9 | 0.405 | 0.345 |
| RlatCa27 | 0.608 | 0.536 |
| RlatCa41 | 0.441 | 0.498 |
| Observed heterozygosity | ||
| All loci | 0.404 (0.075) | 0.547 (0.147) |
| RlatCa9 | 0.423 | 0.379 |
| RlatCa27 | 0.321 | 0.607 |
| RlatCa41 | 0.469 | 0.655 |
Figure 1.
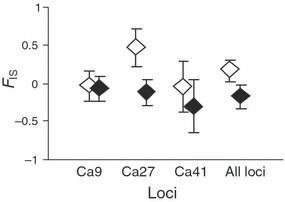
Inbreeding coefficient (FIS) of hatchling (empty diamonds) and metamorphic Rana latastei (solid diamonds) derived from variation at three polymorphic microsatellite loci (RlatCa9, RlatCa27 and RlatCa41). Error bars are 95% CI.
Figure 2.
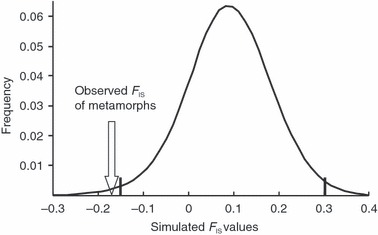
Distribution of simulated FIS values. The two bold vertical lines indicate the 1% percentiles; the arrow indicates the FIS value observed in metamorphs. See text for more details on the simulations.
Genotype frequencies differed significantly among life-history stages (FST = 0.101, P < 0.001), but pairwise FST was not significant if locus RlatCa27 was removed from the analysis (FST = 0.010, P = 0.169). Locus RlatCa27 exhibited an atypically large FST value among life-history stages (P = 0.001, Fig. 3). This result is particularly striking because Fdist2 is expected to have relatively low power for detecting outliers when the number of loci included in the analysis is not large (Beaumont and Nichols 1996; Nielsen et al. 2006).
Figure 3.
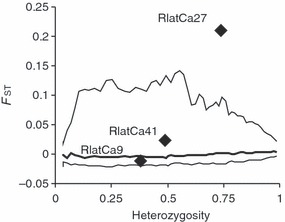
Individuation of loci under selection using Fdist2: plot of FST values against heterozygosity estimates (Beaumont and Nichols 1996) for the comparison tadpoles/metamorphs. Each diamond represents a microsatellite marker; the intermediate line is the expected median; upper and lower lines are the limits of the 99% envelope.
Both the significant decrease in the inbreeding coefficient and change in genotype distribution detected among life-history stages was primarily driven by the disappearance of certain homozygotes in the metamorphic sample that were present in the hatch data set. Specifically, 38% of hatchlings were homozygous for the same allele (154 bp) at locus RlatCa27, while none of the metamorphosing froglets were homozygous for this allele at this locus (Fig. 4). The frequency of this allele dropped from 0.518 in larvae to 0.054 in the metamorphs. One further homozygote was eliminated from the genotype pool as development progressed (166 bp) and no new homozygotes occurred in the metamorphic data pool that were not previously detected in the earlier life-history stage (Fig. 4).
Figure 4.
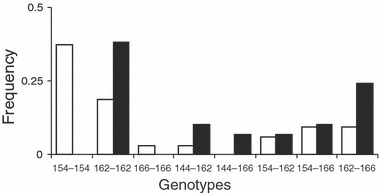
Observed frequencies of genotypes at locus RlatCa27 for the two life-history stages. Alleles are enumerated by length in base pairs. Open bars: hatchlings; solid bars: metamorphs.
The first component extracted from the CA explained 24.3% of the total variation of genotypes and showed clearly how the genotypes of metamorphs constituted a small subset of the hatchling genotypes (Fig. 5). Hatchling scores along the first CA component ranged from +2 to −1, whereas all metamorph scores were <0.5. Genotype variability along the first component significantly decreased from hatch to metamorphosis (Levene's test for homogeneity of variances: F1,59 = 29.0, P < 0.001, Fig. 5).
Figure 5.
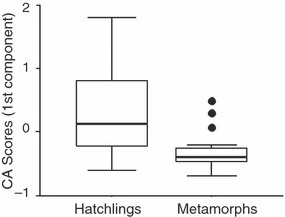
Box-plots representing the distribution of genotypes of hatchlings and metamorphs along the first component extracted by Correspondence Analysis.
Discussion
Our study showed that within a cohort of R. latastei FIS decreases over time while observed heterozygosity increased slightly. One locus was predominantly responsible for atypically large differences among life-history stages (FST) that are considered to be evidence of selection at a locus (Beaumont and Nichols 1996). Further, only a subset of the genotypes present among clutches persisted through to metamorphosis and the frequency of a common allele, present only in hatchlings in the homozygous state, was strongly reduced in the metamorphic cohort. We present arguments below for why we believe these observed patterns are indicative of strong selection against inbreeding.
Microsatellites are usually considered to be neutral loci. Inferring selective processes from changes in neutral markers requires the elimination of the alternative explanations, such as immigration, drift, nonrandom mating and mutation. In our study, nonrandom mating and mutation can be ignored, as we sampled a single, nonreproductive cohort. Immigration had no effect on our sample because of pond isolation and the fact we sampled before metamorphs from nearby wetlands could have emigrated. Stochasticity may have a role in some of the changes observed; for instance, the frequency of some but not all the homozygotes decreased in metamorphs (Fig. 4). However, stochasticity cannot be the sole cause for the observed changes, as simulations showed that it is highly unlikely that the strong decrease of FIS we have observed could be attributed to stochastic processes (Fig. 2). Thus, we conclude that natural selection was the most likely driver of the temporal variation in genetic frequencies we observed; the strong decrease of FIS suggests that selection against inbreeding is the most likely explanation of the observed genetic changes. Nevertheless, it should be remarked only locus RlatCa27 was not in Hardy–Weinberg equilibrium in larvae; other possibilities therefore exist for the pattern at that locus. For example, allele 154 might be associated with mate choice in adults, causing an excess of homozygotes in eggs, then selected against in larvae. We are not the first to observe a relationship between measures of genetic diversity at microsatellite loci and measures of fitness (Chapman et al. 2009). Furthermore, previous research has identified specific patterns in the correlation between marker variability and fitness that are matched by our data set. For example, in red deer, reed warblers, harbor seals and sea lions, heterozygosity at multilocus microsatellite genotypes was strongly correlated with components of fitness, but in all these studies single microsatellite loci were shown to have a significantly greater contribution to interindividual variability and locus-specific correlations with fitness components (Pemberton et al. 1999; Hansson et al. 2004; Acevedo-Whitehouse et al. 2006).
Balloux et al. (2004) showed that, in the absence of strong linkage disequilibrium and complex mating systems, it should be necessary to sample large numbers of microsatellite loci to detect a true fitness/heterozygosity correlation. This has not prevented the relationship between diversity estimated at microsatellites and fitness from being detected frequently in studies using a handful of markers. In our study species, six loci were sufficient for detecting significant variation in genetic diversity across the species range (Garner et al. 2004) and this variation in diversity correlated strongly with fitness measures (Pearman and Garner 2005; Ficetola et al. 2007). The fact that we detected an effect with only half that many loci reflects the pattern of genetic variability inherent to the western part of the species range. Fixation for three of six published loci used in this study was previously detected in the Lombardy Region because of the joint effect of postglacial colonization and human-driven habitat fragmentation (Garner et al. 2004; Ficetola et al. 2007); during the course of PCR primer development and cross-amplification with primers developed for other Rana species, another 31 primer sets were characterized as monomorphic in R. latastei (Garner and Tomio 2001; Primmer and Merilä 2002). Thus, based on a random sample of dinucleotide repeat regions that amplify reliably, the true number of polymorphic loci may be as low as 3 of 37.
FIS of larvae was significantly higher than zero (Fig. 1), which at first glances is puzzling, as FIS is a measure of inbreeding as nonrandom mating (Keller and Waller 2002), and we considered a single population breeding in one small pond, where it is difficult to invoke substructuring (Wahlund effect). Nevertheless, large FIS values are often observed in small, isolated populations of anuran amphibians (e.g., Hitchings and Beebee 1997; Andersen et al. 2004). Increased levels of polygyny are commonly reported for anurans, and have also been described for the study population (Ficetola et al. 2010); polygyny can result in inbreeding coefficients greater than expected under random mating; this effect can be exacerbated in small populations (Balloux et al. 2004). No matter the process originating positive FIS in larvae, the change in FIS was more striking than its absolute values: FIS strongly decreased during larval development, indicating than the less inbred genotypes were favoured by selection.
Linkage between microsatellites and loci under selection can be particularly strong in small, inbred populations carrying a heavy genetic load and after bottlenecks, as in these populations larger portions of the genome are expected to be under balancing selection, and probability is greatest for loci linked to overdominant loci to remain polymorphic after bottleneck events (Ohta 1982; Balloux et al. 2004; Van Oosterhout et al. 2004; Zartman et al. 2006). The few microsatellites retaining heterozygosity can therefore constitute a nonrandom sample of the genome (Santucci et al. 2007). The study population corresponds well to this situation: it is small, isolated and passed through a demographic bottleneck some years before the season in which we sampled for this study (Ficetola and De Bernardi 2005). From this, the relative importance of three loci in our species for describing genetic variability is likely greater than in other, more genetically variable species. A limited number of variable microsatellites is typical of endangered species suffering loss of genetic diversity (Gebremedhin et al. 2009a). These species are often the ones for which studies are more urgent; lack of variable microsatellites should not hamper performing analyses that can provide important management information (Richards-Zawacki 2009).
There is debate if the relationship between inbreeding and fitness is due to the effects of many loci of small effect or the effects of a few loci, or even one, of marked effect (Chapman et al. 2009). The limited panel of available markers available for this study does not allow us to distinguish between a multilocus or single locus model. Even so, although all our polymorphic loci contribute to the observed changes in FIS and heterozygosity, the strong effect of locus RlatCa27 suggests that certain allele(s) at this locus may be linked to lethal or semi-lethal allele(s) at a locus involved in early development. The observation that not all loci decreased their frequency in the homozygote state (e.g., 162 bp: Fig. 4) supports the hypothesis that not all of the microsatellite allele(s) is/are associated with deleterious mutations. Such linkage patterns between microsatellites and genes exposed to strong selection have been inferred and even detected several times (Hansson et al. 2004; Acevedo-Whitehouse et al. 2006; Nielsen et al. 2006; Mäkinen et al. 2008; Amos and Acevedo-Whitehouse 2009), and might be more detectable in small, isolated populations (Balloux et al. 2004; Van Oosterhout et al. 2004).
An increased frequency of recessive homozygotes through inbreeding is expected to increase the efficiency of natural selection against deleterious mutants and reduce inbreeding depression through purging (Crow 1970; Boakes and Wang 2005). If inbreeding depression is mainly caused by deleterious mutations having a strong effect, selection may rapidly eliminate deleterious alleles, and population fitness rebounds (Wang et al. 1999; Wang 2000) while the inbreeding levels of populations are reduced (Moore 2007). However, whether purging actually occurs to an extent that enables the recovery of populations has been strongly debated, primarily due to the equivocal nature of both field and experimental evidence (Leberg and Firmin 2008; Chapman et al. 2009). The disappearance of one common allele occurring primarily in the homozygous state among life-history stages (154 bp at locus RlatCa27) fits the signature of strong selection against inbreeding. Theoretical models suggest that this type of selection can be particularly effective in species with large reproductive ability (Wang 2000). In many anuran species, including R. latastei, fecundity is on the order of thousands of eggs. Moreover, larval mortality is high and density dependent, creating ideal conditions for selection to take effect early during development. If a sufficient number of females reproduce, the death of a large proportion of tadpoles may not threaten population persistence in the short term (Vonesh and De la Cruz 2002). This suggests that purging could be more effective in anuran populations than in taxa with limited individual reproductive effort, such as larger vertebrates (Jamieson et al. 2006; Chapman et al. 2009).
Under these conditions, selection may affect population dynamics by reducing inbreeding depression and the risk of an extinction vortex. Relationships between evolutionary and population dynamics can have important consequences for management and in population viability analyses, and when alternative options (e.g., translocations versus in situ management) are compared (Ficetola and De Bernardi 2005; Ficetola et al. 2007). For instance, actions allowing for a rapid demographic rebound (Wang et al. 1999), such as habitat reclamation, can be particularly effective for the recovery of populations. Our knowledge of the dynamics of inbreeding depression in the wild is far from complete, however, and more studies are required to improve decision making that account for the genetic responses that may or may not be associated with population recovery. Long-term studies of amphibian populations are increasingly available, and will be necessary to evaluate the relative importance of selection and inbreeding in determining population dynamics, and whether selection is strong enough to rescue isolated populations from inbreeding depression (Bensch et al. 2006). Our analysis considered only one population. Populations often experience different histories, conditions and selective forces; analyses including a larger number of populations are better for describing general patterns (Halverson et al. 2006).
To date, many studies on temporal variation of genetic features in wild, nonmodel species have been performed over long temporal scales. Our analysis showed a striking change in genetic features during a very short time, which might affect population dynamics. The fate of the population will be therefore determined not only by the interaction between environment, demographic processes and inbreeding depression. Selective forces can act over the functional variability among individuals, promoting changes influencing the future of endangered species, and should be considered in the analysis of population dynamics (Bensch et al. 2006; Saccheri and Hanski 2006). The short time scale over which these forces can operate may increase the complexity of interactions between environment, demography and genetics; molecular genetics can help to elucidate these processes in the wild, also at short temporal scales. The panel of tools available for conservation genetics is quickly broadening. The possibility to detect the signature of selection in natural populations using both neutral and non-neutral markers may increase our understanding of evolutionary processes in threatened populations (Gebremedhin et al. 2009b), providing a widening range of management options for conservation.
Acknowledgments
We thank R. Martinelli, L. Trotta and R. Pennati for helping during laboratory work, and P. Taberlet and three anonymous referees for comments on previous draft of the manuscript. G.F.F. was partially funded by a scholarship of the University of Milano-Bicocca.
Literature cited
- Acevedo-Whitehouse K, Spraker TR, Lyons E, Melin SR, Gulland F, Delong RL, Amos W. Contrasting effects of heterozigosity on survival and hookworm resistence in California sea lion pups. Molecular Ecology. 2006;15:1973–1982. doi: 10.1111/j.1365-294X.2006.02903.x. [DOI] [PubMed] [Google Scholar]
- Amos W, Acevedo-Whitehouse K. A new test for genotype-fitness associations reveals a single microsatellite allele that strongly predicts the nature of tuberculosis infections in wild boar. Molecular Ecology Resources. 2009;9:1102–1111. doi: 10.1111/j.1755-0998.2009.02560.x. [DOI] [PubMed] [Google Scholar]
- Andersen LW, Fog K, Damgaard C. Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea. Proceedings of the Royal Society B: Biological Sciences. 2004;271:1293–1302. doi: 10.1098/rspb.2004.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone F, Garner TWJ, Schmidt BR, Corti C, Vogrin M. Rana latastei. In: Stuart SN, Hoffman M, Chanson JS, Cox NA, Berridge RJ, Ramani P, Young BE, editors. Threatened Amphibians of the World. Barcelona: Lynx Editions, IUCN and Conservation International; 2008. p. 506. [Google Scholar]
- Balloux F, Amos W, Coulson T. Does heterozygosity estimate inbreeding in real populations? Molecular Ecology. 2004;13:3021–3031. doi: 10.1111/j.1365-294X.2004.02318.x. [DOI] [PubMed] [Google Scholar]
- Barbieri F, Mazzotti S. Rana latastei Boulenger, 1879. In: Sindaco R, Doria G, Razzetti E, Bernini F, editors. Atlas of Italian Amphibians and Reptiles. Firenze: Polistampa; 2006. pp. 362–367. [Google Scholar]
- Beaumont MA. 2000. Fdist2 – a program to detect loci that might be under selection in samples from structured populations http://www.rubic.rdg.ac.uk/~mab/software.html. (Accessed April 2010)
- Beaumont MA. Adaptation and speciation: what can FST tell us? Trends in Ecology & Evolution. 2005;20:435–440. doi: 10.1016/j.tree.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society B: Biological Sciences. 1996;263:1619–1626. [Google Scholar]
- Belkir K. GENETIX, Logiciel Sous WindowsTM Pour la Génétique des Populations. Monpellier: Université de Montpellier II; 2004. [Google Scholar]
- Bensch S, Andrén H, Hansson B, Pedersen HC, Sand H, Sejberg D, Wabakken P, et al. Selection for heterozygosity gives hope to a wild population of inbred wolves. PLoS ONE. 2006;1:e72. doi: 10.1371/journal.pone.0000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlsma R, Bundgaard J, Van Putten WF. Environmental dependence of inbreeding depression and purging in Drosophila melanogaster. Journal of Evolutionary Biology. 1999;12:1125–1137. [Google Scholar]
- Boakes E, Wang J. A simulation study on detecting purging of inbreeding depression in captive populations. Genetical Research. 2005;86:139–148. doi: 10.1017/S001667230500772X. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Nakagawa S, Coltman DW, Slate J, Sheldon BC. A quantitative review of heterozygosity-fitness correlations in animal populations. Molecular Ecology. 2009;18:2746–2765. doi: 10.1111/j.1365-294X.2009.04247.x. [DOI] [PubMed] [Google Scholar]
- Crow JF. Genetic loads and the cost of natural selection. In: Kojima KJ, editor. Mathematical Topics in Population Genetics. Berlin: Springer; 1970. pp. 128–177. [Google Scholar]
- Edmands S. Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Molecular Ecology. 2007;16:463–475. doi: 10.1111/j.1365-294X.2006.03148.x. [DOI] [PubMed] [Google Scholar]
- Ficetola GF, De Bernardi F. Amphibians in an human-dominated landscape: the community structure is related to habitat features and isolation. Biological Conservation. 2004;119:219–230. [Google Scholar]
- Ficetola GF, De Bernardi F. Supplementation or in situ conservation? Evidence of local adaptation in the Italian agile frog Rana latastei and consequences for the management of populations. Animal Conservation. 2005;8:33–40. [Google Scholar]
- Ficetola GF, De Bernardi F. Offspring size and survival in the frog Rana latastei: from among-population to within-clutch variation. Biological Journal of the Linnean Society. 2009;97:845–853. [Google Scholar]
- Ficetola GF, Garner TWJ, De Bernardi F. Genetic diversity, but not hatching success, is jointly affected by post glacial colonization and isolation in the threatened frog, Rana latastei. Molecular Ecology. 2007;16:1787–1797. doi: 10.1111/j.1365-294X.2006.03198.x. [DOI] [PubMed] [Google Scholar]
- Ficetola GF, Padoa-Schioppa E, Wang J, Garner TWJ. Polygyny, census and effective population size in the threatened frog, Rana latastei. Animal Conservation. 2010 doi: 10.1111/j.1469-1795.2009.00306.x. [Google Scholar]
- Frankham R. Stress and adaptation in conservation genetics. Journal of Evolutionary Biology. 2005;18:750–755. doi: 10.1111/j.1420-9101.2005.00885.x. [DOI] [PubMed] [Google Scholar]
- Garner TWJ, Tomio G. Microsatellites for use in studies of the Italian Agile Frog, Rana latastei (Boulenger) Conservation Genetics. 2001;2:77–80. [Google Scholar]
- Garner TWJ, Angelone S, Pearman PB. Genetic diversity across a vertebrate species’ range: a test of the central-peripheral hypothesis. Molecular Ecology. 2004;13:1047–1053. doi: 10.1111/j.1365-294X.2004.02119.x. [DOI] [PubMed] [Google Scholar]
- Gebremedhin B, Ficetola GF, Naderi S, Rezaei H-R, Maudet C, Rioux D, Luikart G, et al. Combining genetic and ecological data to assess the conservation status of the endangered Ethiopian Walia ibex. Animal Conservation. 2009a;12:89–100. [Google Scholar]
- Gebremedhin B, Ficetola GF, Naderi S, Rezaei H-R, Maudet C, Rioux D, Luikart G, et al. Frontiers in identifying conservation units: from neutral markers to adaptive genetic variation. Animal Conservation. 2009b;12:107–109. [Google Scholar]
- Gilpin ME, Soulé ME. Minimum viable populations: processes of extinction. In: Soulé ME, editor. Conservation Biology: The Science of Scarcity and Diversity. Sunderland, MA: Sinauer Associates; 1986. pp. 19–34. [Google Scholar]
- Glemin S. How are deleterious mutations purged? Drift versus nonrandom mating. Evolution. 2003;27:2678–2687. doi: 10.1111/j.0014-3820.2003.tb01512.x. [DOI] [PubMed] [Google Scholar]
- Gosner KL. A simplified table for staging Anuran embryos and larvae with notes on identification. Herpetologica. 1960;16:183–190. [Google Scholar]
- Halverson MA, Skelly DK, Caccone A. Inbreeding linked to amphibian survival in the wild but not in the laboratory. Journal of Heredity. 2006;97:499–507. doi: 10.1093/jhered/esl019. [DOI] [PubMed] [Google Scholar]
- Hansson B, Westerdahl E, Hasselquist D, Akesson M, Bensch S. Does linkage disequilibrium generate heterozigosity-fitness correlations in great reed warblers? Evolution. 2004;58:870–879. doi: 10.1111/j.0014-3820.2004.tb00418.x. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. Conservation genetics: where are we now? Trends in Ecology & Evolution. 2001;16:629–636. [Google Scholar]
- Hitchings SP, Beebee TJC. Genetic substructuring as a result of barriers to gene flow in urban Rana temporaria (common frog) populations: implications for biodiversity conservation. Heredity. 1997;79:117–127. doi: 10.1038/hdy.1997.134. [DOI] [PubMed] [Google Scholar]
- Jamieson IG, Wallis GP, Briskie JV. Inbreeding and endangered species management: is New Zealand out of step with the rest of the world? Conservation Biology. 2006;20:38–47. doi: 10.1111/j.1523-1739.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- Joron M, Brakefield PM. Captivity masks inbreeding effects on male mating success in butterflies. Nature. 2003;424:191–194. doi: 10.1038/nature01713. [DOI] [PubMed] [Google Scholar]
- Keller LF, Waller DM. Inbreeding effects in wild populations. Trends in Ecology & Evolution. 2002;17:230–241. [Google Scholar]
- Kruuk LEB, Clutton-Brock TH. Introduction. Evolutionary dynamics of wild populations: the use of long-term pedigree data. Proceedings of the Royal Society B: Biological Sciences. 2008;275:593–596. doi: 10.1098/rspb.2007.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latta RG. Conservation genetics as applied evolution: from genetic pattern to evolutionary process. Evolutionary Applications. 2008;1:84–94. doi: 10.1111/j.1752-4571.2007.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberg PL, Firmin BD. Role of inbreeding depression and purging in captive breeding and restoration programmes. Molecular Ecology. 2008;17:334–343. doi: 10.1111/j.1365-294X.2007.03433.x. [DOI] [PubMed] [Google Scholar]
- Lodé T, Holveck M-J, Lesbarrères D. Asynchronous arrival pattern, operational sex ratio and occurrence of multiple paternities in a territorial breeding anuran, Rana dalmatina. Biological Journal of the Linnean Society. 2005;86:191–200. [Google Scholar]
- Lynch M, Conery J, Burger R. Mutation accumulation and the extinction of small populations. The American Naturalist. 1995;146:489–518. [Google Scholar]
- Mäkinen HS, Shikano T, Cano JM, Merilä J. Hitchhiking mapping reveals a candidate genomic region for natural selection in three-spined stickleback chromosome VIII. Genetics. 2008;178:453–465. doi: 10.1534/genetics.107.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J. Phenotype matching and inbreeding avoidance in African elephants. Molecular Ecology. 2007;16:4421–4423. doi: 10.1111/j.1365-294X.2007.03560.x. [DOI] [PubMed] [Google Scholar]
- Nielsen EE, Hansen MM, Meldrup D. Evidence of microsatellite hitch-hiking selection in Atlantic cod (Gadus morhua L.): implications for inferring population structure in nonmodel organisms. Molecular Ecology. 2006;15:3219–3229. doi: 10.1111/j.1365-294X.2006.03025.x. [DOI] [PubMed] [Google Scholar]
- Ohta T. Linkage disequilibrium due to random genetic drift in finite subdivided populations. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:1940–1944. doi: 10.1073/pnas.79.6.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearman PB, Garner TWJ. Susceptibility of Italian Agile Frog populations to an emerging Ranavirus parallels population genetic diversity. Ecology Letters. 2005;8:401–408. [Google Scholar]
- Pemberton JM, Coltman DW, Coulson TN, Slate J. Using microsatellites to measure the fitness consequences of inbreeding and outbreeding. In: Goldstein DB, Schlötterer C, editors. Microsatellites: Evolution and Applications. Oxford: Oxford University Press; 1999. pp. 151–164. [Google Scholar]
- Primmer CR, Merilä J. A low rate of cross-species microsatellite amplification success in Ranid frogs. Conservation Genetics. 2002;3:445–449. [Google Scholar]
- Raymond M, Rousset F. genepop version 1.2: population genetics software for exact tests and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Richards-Zawacki CL. Effects of slope and riparian habitat connectivity on gene flow in an endangered Panamanian frog, Atelopus varius. Diversity and Distributions. 2009;15:796–806. [Google Scholar]
- Rowe G, Beebee TJC. Population on the verge of a mutational meldtown? Fitness cost of genetic load for an amphibian in the wild. Evolution. 2003;57:177–181. doi: 10.1111/j.0014-3820.2003.tb00228.x. [DOI] [PubMed] [Google Scholar]
- Saccheri I, Hanski I. Natural selection and population dynamics. Trends in Ecology & Evolution. 2006;21:341–347. doi: 10.1016/j.tree.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, Hanski I. Inbreeding and extinction in a butterfly metapopulation. Nature. 1998;392:491–494. [Google Scholar]
- Salvidio S. Detecting amphibian population cycles: the importance of appropriate statistical analyses. Biological Conservation. 2009;142:455–461. [Google Scholar]
- Santucci F, Ibrahim KM, Bruzzone A, Hewit GM. Selection on MHC-linked microsatellite loci in sheep populations. Heredity. 2007;99:340–348. doi: 10.1038/sj.hdy.6801006. [DOI] [PubMed] [Google Scholar]
- Van Oosterhout C, Van Heuven MK, Brakefield PM. On the neutrality of melecular genetic markers:pedigree analysis of genetic variation in fragmented populations. Molecular Ecology. 2004;13:1025–1034. doi: 10.1111/j.1365-294X.2004.02114.x. [DOI] [PubMed] [Google Scholar]
- Vieites DR, Nieto-Roman S, Barluenga M, Palanca A, Vences M, Meyer A. Post-mating clutch piracy in an amphibian. Nature. 2004;431:305–308. doi: 10.1038/nature02879. [DOI] [PubMed] [Google Scholar]
- Vonesh JR, De la Cruz O. Complex life cycles and density dependance: assessing the contribution of egg mortality to amphibian declines. Oecologia. 2002;133:325–333. doi: 10.1007/s00442-002-1039-9. [DOI] [PubMed] [Google Scholar]
- Wang J. Effects of population structures and selection strategies on the purging of inbreeding depression due to deleterious mutations. Genetical Research. 2000;76:75–86. doi: 10.1017/s0016672399004450. [DOI] [PubMed] [Google Scholar]
- Wang J, Hill WG, Charlesworth D, Charlesworth B. Dynamic of inbreeding depression due to deleterious mutations in small populations: mutation parameters and inbreeding rate. Genetical Research. 1999;74:165–178. doi: 10.1017/s0016672399003900. [DOI] [PubMed] [Google Scholar]
- Wright S. Evolution and the Genetic of Populations. Vol. 2: The Theory of Gene Frequencies. Chicago: University of Chicago Press; 1969. [Google Scholar]
- Wright LI, Tregenza T, Hosken DJ. Inbreeding, inbreeding depression and extinction. Conservation Genetics. 2008;9:833–843. [Google Scholar]
- Zartman C, McDaniel SF, Shaw AJ. Experimental habitat fragmentation increases linkage disequilibrium but does not affect genetic diversity or population structure in the Amazonian liverworth Radula flaccida. Molecular Ecology. 2006;15:2305–2315. doi: 10.1111/j.1365-294X.2006.02929.x. [DOI] [PubMed] [Google Scholar]