

Abstract
The recurrent evolution of crop-related weeds during agricultural history raises serious economic problems and challenging scientific questions. Weedy forms of sunflower, a species native from America, have been reported in European sunflower fields for a few decades. In order to understand their origin, we analysed the genetic diversity of a sample of weedy populations from France and Spain, and of conventional and ornamental varieties. A crop-specific maternally inherited marker was present in all weeds. At 16 microsatellite loci, the weedy populations shared most of their diversity with the conventional varieties. But they showed a large number of additional alleles absent from the cultivated pool. European weedy populations thus most probably originated from the unintentional pollination of maternal lines in seed production fields by wild plants growing nearby, resulting in the introduction of crop-wild hybrids into the farmers’ fields. The wide diversity and the low population structure detected were indicative of a multiplicity of introductions events rather than of field-to-field propagation. Further studies are required to understand the local evolutionary dynamics of a weedy population, and especially the respective roles of crop-to-weed gene flow and selection in the fate of an initial source of crop-wild hybrids.
Keywords: crop-wild complexes, genetic diversity, invasive species, sunflower, weediness
Introduction
Crop plants stem from wild species through the process of domestication, the adaptation to cultivation and use by humans (Zeder et al. 2006). During agricultural history, the continuous adaptation of the crop to new environments, new uses and new cultural practices, the constant evolution of the wild populations, as well as gene flow between crops and their wild relatives led to the constitution of a continuum between typical ancestral wild plants and current elite crop varieties (i.e. crop-wild-weed complexes, Barnaud et al. 2009; Van Raamsdonk and Van der Maesen 1996). Some crop-related populations have adapted in an undesired way to the new environment constituted by agricultural fields, the agro-ecosystem, giving rise to agricultural weeds (Arnold 2004). Weeds can be defined as plants that have the capacity to compete with the crop and to lead to more or less severe yield losses (Basu et al. 2004). Crop-related weeds have evolved through different kinds of processes (Londo and Schaal 2007): colonization of cultivated fields by plants from wild populations (e.g. wild rice taxa in Asia, Ellstrand 2003), crop-wild hybridization (Arnold 2004; Boudry et al. 1993), reversion of cultivated plants to wild habits (e.g. some weedy rice, Vaughan et al. 2005). Among these processes, crop-wild and crop-weed hybridization is recognized as very important in generating more noxious weeds (Arnold 2004; Campbell et al. 2006). Understanding how crop-related weeds originate and spread, and how they adapt to the agro-ecosystem constraints is a fundamental topic of research, as an example of recent and rapid evolution (Kane and Rieseberg 2008). It may also help prevent the evolution of new weeds as well as design appropriate management methods.
Common sunflower (Helianthus annuus L.) is an annual self-incompatible insect-pollinated plant. It is native to North America where it has been domesticated at least 4000 years ago (Harter et al. 2004). Nowadays cultivated sunflower markedly differs from typical wild sunflower on a set of characters: absence of anthocyan pigmentation in stem and head, absence of seed dormancy and seed shattering, self-compatibility, absence of branching, bigger heads and seeds (Burke et al. 2002). Wild and domesticated sunflower belong to the same species and are interfertile (Snow et al. 1998). When unharvested seeds of the crop germinate, they give rise to volunteers: such plants grow within fields, or in fallows and field margins; they display the morphological traits of the cultivated form, except for the segregation of a recessive branching trait (Reagon and Snow 2006; Rojas-Barros et al. 2008) and have never been reported to constitute self-perpetuating populations nor to raise serious agronomic problems (Ostrowski et al. 2010). By contrast, in America, weedy forms of sunflower strongly affect the yield of crops such as soybean and corn (Kane and Rieseberg 2008). Sunflower is even listed as noxious in some states and has evolved resistance to herbicide (Massinga et al. 2003). Based on an analysis of microsatellite diversity, Kane and Rieseberg (2008) showed that weedy sunflower populations are genetically close to the natural wild populations occurring in the same region, suggesting that weediness can evolve multiple times independently from wild H. annuus. No phenotypic traits distinguishing weedy and wild forms have been described in published studies.
In Europe, where H. annuus is not native, volunteers are also commonly observed in cultivated areas (Ostrowski et al. 2010). Weedy sunflower populations have been reported since the 1970s (Faure et al. 2002; Holec et al. 2005; Vischi et al. 2006), and have been more precisely described through field surveys in France and Spain (Muller et al. 2009): they show typical wild traits (e.g. pigmentation, seed dormancy, strong branching), in combination with domesticated traits. Weedy populations are thus made of a wide diversity of phenotypes, constituting a continuum between wild and cultivated morphotypes. French and Spanish weedy sunflowers are mostly observed within sunflower fields, and more rarely in other summer crops such as soybean and sorghum; in Spain, they sometimes occur on roadsides and ditches. Around 20% of sunflower fields are affected in the surveyed areas; in 1–4% of the fields, local density can reach 15 plants per m2, which strongly impedes the yield and the harvest by farmers. Achenes can persist in the seed banks for several years (Muller et al. 2009). The only management technique currently available is the manual eradication of the weeds, as long as the infestation is not too strong.
Different scenarios can account for the origin of these weedy populations. First, seeds of wild forms could have been introduced unintentionally by travellers, or have escaped from research nurseries (S1). Second, during the seed production process, pollination of seed bearers by wild sunflowers could have introduced crop-wild hybrids in the seed lots sold to farmers (S2, as described for weed beets, Boudry et al. 1993). Third, weedy sunflower could result from the spontaneous evolution of volunteer populations (S3). Finally, ornamental sunflowers commonly grown in gardens and showing traits such as branching and anthocyan pigmentation could have escaped and/or pollinated volunteer populations (S4).
To discriminate among these hypothesis, we analysed the genetic diversity of a sample of French and Spanish weedy populations, volunteer populations, conventional and ornamental varieties, with 16 microsatellite loci and a mitochondrial crop-specific DNA marker (Rieseberg et al. 1994). Under scenarios S2 and S3, weeds are expected to all carry the crop-specific maternally inherited marker, whereas it should be absent for scenario S1. Because microsatellite diversity is known to be greatly reduced in the cultivated pool compared to the wild populations (Tang and Knapp 2003), the scenarios S1 and S2 imply that a large diversity is observed in weedy populations, compared to scenario S3 where the diversity of weeds is expected to mirror the diversity of the conventional varieties. The outcome of S4 depends on the pattern of diversity within the ornamentals, for which no data is available yet.
Based on our results, we compare the validity of the scenarios and discuss the contribution of different factors to the further evolution of weedy sunflowers: recurrent introductions, dispersal from one field to another, and gene flow from the crop towards the weedy populations.
Material and methods
Plant material
A summary of the sample analysed in this study is given in Table 1. We collected 38 weedy populations in three regions: Andalusia in Spain, and two French regions, Poitou-Charentes and Lauragais (Fig. 1). Spanish populations were sampled in summer 2005 during a survey of the occurrence and distribution of weedy sunflowers (Muller et al. 2009). Populations located within sunflower fields and roadside populations were sampled. French populations were collected during three separate surveys, in 2004, 2005 and 2006. Populations sampled in 2005 and 2006 were described in Muller et al. (2009). One population was sampled both in 2004 and in 2006 (Odars, samples further denoted Odars04 and Odars06). Sampled French populations were all located in moderately to highly infested sunflower fields. We collected three volunteer populations in 2004. One population was located in the Gard (described in Ostrowski et al. 2010) and the two others in the Lauragais. Weedy and volunteer populations were mapped using global positioning system. They have in common to grow spontaneously, without having been sown, and will be referred to as ‘the natural populations’ in the following. In all situations, independent, open-pollinated, progenies of seeds were collected.
Table 1.
Summary of the sample analysed in the present study
| Number of populations | Total sample size | Average sample size per population | |
|---|---|---|---|
| Conventional F1-hybrids | 18 | 86 | 4.8 |
| Ornamental varieties | 6 | 31 | 5.2 |
| Volunteers | 3 | 64 | 21.3 |
| Weedy populations | 38 | 308 | |
| Spain | 14 | 58 | 4.1 |
| Lauragais A | 18 | 71 | 3.9 |
| Lauragais B | 6 | 179 | 29.8 |
| Charentes | 3 | 7 | 2.3 |
| Wild American populations | 4 | 12 | 3 |
Italics: details for the sample of weedy populations (see text).
Figure 1.
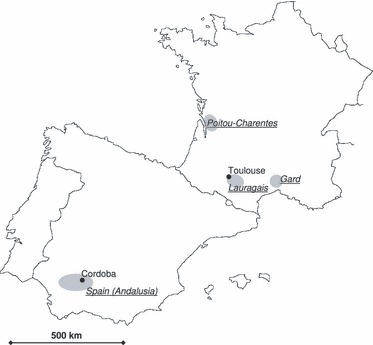
Location of the regions where volunteer and weedy populations have been sampled.
We constituted a sample of 18 conventional F1-hybrids including some of the main varieties cultivated in the past years. Six ornamental varieties including commercial F1-hybrid and populations as well as seeds sold for bird feeding were obtained from garden centres (Table 2). Finally, we included four American wild H. annuus populations stored in the laboratory as an external reference.
Table 2.
Name, producer and sample size of the conventional and ornamental varieties
| Name | Seed company/producer | Sample size |
|---|---|---|
| Conventional varieties | ||
| Albena | Euralis | 5 |
| All stars | Euralis | 6 |
| Atomic | Euralis | 5 |
| Aurasol | Monsanto | 4 |
| Boogy | Syngenta | 5 |
| Country | Syngenta | 3 |
| Filia | RAGT | 4 |
| Heliasol | Semences de France | 7 |
| LG5665M | Limagrain | 3 |
| Melody | Syngenta | 5 |
| Pegasol | Monsanto | 4 |
| PR64H41 | Pioneer | 6 |
| Prodisol | Monsanto | 4 |
| Rigasol | Monsanto | 5 |
| Salsa | Euralis | 4 |
| Santiago | Syngenta | 4 |
| Tekny | Syngenta | 7 |
| Tellia | RAGT | 5 |
| Ornamental varieties | ||
| Beaute d'automne | Royalfleur | 4 |
| Melange | Kokopelli | 9 |
| Oiseaux | Jardiland | 5 |
| Ring flowers | Royalfleur | 3 |
| Soleilsimple | Royalfleur | 5 |
| Sunrich F1 | Royalfleur | 5 |
Molecular data
We randomly chose around five progenies per natural population, except for six French weedy populations (subsample denoted Lauragais B in Table 1) and one volunteer population (FR001, Ostrowski et al. 2010) where at least 25 progenies per population were sampled. One seed per progeny was germinated. Between 3 and 10 seeds per variety, and between two and four seeds per American population were germinated (Table 2). As a whole, 501 individuals were included in the DNA analysis. As a result of extraction or amplification failures, the sample sizes per population varied between 1 and 44.
DNA was isolated from about 100 mg of plant leaves according to the Dneasy Plant Mini kit (Qiagen, GmbH, Hilden, Germany) with the following modification: 1% of Polyvinylpyrrolidone (PVP 40 000) was added to buffer AP1.
Microsatellites
Sixteen microsatellite loci were selected from Tang et al. (2002). Care was taken to choose loci with no previous evidence of null allele (Table 3, Tang et al. 2003; Tang and Knapp 2003). The amplification reaction consisted of 50 ng DNA, 4 pmol of unlabelled reverse primer, 2 pmol of forward primer, fluorescently labelled with NED, HEX or FAM, 1× reaction buffer, 2 mm MgCl2, 200 μm dNTP, 0.25 U Taq DNA polymerase, in a total volume of 25 μL. The amplification method was as follows: 95°C for 2 min, 36 cycles of 94°C for 30 s, Tx for 30 s (Tx is initially 63°C and decreases of 1°C per cycle for the six first cycles, until it reaches 57°C), and 72°C for 45 s, followed by a final extension for 20 min at 72°C. Electrophoresis was performed on an ABI 3130xl Genetic Analyser (Applied Biosystems, Foster City, CA, USA). Samples were prepared by adding 3 μL of diluted PCR products to 6.875 μL formamide and 0.125 μL of GenScan 400HD Rox size standard. The GENEMAPPER software (Applied Biosystems) was used to analyse the DNA fragments and to score the genotypes.
Table 3.
Microsatellite loci, linkage group, core motif, fluorescent dye, allele size range and summary diversity statistics over the whole sample (excluding wild American populations)
| Locus | Linkage group | Position (cM) | Core motif | Fluorescent dye | Allele size range | No. of alleles | He |
|---|---|---|---|---|---|---|---|
| ORS297 | 17 | 29.1 | GT | FAM | 214–237 | 18 | 0.709 |
| ORS309 | 4 | 75.5 | A | FAM | 116–130 | 8 | 0.526 |
| ORS337 | 4 | 62.2 | AC | FAM | 165–197 | 15 | 0.394 |
| ORS342 | 2 | 65 | GT | NED | 305–361 | 26 | 0.665 |
| ORS344 | 15 | 71.4 | AC | FAM | 205–241 | 15 | 0.375 |
| ORS371 | 1 | 44.2 | GT | HEX | 234–264 | 17 | 0.666 |
| ORS380 | 10 | 69.7 | GT | NED | 380–434 | 25 | 0.721 |
| ORS432 | 3 | 42.3 | AC | HEX | 155–167 | 7 | 0.602 |
| ORS610 | 1 | 3.4 | AG | NED | 128–167 | 19 | 0.642 |
| ORS620 | 4 | 57.1 | AG | FAM | 224–266 | 18 | 0.659 |
| ORS656 | 16 | 26.1 | CT | NED | 181–254 | 23 | 0.792 |
| ORS674 | 4 | 100.8 | CT | NED | 331–372 | 23 | 0.805 |
| ORS735 | 17 | 62.6 | AG | HEX | 352–385 | 16 | 0.698 |
| ORS788 | 16 | 46.2 | AG | FAM | 252–296 | 25 | 0.628 |
| ORS887 | 9 | 38.4 | AC | HEX | 224–249 | 16 | 0.639 |
| ORS925 | 2 | 5.9 | AC | FAM | 165–251 | 22 | 0.834 |
He: Nei's genetic diversity.
Mitochondrial marker
The quasi totality of the varieties cultivated in Europe are F1-hybrids, which result from the cross between two inbred lines. The maternal lines carry a cytoplasm conferring male-sterility, and until now, a single cytotype, named CMS89 or PET1, has been used. Because of its maternal inheritance, this cytotype is thus present in all commercial F1-hybrid varieties (Horn 2002). Using a PCR-based strategy, Rieseberg et al. (1994) showed that PET1 was absent from wild H. annuus populations, suggesting that it is diagnostic of the maternal lines used for hybrid-seed production. We used the combination of the three PCR-primers described in Rieseberg et al. (1994) to detect the occurrence of PET1 in our sample. Our amplification procedure consisted of 10 ng DNA, 10 pmol of each primer, 1× reaction buffer, 2 mm MgCl2, 100 μm dNTP, 0.5 U Taq DNA polymerase, in a total volume of 25 μL. The amplification method was as follows: 95°C for 4 min, 36 cycles of 94°C for 45 s, Tx for 45 s (Tx is initially 65°C and decreases of 1°C per cycle for the 12 first cycles, until it reaches 56°C), and 72°C for 2 min and 30 s. Amplification products were separated by electrophoresis on 1.5% agarose gels, stained with ethidium bromide and photographed under UV light. This procedure was applied to the whole sample of varieties, wild and weedy populations, except for a part of weedy population F06bis, for which DNA was not available anymore. Only eight volunteer individuals were included.
Data analysis
Genotypic disequlibrium
We analysed genotypic disequilibrium for all pairs of loci in each sufficiently sampled population of weedy sunflowers and volunteers (seven populations, see Molecular data) with exact tests with GENEPOP (Raymond and Rousset 1995) and applied a sequential Bonferroni correction (Rice 1989).
Group level analysis and distinction between different classes of alleles
Standard statistics of diversity were computed using GENETIX (Belkhir et al. 2001) and FSTAT (Goudet 2001): Nei's expected heterozygosity (He, Nei 1987), the number of different alleles and the allelic richness standardized for similar sample sizes (Ra, Petit et al. 1998). Because the genetic structures of the different groups were highly heterogeneous – F1-hybrids theoretically genetically homogeneous, ornamental varieties of unknown genetic structure, mixture of natural populations of different sample sizes, we first computed these statistics at the group level (Table 1). The significance of the differences in genetic diversity statistics was tested using Wilcoxon signed-rank tests, comparing values for the same loci in different groups using R (R Development Core Team 2008).
We denoted as ‘cultivated’ alleles (C), the alleles that were observed in the conventional varieties or in the volunteer populations. All the other alleles were called ‘original’ alleles (O). For each pool, we computed the frequencies of original and cultivated alleles (forig and fcult), separately for each locus and over loci. The difference on forig between groups was tested using Wilcoxon signed-rank test as described earlier.
Rarefaction curves were built for conventional varieties, ornamental varieties and weedy populations to assess to what extent our sample caught the allelic diversity existing in each group. This especially allows assessing if the O alleles are original because of an incomplete sampling of the conventional varieties or because of a true originality of the weedy populations. For a given group comprising N varieties (or populations), and for each value of k varying between 2 and N − 1, we constituted 200 random subsamples of k varieties. We computed the total number of alleles observed over loci for each sample, and on average over the 200 subsamples of size k. For k = 1, we used the N observed values. For the ornamental group (six varieties), all combinations of k varieties among six were made as it was computationally feasible. For population Odars, only the sample Odars06 was included in the computations.
All C alleles were shared between weedy populations and conventional varieties (see Results). In order to compare their pattern of allele frequency in these two groups, we computed for each locus allelic frequencies within the ‘C allele pool’ of the weedy populations, i.e. excluding O alleles. We computed average within-population frequencies over all weedy populations, and separately over Spanish and Lauragais weedy populations. Using R, we then performed a linear regression of C allele frequencies in the weedy pools on C allele frequencies in the pool of conventional varieties. Additionally, we used an analysis of covariance to include the loci as a cofactor. Allele frequencies were transformed by the function 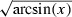 prior to analysis.
prior to analysis.
Population-level analysis: diversity and differentiation
Population-level analyses were conducted only for populations with a sample size of at least three individuals: this corresponds to 33 weedy populations, three volunteer populations, three wild H. annuus populations, and all conventional and ornamental varieties. The sample Odars04 was not included, except for the neighbor-joining tree (see below). Population statistics (He, Ra and forig) were computed for each weedy and volunteer population. Overall FST values and their significance were computed using GENETIX. AMOVA between groups was performed with Arlequin (Excoffier et al. 2005).
A neighbour-joining tree (Saitou and Nei 1987) based on pairwise Cavalli-Sforza chord distances (Dc, Cavalli-Sforza and Edwards 1967) was built using PHYLIP version 3.65 (phylogenetic inference package, Felsenstein 2005). The confidence of the tree was assessed through 1000 bootstraps of allele frequency data: the majority rule tree and the bootstrap values were obtained with CONSENSE.
To determine whether weedy population genetic structure followed a pattern of isolation by distance, Dc genetic distance matrices were correlated with geographical distance matrices using a Mantel test in FSTAT version 2.9.3.2. Tests were performed for two subsets of the data: Spanish weedy populations and Lauragais weedy populations.
For the seven natural populations (six weedy and one volunteer) for which more than 25 genotypes were available, FIS values were computed and their significance was assessed by 1000 permutations of alleles among individuals with GENETIX version 4.05.2. The significance of the differences of He, Ra and forig between populations was assessed as above for group-level comparisons. forig was also computed at the individual level.
Population structure on individual alleles
We used permutations to look for a geographical structure of cultivated or original alleles, i.e. to detect if some alleles were more geographically clustered than expected at random. First, for each allele, we computed the average distance of an allele copy to the barycentre of all copies of that allele: this value was called the dispersion of an allele. We computed the average over loci and/or over allelic class (O or C) of the dispersion values. Singletons were excluded from the data set.
Then for each locus, two kinds of permutations were performed: first, 1000 permutations of allele copies among all geographical locations; second, 1000 permutations of populations among geographical locations. Permutations of alleles allow detecting a geographical clustering which can be due both to differentiation between populations and to resemblance between geographically closed populations; permutations of populations allow detecting specifically the contribution of geographical distance between populations to the clustering of alleles. For each permutation, the dispersion values and their averages were computed. The significance of observed values was obtained by computing the proportion of simulated values that were lower than the observed one. Significance was assessed at the allelic level (for the individual allelic values), at the locus level (for the averages over alleles at a locus), at the allele class level (for the averages over O and C alleles) and crossing the locus and allele class levels. Permutations were programmed in R.
Genetic clustering and admixture analysis
To strengthen our descriptive results relying on the distinction between C and O alleles, we used STRUCTURE version 2.3.3 (Falush et al. 2003; Hubisz et al. 2009). This software uses multilocus genotypes to infer clusters of genetically similar individuals and to estimate admixture proportion of individuals between clusters. We ran the program for values of K = 1 to K = 10 clusters. Each run consisted of a burn-in of 100 000 steps followed by 100 000 steps of data collection and was repeated five times. We used the model of correlated allele frequencies between clusters. We incorporated sampling locations as priors in the clustering (Hubisz et al. 2009): we defined as locations every natural population, American population and ornamental variety but grouped all conventional varieties as a single location. Following Evanno et al. (2005), we calculated ΔK, the second-order change of the likelihood function divided by the standard deviation of the likelihood, to discuss the optimal value of K. Results were visualized using the program DISTRUCT (Rosenberg 2004).
Results
Cytoplasmic analysis
Results were clear-cut. For conventional varieties, for volunteer populations and for one ornamental variety (Sunrich F1, which is an F1-hybrid), all plants analysed showed as expected the PET1 cytotype. By contrast, this cytotype was absent from all other ornamental varieties and all wild individuals. Finally, all weedy individuals showed the PET1 cytotype.
Nuclear diversity
Among 840 tests for genotypic disequilibrium between all pairs of loci, 63 (7.5%) were significant at the level of P < 0.05, 19 of which concerning the volunteer population FR001. Overall, only three tests were significant when applying Bonferroni correction. Two of these tests involved ORS337 and respectively ORS620 (linked on LG4, Table 3) and ORS735. Although linkage does not seem to strongly affect genotypic disequilibrium, the effect of the presence or absence of ORS337 was assessed in the statistical tests to avoid any misconclusion.
Summary statistics are given in Table 3 (sample-wide values for individual locus) and Table 4 (group level values). Between 7 and 26 alleles per locus were scored over the whole sample. Conventional varieties and volunteer populations showed the lowest diversity statistics, whereas ornamental varieties, French and Spanish weedy populations were always significantly more diverse. Spanish populations showed a greater genetic diversity than French populations. Ornamental varieties were intermediate either between conventional varieties and weedy populations with respect of allelic richness, and between French and Spanish weedy population with respect of total genetic diversity. American wild populations exhibited very high levels of diversity, despite a very small sample size.
Table 4.
Population genetic statistics in the different groups of varieties, wild and weedy sunflower
| A | Ra | He | No. of original alleles | forig | |
|---|---|---|---|---|---|
| Conventional varieties | 4.19 | 4.05 b | 0.506 a | 0 | 0.000 |
| Ornemental varieties | 6.44 | 6.31 c | 0.701 bc | 43 | 0.224 ab |
| Volunteer populations | 3.63 | 3.30 a | 0.475 a | 0 | 0.000 |
| Weeds | |||||
| Spain | 13.81 | 10.66 e | 0.753 c | 152 | 0.261 b |
| France | 14.00 | 8.25 d | 0.665 b | 151 | 0.147 a |
| Overall | 17.75 | 9.04 | 0.687 | 211 | 0.168 |
| Wild Helianthus annuus | 8.56 | – | 0.833 | 93 | 0.584 |
A: Average number of alleles per locus.
Ra: Average allelic richness per locus, standardized for a sample size of 26 diploid individuals.
He: Nei's genetic diversity over loci.
The number of original alleles per group is the sum over loci.
Values with the same letters are not significantly different (P < 0.05). ORS337 is included; including it or not does not change the significance of the tests. Wild populations were not included in the statistical tests.
Conventional varieties and ornamental varieties showed intravarietal genotypic diversity. This was unexpected for conventional varieties as they are supposedly composed of a single genotype. However, this diversity was almost caused by differences involving between one and four loci, and the number of alleles per loci never exceeded three, except for one seed lot where a mixing may have occurred (Tekny). The alleles added by the outlier genotypes were never original relative to the other varieties, i.e. were always present in an other variety. Seed lot contamination was probably involved in this polymorphism. Polymorphism was not unexpected in the ornamental varieties, as their genetic structure was not supposed to be homogenous.
The volunteer populations shared all their alleles with the conventional varieties except for six rare ones among a total of 58. Over all loci, the cultivated pool showed 67 different alleles, 73 when including the volunteer populations that were supposedly derived from cultivated fields.
In the following, we will distinguish two classes of alleles: the ‘cultivated’ ones (C) are those observed either in the conventional varieties or in the volunteer populations. They represent the diversity of the European cultivated pool. The ‘cultivated’ alleles were all shared with the weedy populations (Fig. 2 for three representative loci). All the other alleles will be referred to as the ‘original’ alleles (O).
Figure 2.
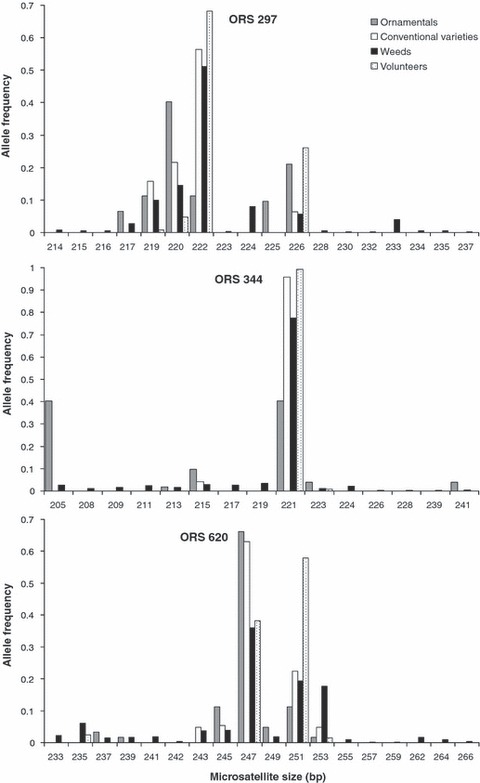
Allele frequency distribution in the different groups for three representative loci.
Two hundred and eleven O alleles occurred in the weedy pool (Table 4), 209 when we considered only the 33 weedy populations retained for further analysis. This value was high compared to the 73 cultivated alleles. But these O alleles were always rare relative to the C alleles. Fifty-six of these alleles were observed only once, and 90 were present in a single population. On average over loci, O alleles represented 16.8% of the diversity of the weedy pool. This was reflected in the patterns of allelic frequencies in the different pools (Fig. 2). The main alleles in the weedy populations were always C alleles. The ornamental varieties, although less diverse, also displayed O alleles, some absent from the weedy populations. The main alleles in the ornamentals are not always C ones.
C alleles not only represent the largest part of the diversity of the weedy populations, they also have similar frequencies in the weedy and the cultivated pool. Linear regressions of C allele frequencies in the weedy pools on frequencies in the cultivated pool were always highly significant. The locus effect was not significant, e.g. no difference on the slope or on the y-intercept was detected. The best fit was obtained with the y-intercept set to zero (R2 = 0.93, P < 0.0001 at the level of the whole weedy pool). For each locus, the most common alleles in the cultivated pool are always the most common alleles in the weedy populations (Fig. 2).
Because we analysed only respectively 6 and 18 ornamental and conventional varieties, we assessed if our sample was representative of the diversity of the cultivated pool. As shown in Fig. 3, the total number of alleles scored in k conventional varieties reached a plateau: for k = 18 varieties, an added variety hardly added new allele to the sample. We can then assume that we sampled almost if not all the diversity of the pool of conventional varieties at the genotyped loci, and that, to a large extent, we did not falsely qualify alleles as original. By contrast, the number of alleles in the ornamental sample did not reach a plateau showing that we did not catch a significant part of the whole diversity of ornamentals. The curves of the weedy populations did not reach a plateau either; they are always higher than the two others curves, which suggests that neither cultivated nor ornamental varieties can account for the alleles observed in the weedy populations: these alleles must have a different origin.
Figure 3.
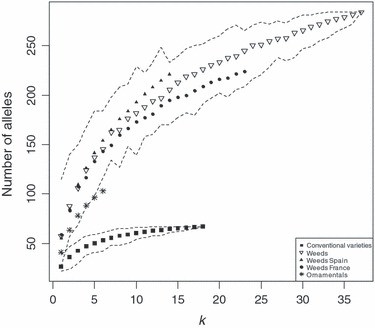
Total number of alleles over the 16 loci for subsamples of different sizes (k) within each group. Each point is the average over 200 random subsamples, except for the ornamentals where all combinations of subsamples (<200) have been constructed. Dashed lines show the minimum and maximum values for the weeds and for the conventional varieties.
Population variability and structure
There was a wide variability among natural populations for diversity statistics (Fig. 4). Spanish weedy populations were among the most diverse and those showing the highest frequency of O alleles, but a large overlap exist with French weedy populations. By contrast, low values of He and Ra markedly distinguished the volunteers from the weedy populations (Fig. 4).
Figure 4.
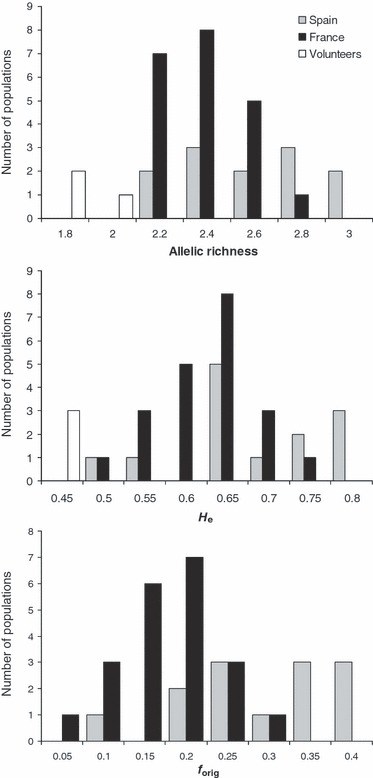
Distribution of diversity statistics across weedy populations from France and Spain and volunteer populations. Allelic richness is the mean allelic richness over loci, standardized for two diploid individuals per population.
A similar level of variation was observed between the seven intensively sampled natural populations (Table 5). The frequency of O alleles per individual showed a wide range of variation from individuals carrying only C alleles to individuals showing more than 50% of O alleles, especially in Villefranche, the most diverse and most original population (not shown). Significant FIS values and high variance of these values among locus were detected. The number of C alleles per population exceeded what is observed in the volunteer populations, and the number of different O alleles reached 5 for some locus in three populations (Table 5).
Table 5.
Diversity statistics within the intensively sampled natural populations
| He | Ra | forig | FIS | O allele per locus | C allele per locus | |
|---|---|---|---|---|---|---|
| Baziege | 0.540 cd | 3.96 c | 0.061 c | 0.156*** | 0.81 [0,2] | 3.81 [2,6] |
| Fourquevaux | 0.612 bc | 4.94 b | 0.134 bc | 0.192*** | 2.19 [0,5] | 3.75 [1,6] |
| F06bis | 0.597 bc | 4.07 c | 0.103 bc | 0.311*** | 1.06 [0,3] | 3.37 [1,6] |
| Gardouch | 0.640 ab | 4.86 b | 0.170 ab | 0.257*** | 1.87 [0,5] | 3.94 [1,7] |
| Odars 06 | 0.640 b | 5.05 b | 0.160 b | 0.233*** | 2.44 [0,4] | 3.69 [2,7] |
| Villefranche | 0.698 a | 5.86 a | 0.239 a | 0.152*** | 3.44 [1,5] | 3.75 [2,7] |
| FR001 | 0.450 d | 2.53 d | 0 | 0.252*** | 0 | 2.87 [2,5] |
Values with the same letters are not significantly different from each other.
P < 0.0001 O (resp. C) alleles per locus gives the average over loci of the number of different original (resp. C) alleles within the population, with its minimum and maximum values. FR001 is a volunteer population.
Significant FST values were detected within each group of natural populations: Spanish weeds (FST = 0.120; P < 0.001), French weeds (FST = 0.080; P < 0.001) and volunteer populations (FST = 0.167; P < 0.001). AMOVA revealed a significant differentiation between these groups, even if much of the variation lies within groups and populations (Table 6). Apart from this, no clear pattern of geographical differentiation between populations arose: evidence for isolation by distance was only detected in the Lauragais (R2 = 33.16; P = 0.0005), but it was almost only because of F09 (R2 = 4.24; P = 0.017 without F09) a weedy population distant from the other populations.
Table 6.
AMOVA and hierarchical analysis
| France/Spain | Weeds/volunteers | |
|---|---|---|
| Source of variation (%) | ||
| Among groups | 2.5 | 5.4 |
| Among populations within groups | 10.1 | 11.8 |
| Within populations | 87.4 | 82.8 |
| Fixation indices | ||
| FST | 0.126*** | 0.176*** |
| FSC | 0.104*** | 0.124*** |
| FCT | 0.025*** | 0.054* |
P < 0.05
P < 0.001.
Based on Cavalli-Sforza chord distance (Fig. 5), only wild populations clustered notably together (i.e. with bootstrap values >50%). Volunteer, weedy populations, conventional and ornamental varieties were intermixed. There was some clustering of varieties, Spanish and Lauragais weedy populations, but few bootstrap values exceeded 50%. Two clusters included varieties from the same seed company (V1 for Monsanto, and V2 for Syngenta, Fig. 5). Interestingly, each volunteer population clustered significantly with a conventional variety. Finally, three pairs of weedy populations clustered, one corresponding to the two samples of population Odars. Drawing a NJ tree of individuals did not show clear clustering patterns except for wild populations and did not provide further information on weedy populations’ relationships (not shown).
Figure 5.
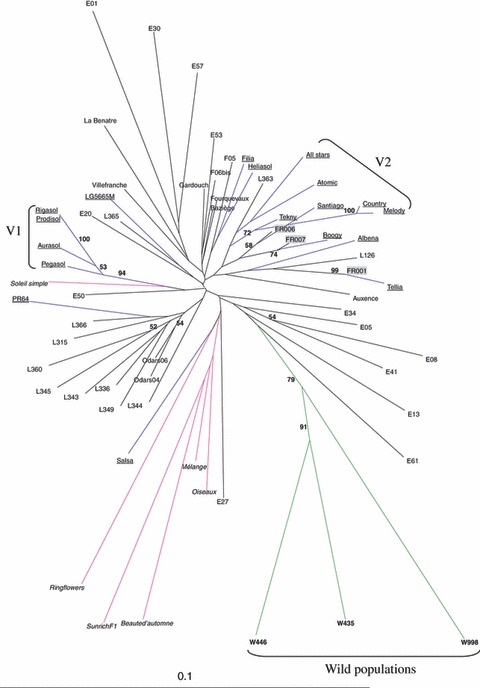
Consensus neighbour-joining tree depicting the relationships between sampled natural populations and varieties, based on Cavalli-Sforza's genetic distance. Percentages on each branch indicate the proportion of bootstrap replicates in which the two sets separated by that branch appear. Only values over 50 are reported. Italics: ornamental varieties. Underlined: conventional varieties. Shadowed: volunteer populations. Bold: American wild populations. Other: weedy populations. V1, V2: cluster of varieties sold by the same seed company (see text).
Permutations of allele copies between populations showed that observed dispersion values are smaller than expected, on average for C and O alleles, as well as for the average locus values, when distinguishing O and C. This shows that populations are differentiated significantly from each other when considering separately O and C alleles (Table 7). By contrast, permutations of populations yielded significant results only for cultivated alleles, and mostly for Lauragais populations. This is not only because of a lack of power of our sampling design as there are also less loci for which the observed values were lower than the average of permutations. As a whole, this suggests that the geographical location of populations did not explain much of the differentiation observed.
Table 7.
Summary of the results of the permutations
| Permutations of alleles | Permutations of populations | |||||
|---|---|---|---|---|---|---|
| Allele type | General | Lauragais | Spain | General | Lauragais | Spain |
| P-value of average dispersion values per allele type | ||||||
| C | <0.001 | 0.001 | 0.004 | 0.002 | 0.028 | 0.221 |
| O | 0.288 | <0.001 | <0.001 | 0.177 | 0.084 | 0.277 |
| Significant loci (loci with lower than expected dispersion value) | ||||||
| C | 6 (11) | 8 (13)* | 2 (13) | 14 (16) | 9 (16) | 0 (13) |
| O | 3 (6)** | 14 (15) | 5 (12) | 4 (9)* | 3 (13)† | 1 (8) |
P-value of average dispersion values per allele type: proportion of simulated values of dispersion that were lower than the observed ones. P-values less than 0.05 denotes a significantly lower than expected dispersion of alleles copy within a given allele type.
Significant loci: number of loci for which the observed average dispersion values were significantly lower than the simulated ones (P-value < 0.05). The number of loci for which observed dispersion values were lower than the average of simulated values, significantly or not, is given between parentheses.
*,**One or two loci for which the average dispersion value was significantly higher than expected (i.e. P-value > 0.95).
Only 15 loci available.
Including or not ORS337 in the computation did not change the P-values.
Genetic clustering and admixture analysis
The variation of the likelihood of the data and the statistic ΔK supported the number of clusters K = 2 as the most probable (Fig. 6). For this value, a clear distinction between conventional varieties and wild American populations was observed. Volunteer populations clustered with the conventional varieties (Fig. 7), confirming that they arose from the escape of cultivated plants. Weedy and ornamental individuals showed varying levels of admixture between the two clusters. Interestingly, for weedy populations, the proportion of ancestry to the wild cluster is strongly correlated with the frequency of original alleles, at the population level (R2 = 0.8955) and at the individual level (R2 = 0.8063). Increasing K did not give rise to clear and strongly repeatable results; the patterns observed revealed mainly (i) subdivisions within the cluster of conventional varieties and (ii) strong admixture within almost all weedy populations: in some of them appeared a new cluster, which was absent or extremely rare from the wild American populations and from all conventional and ornamental varieties (not shown).
Figure 6.
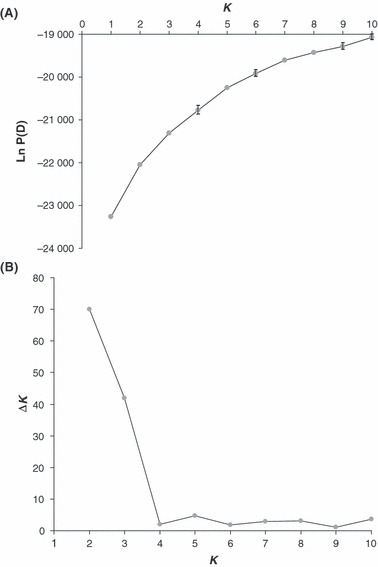
(A) Average and standard deviation of the likelihood of the data vs. the number of clusters (K) assumed under the software STRUCTURE. Standard deviations are plotted when they are larger than the symbol used for the average. (B) Second-order rate of change in K (ΔK) vs. K.
Figure 7.
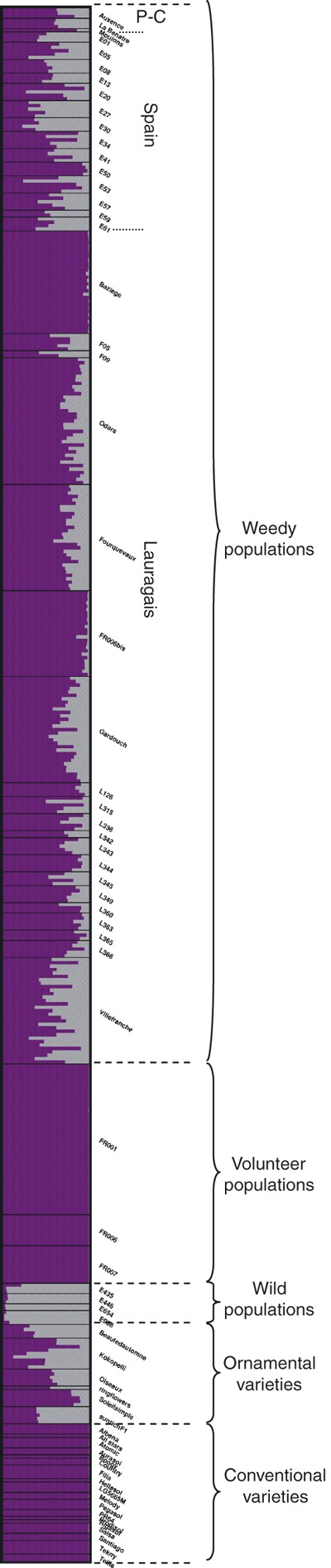
Bayesian analysis of population structure and admixture in our data set, for a number of K = 2 clusters. Each individual is represented by a thin horizontal line, which is partitioned into two coloured segments that indicate the individual's ancestry into the two clusters.
Discussion
Origin of weedy populations
We analysed the genetic diversity and population structure of weedy sunflowers in Europe, in reference to cultivated varieties. According to our results, we can conclude that the most probable origin of these weedy populations is the unintentional pollination of the maternal lines of F1-hybrid seed by wild plants growing nearby and the resulting introduction of crop-wild hybrids into the cultivated fields. First, all weedy plants carry the mitochondrial DNA diagnostic for the maternal line used in hybrid seed production. This is in accordance with the observation of male-sterile weeds during field surveys (Muller et al. 2009) and discards the hypothesis of wild seeds being a source for weedy populations (scenario S1) Second, the main part of the diversity of the weeds is made of the alleles present in the conventional varieties. The frequencies of these alleles within the weedy pool are strongly correlated with their frequencies within the pool of conventional varieties: it suggests that the main part of these ‘cultivated’ alleles come from the varieties, either through the initial crop-wild hybrids introduced in the field or from gene flow from the varieties cultivated in the field towards the weedy populations.
Last, at nuclear microsatellite loci, weedy populations display a large diversity of original alleles (denoted as O alleles), absent from our sample of conventional varieties. Although this sample is not exhaustive, the curves in Fig. 3 showed that it probably caught the vast majority of cultivated alleles. Moreover, Tang and Knapp (2003), Tang et al. (2003) and Zhang et al. (2005) analysed respectively 19, 24 and 124 inbred lines at microsatellite loci: For the fourteen loci that we have in common with at least one of these studies, only three more alleles as a whole have been detected. Even if previously grown landraces are expected to be more diverse than elite inbred lines and might explain the occurrence of some original alleles, the number of scored O alleles is still too big to be accounted for by the diversity of the cultivated pool.
Ornamental varieties shared O alleles with the weedy populations, but also displayed alleles absent both from the weeds and from the conventional varieties. Even if Fig. 3 suggests that we missed alleles from the ornamental varieties and even it cannot be excluded that they have contributed to some weedy populations, it seems that they represent a distinct gene pool that again cannot account for the extraordinary diversity of alleles present in the weeds (Fig. 2). The position of ornamental varieties in breeding schemes relative to conventional varieties, wild forms and even related Helianthus species is unclear. A more thorough investigation of the diversity of the ornamentals would be illuminating first in this respect, and as a by-product may bring some light on their actual role in the evolution of weedy populations.
Our small sample of four wild American populations illustrates the enormous allelic richness occurring in the area of origin of H. annuus. Some O alleles are found in this sample, and a preliminary analysis of a bigger data set including 77 wild populations shows that grossly all C and O alleles are present in the wild. A wild origin of these alleles is thus a reasonable hypothesis. Helianthus annuus is a bee-pollinated outcrosser, and pollination distance can reach at least 1000 m (Arias and Rieseberg 1994) which is more than the isolation distance required for commercial seed production (500 m in the European Community). In America, off-type plants showing wild-type traits and supposedly derived from wild pollen contamination during seed production are found in most sunflower fields (Reagon and Snow 2006). In Europe, F1-hybrid seeds have been introduced from America in the 1970s, in the beginning of F1-hybrid seed development. These importations have been suggested as the probable origin for weeds occurring in Spain and Italy (Faure et al. 2002), although no studies have been conducted to ascertain this hypothesis. Nowadays, commercial seeds are still imported from America, although it is difficult to say from import and export data if these seeds are sown in France or re-exported to other countries (C. Lascrombes, personal communication).
Analysis of the population structure existing in American populations (Harter et al. 2004) could potentially allow identifying a geographical origin of the contaminants. However, as weedy plants are now present in Europe, they can serve as pollen contaminants in seed production fields located in Europe. In France, no specific area is devoted to seed production; seed production fields occur in the regions where weedy populations have been observed (http://www.gnis.fr), even if no survey has been conducted to assess precisely their proximity. The rate of impurity allowed in certified seed is 5%: with a seed density of 60 000 plants per hectare, even 1% of contaminants will result in 600 crop-wild hybrids sown in a field. In Lauragais, we saw every now and then, off-type plants, showing wild traits growing on the rows of otherwise uninfested sunflower fields. Even if the fate of these sparse plants is not certain, it suggests that new weedy populations might now arise from European crop-weed hybrids and blur the information on the original introductions. This, in addition to the rarity and the wide diversity of original alleles, makes rather unrealistic a full elucidation of the scenario of introduction and diffusion of these weeds.
Our Bayesian analysis validated our conclusions based on the analyses on C and O alleles. It additionally highlighted the complexity of the genetic structure of weedy populations and of ornamental varieties, which appear as strongly admixed even when increasing the number of clusters K. Such analysis looks promising if a more exhaustive sample of wild American populations and ornamental varieties is to be constituted.
Evolutionary history of the weedy populations: multiple introduction events, dispersal and crop-weed gene flow
The structure of genetic diversity within and between weedy populations results from the interactions between different factors: (i) the initial introduction event in a field, involving a given seed lot or a given contaminating wild population. The source can be shared or not between populations. (ii) Dispersal from an infested field, giving rise to a new weedy population; and (iii) gene flow from the varieties cultivated in the field towards the weedy populations.
The outcome of such history is expected to be complex and difficult to interpret. As an illustration, within population, the number of different cultivated and original alleles can reach 7 for C alleles and 5 for O alleles (Table 5). Even if the status and origin of cultivated and original alleles is not clear, this pattern at least suggests a diversity of contributors for a given weedy population: an unknown number of initial crop-wild hybrids introduced in the field, a possible multiplicity of introductions, and different varieties further cultivated in this field. By contrast, the volunteer populations have a simpler genetic constitution and cluster each with a single conventional variety in Fig. 5; this variety may be very close to the one or the one itself from which this volunteer population originated.
Despite this complexity, our results can still bring a few insights in two respects: the dynamics of new infestations, and the occurrence of crop to weed gene flow within infested fields.
Population structure and introduction events
Weedy populations were significantly differentiated from each other, with a small but significant differentiation between France and Spain (Table 6). But apart from this, no clear geographical structure was detected: no significant clustering in the neighbour-joining tree (Fig. 5) and no evidence of isolation by distance. On the assumption that the original alleles, contrary to the cultivated ones, could carry the footprints of an introduction event, and of its dispersal, we conducted separate analysis on the geographical structure of these alleles only: permutations of alleles between populations showed that these alleles were significantly more clustered than expected at random. This could result from differentiation of single populations, because of different introduction events and/or from strong genetic drift since the introduction. By contrast, permutation of populations led to much less significant results, showing that original alleles are not shared by geographically close populations, but are rather randomly distributed in space.
Although our sampling design is not sufficiently powerful to demonstrate any detailed history (original alleles are all rare and population sample sizes are small), our results are rather compatible with multiple, uncorrelated introductions of the weedy populations. Dispersal of weeds from infested fields to neighbouring fields seems rare, which is compatible with field observations (Muller et al. 2009): strongly infested fields often neighboured fields without any trace of weeds. New apparitions of infested fields through contaminated seed lots could be more frequent than new apparitions through seed dispersal.
Crop to weed gene flow
Crop to weed gene flow is possible, as the weeds and the crop belong to the same species, grow in the same location and overlap partially in flowering time (Muller et al. 2009). Weedy populations showed variable proportions of original alleles (Fig. 4, Table 5). Phenotypic data is available on the populations sampled in 2006. Interestingly, the populations showing the highest frequencies of original alleles are those that show the highest frequencies of wild phenotypic traits and reciprocally (anthocyan pigmentation, self-incompatibility, seed dormancy, Muller et al. 2009), suggesting original alleles are indeed and grossly indicative of a variable proportion of wild genome. Significant FIS values as well as their high variance across loci denoted nonrandom mating within the population: partial selfing and gene flow variable in space and time. Therefore, the observed variation across populations of the frequencies of original alleles depends (i) on the actual rates of crossing and actual flowering time of the sampled heads at the present generation and (ii) on the evolutionary history within each field in the preceding generations, including the age and size of the initial introduction event and recurrent gene flow from the cultivated varieties. Both factors could also explain the differences observed at the country level: Spanish weedy populations indeed showed a higher allelic richness, a higher frequency of original alleles and a higher level of admixture than their French counterparts. Further studies are required to study independently these different factors.
Perspectives
Our study pointed out the most probable hypothesis for the origin of the weedy populations, namely seed lot contaminations through crop-wild hybridization in seed producing fields. Such hypothesis has already been demonstrated for the origin and evolution of weed beets invading sugar beet producing fields in Northern Europe (Boudry et al. 1993) and for some American weedy rice populations (Londo and Schaal 2007). Wild carrots are also known to pollinate seed plants in seed production fields (Magnussen and Hauser 2007). Nowadays, as new contaminations seem to be more rapid than seed dispersal across the agro-ecosystem, attention has to be paid to the isolation of seed producing fields. For instance, in the case of weed beets, a shift of the seed production area, together with an increased care taken to the elimination of wild beets flowering around seed production fields has helped to reduce the contamination rate to 0.04% (Arnaud et al. 2009).
Further studies are now needed especially at the field level to disentangle the interacting factors that may (or may not) lead from an introduction event (a few crop-wild hybrids) to a strongly infested field: size of the initial source, importance of crop-weed flowering overlap, matting patterns and selective pressures acting on wild vs. domesticated traits. This is of interest to understand the evolution of invasiveness in presence of gene flow. A major question is especially on how gene flow from the crop may or not promote adaptive divergence of the weedy populations, and further the evolution of more aggressive or less controllable weeds. The interaction between selection and gene flow is a basic field of research (Räsänen and Hendry 2008) as well as a key issue when investigating the spread of crop alleles into wild populations (Chapman and Burke 2006). More particularly, such studies could help predict the impact of new varieties carrying an herbicide resistance recently released in Europe (Sala et al. 2008).
Acknowledgments
We thank Florent Pourageaux for contribution to molecular analysis and Marie Roumet and Laurène Gay for careful reading of the manuscript. We thank the reviewers for their helpful comments. This work was funded by the Bureau des Ressources Génétiques and by the Centre technique interprofessionnel des oléagineux métropolitains (CETIOM).
Literature cited
- Arias DM, Rieseberg LH. Gene flow between cultivated and wild sunflowers. Theoretical and Applied Genetics. 1994;89:655–660. doi: 10.1007/BF00223700. [DOI] [PubMed] [Google Scholar]
- Arnaud J-F, Fenart S, Godé C, Deledicque S, Touzet P, Cuguen J. Fine-scale geographical structure of genetic diversity in inland wild beet populations. Molecular Ecology. 2009;18:3201–3215. doi: 10.1111/j.1365-294X.2009.04279.x. [DOI] [PubMed] [Google Scholar]
- Arnold ML. Natural hybridization and the evolution of domesticated, pest and disease organisms. Molecular Ecology. 2004;13:997–1007. doi: 10.1111/j.1365-294X.2004.02145.x. [DOI] [PubMed] [Google Scholar]
- Barnaud A, Deu M, Garine E, Chantereau J, Bolteu J, Koïda EO, McKey D, et al. A weed-crop complex in sorghum: the dynamics of genetic diversity in a traditional farming system. American Journal of Botany. 2009;96:1869–1879. doi: 10.3732/ajb.0800284. [DOI] [PubMed] [Google Scholar]
- Basu C, Halfhill MD, Mueller TC, Stewart CN., Jr Weed genomics: new tools to understand weed biology. Trends in Plant Science. 2004;9:391–398. doi: 10.1016/j.tplants.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F. GENETIX 4.02, logiciel sous WindowsTM pour la génétique des populations. Laboratoire Génome, Populations, Interactions. Montpellier, France: CNRS UMR 5000, Université de Montpellier II; 2001. [Google Scholar]
- Boudry P, Mörchen M, Saumitou-Laprade P, Vernet P, Van Dijk H. The origin and evolution of weed beets: consequences for the breeding and release of herbicide-resistant transgenic sugar beets. Theoretical and Applied Genetics. 1993;87:471–478. doi: 10.1007/BF00215093. [DOI] [PubMed] [Google Scholar]
- Burke JM, Tang S, Knapp SJ, Rieseberg LH. Genetic analysis of sunflower domestication. Genetics. 2002;161:1257–1267. doi: 10.1093/genetics/161.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LG, Snow AA, Ridley CE. Weed evolution after crop gene introgression: greater survival and fecundity of hybrids in a new environment. Ecology Letters. 2006;9:1198–1209. doi: 10.1111/j.1461-0248.2006.00974.x. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Edwards AWF. Phylogenetic analysis: models and estimation procedures. American Journal of Human Genetics. 1967;19:233–257. [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Burke JM. Letting the gene out of the bottle: the population genetics of genetically modified crops. New Phytologist. 2006;170:429–443. doi: 10.1111/j.1469-8137.2006.01710.x. [DOI] [PubMed] [Google Scholar]
- Ellstrand NC. Current knowledge of gene flow in plants: implications for transgene flow. Philosophical Transactions of the Royal Society, Series B. 2003;358:1163–1170. doi: 10.1098/rstb.2003.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Laval LG, Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure N, Serieys H, Bervillé A. Potential gene flow from cultivated sunflower to volunteer, wild Helianthus species in Europe. Agriculture, Ecosystems and Environment. 2002;89:183–190. [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny inference package) version 3.6. Distributed by the author. University of Washington, Seattle: Department of Genome Sciences; 2005. [Google Scholar]
- Goudet J. 2001. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3) http://www.unil.ch/izea/softwares/fstat.html (accessed on 6 April 2006)
- Harter A, Gardner KA, Falush D, Lentz DL, Bye RA, Rieseberg LH. Origin of extant domesticated sunflowers in eastern North America. Nature. 2004;430:201–205. doi: 10.1038/nature02710. [DOI] [PubMed] [Google Scholar]
- Holec J, Soukup J, Cerovska M, Novakova K. Proceedings of the 2nd European Conference on Co-Existence between GM and non-GM based agricultural supply chain. Montpellier, France: 2005. Common sunflower (Helianthus annuus var. annuus) – potential threat to coexistence of sunflower crops in Central Europe; pp. 271–272. [Google Scholar]
- Horn R. Molecular diversity of male sterility inducing and male-fertile cytoplasms in the genus Helianthus. Theoretical and Applied Genetics. 2002;104:562–570. doi: 10.1007/s00122-001-0771-6. [DOI] [PubMed] [Google Scholar]
- Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources. 2009;9:1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane NC, Rieseberg LH. Genetics and evolution of weedy Helianthus annuus populations: adaptation of an agricultural weed. Molecular Ecology. 2008;17:384–394. doi: 10.1111/j.1365-294X.2007.03467.x. [DOI] [PubMed] [Google Scholar]
- Londo JP, Schaal BA. Origins and population genetics of weedy rice in the USA. Molecular Ecology. 2007;16:4523–4535. doi: 10.1111/j.1365-294X.2007.03489.x. [DOI] [PubMed] [Google Scholar]
- Magnussen LS, Hauser TP. Hybrids between cultivated and wild carrots in natural populations in Denmark. Heredity. 2007;99:185–192. doi: 10.1038/sj.hdy.6800982. [DOI] [PubMed] [Google Scholar]
- Massinga RA, Al-Khatib K, St. Amand P, Miller JF. Gene flow from imidazolinone-resistant domesticated sunflower to wild relatives. Weed Science. 2003;51:854–862. [Google Scholar]
- Muller M-H, Delieux F, Fernandez-Martinez JM, Garric B, Lecomte V, Anglade G, Leflon M, et al. Occurrence, distribution and distinctive morphological traits of weedy Helianthus annuus L. populations in Spain and France. Genetic Resources and Crop Evolution. 2009;56:869–877. [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia Univeristy Press: New York; 1987. [Google Scholar]
- Ostrowski MF, Rousselle Y, Tsitrone A, Santoni S, David J, Reboud X, Muller M-H. Using linked markers to estimate the genetic age of a volunteer population: a theoretical and empirical approach. Heredity. 2010;105:384–393. doi: 10.1038/hdy.2009.156. [DOI] [PubMed] [Google Scholar]
- Petit RJ, El Mousadik A, Pons O. Identifying populations for conservation on the basis of genetic markers. Conservation Biology. 1998;12:844–855. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. ISBN 3-900051-07-0. http://www.R-project.org. [Google Scholar]
- Räsänen K, Hendry AP. Disentangling interactions between adaptive divergence and gene flow when ecology drives diversification. Ecology Letters. 2008;11:624–636. doi: 10.1111/j.1461-0248.2008.01176.x. [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. Population genetics software for exact test and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Reagon M, Snow AA. Cultivated Helianthus annuus (Asteraceae) volunteers as a genetic “bridge” to weedy sunflower populations in North America. American Journal of Botany. 2006;93:127–133. [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Van Fossen C, Arias DM, Carter RL. Cytoplasmic male sterility in sunflower: origin, inheritance, and frequency in natural populations. Journal of Heredity. 1994;85:233–238. doi: 10.1093/oxfordjournals.jhered.a111443. [DOI] [PubMed] [Google Scholar]
- Rojas-Barros P, Hu J, Jan CC. Molecular mapping of an apical branching gene of cultivated sunflower (Helianthus annuus. Theoretical and Applied Genetics. 2008;117:19–28. doi: 10.1007/s00122-008-0748-9. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Molecular Ecology Notes. 2004;4:137–138. [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenic trees. Molecular Biology and Evolution. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sala CA, Bulos M, Echarte M, Whitt SR, Ascenzi RA. Molecular and biochemical characterization of an induced mutation conferring imidazolinone resistance in sunflower. Theoretical and Applied Genetics. 2008;118:105–112. doi: 10.1007/s00122-008-0880-6. [DOI] [PubMed] [Google Scholar]
- Snow AA, Moran-Palma P, Rieseberg LH, Wszelaki A, Seiler GJ. Fecundity, phenology, and seed dormancy of F1 wild-crop hybrids in sunflower (Helianthus annuus, Asteraceae) American Journal of Botany. 1998;85:794–801. [PubMed] [Google Scholar]
- Tang S, Knapp SJ. Microsatellites uncover extraordinary diversity in native American landraces and wild populations of cultivated sunflower. Theoretical and Applied Genetics. 2003;106:990–1003. doi: 10.1007/s00122-002-1127-6. [DOI] [PubMed] [Google Scholar]
- Tang S, Yu J-K, Slabaugh MB, Shintani DK, Knapp SJ. Simple sequence repeat map of the sunflower genome. Theoretical and Applied Genetics. 2002;105:1124–1136. doi: 10.1007/s00122-002-0989-y. [DOI] [PubMed] [Google Scholar]
- Tang S, Kishore VK, Knapp SJ. PCR-multiplexes for a genome-wide framework of simple sequence repeat marker loci in cultivated sunflower. Theoretical and Applied Genetics. 2003;107:6–19. doi: 10.1007/s00122-003-1233-0. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk LWD, Van der Maesen LJG. Crop-weed complexes: the complex relationship between crop plants and their wild relatives. Acta Botanica Neerlandica. 1996;45:135–155. [Google Scholar]
- Vaughan DA, Sanchez PL, Ushiki J, Kaga A, Tomooka N. Asian rice and weedy rice – evolutionary perspectives. In: Gressel J, editor. Crop Ferality and Volunteerism. Taylor and Francis Group, Boca Raton, Florida, USA: CRC Press; 2005. pp. 257–277. [Google Scholar]
- Vischi M, Cagiotti ME, Cenci CA, Seiler GJ, Olivieri AM. Dispersal of wild sunflower by seed and persistant basal stalks in some areas of central Italy. Helia. 2006;45:89–94. [Google Scholar]
- Zeder MA, Emshwiller E, Smith BD, Bradley DG. Documenting domestication: the intersection of genetics and archaeology. Trends in Genetics. 2006;22:139–155. doi: 10.1016/j.tig.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Zhang LS, Le Clerc V, Li S, Zhang D. Establishment of an effective set of simple sequence repeat markers for sunflower variety identification and diversity assessment. Canadian Journal of Botany. 2005;83:66–72. [Google Scholar]