

Abstract
Gene escape from crops has gained much attention in the last two decades, as transgenes introgressing into wild populations could affect the latter's ecological characteristics. However, different genes have different likelihoods of introgression. The mixture of selective forces provided by natural conditions creates an adaptive mosaic of alleles from both parental species. We investigated segregation patterns after hybridization between lettuce (Lactuca sativa) and its wild relative, L. serriola. Three generations of hybrids (S1, BC1, and BC1S1) were grown in habitats mimicking the wild parent's habitat. As control, we harvested S1 seedlings grown under controlled conditions, providing very limited possibility for selection. We used 89 AFLP loci, as well as more recently developed dominant markers, 115 retrotransposon markers (SSAP), and 28 NBS loci linked to resistance genes. For many loci, allele frequencies were biased in plants exposed to natural field conditions, including over-representation of crop alleles for various loci. Furthermore, Linkage disequilibrium was locally changed, allegedly by selection caused by the natural field conditions, providing ample opportunity for genetic hitchhiking. Our study indicates that when developing genetically modified crops, a judicious selection of insertion sites, based on knowledge of selective (dis)advantages of the surrounding crop genome under field conditions, could diminish transgene persistence.
Keywords: crop breeding, gene flow, genetic modifications, hitchhiking, introgression, Lactuca, linkage disequilibrium, transgenic plants
Introduction
New crop cultivars are continuously generated to deal with the challenges we face as a consequence of human population growth and a changing environment (Beddington 2010). In case of genetically modified (‘transgenic’) crops, one of the main foci is on developing cultivars tolerant not only to biotic but also to various abiotic stresses (Bhatnagar-Mathur et al. 2008; Ashraf 2010). At the same time, there is considerable concern that genes escaping from crops could result in establishment (‘introgression’) of transgenes from crops into wild relatives. Potentially, this would facilitate range expansions of hybrid taxa and thus affect natural plant populations and habitats (Ellstrand 2003; Chandler and Dunwell 2008). Therefore, assessing ecological consequences of crop-wild hybridization has become an important aspect in the development of risk assessment strategies for transgenic crops (Snow et al. 2005; Chapman and Burke 2006).
Not all genes that escape from crops have the same likelihood of introgressing into wild relatives (Stewart et al. 2003; Baack et al. 2008). Linkage Disequilibrium (LD) – the nonrandom association of alleles at separate loci – in hybrid genomes could lead to genetic hitchhiking of transgenes with native crop genes that do confer a selective advantage under natural field conditions (Mackay and Powell 2007). On the other hand, LD could be directionally disrupted through experiencing opposing selective pressure upon physically linked loci (Stewart et al. 2003). Under field conditions, the result would be a segregation pattern that is systematically different from neutral expectations in progeny as many, less fit, genotypes are purged from populations (Burke and Arnold 2001). The extra-agricultural environment, into which crop genes escape, does likely provide a variety of strong stresses. Depending on the selective strength exerted by the environment, distinct changes in LD could rapidly take place around selected genes. At the same time, other un- or less-selected genes in a positively selected area of the genome could introgress, through genetic hitchhiking, at higher than expected rates (Barton 2000; Tanaka 2007). Therefore, genomic segments under strong selection as a whole would behave like basic units to be considered when assessing the potential of introgression of crop alleles into wild relatives, rather than individual genes. Identifying hotspots and cold areas within the genome related to the likelihood of crop gene introgression could be the first step in employing such information for targeting transgene insertion to the safest genomic areas with regard to transgene persistence in wild populations (Stewart et al. 2003).
Gene flow is seen as providing evolutionary useful new variation to plant populations (Schierenbeck and Ellstrand 2009). This adaptive potential of introgressed hybrid swarms has often been emphasized as a large-scale phenomenon (Burke and Arnold 2001; Chapman and Burke 2006). The latter process is suggested to have happened in just tens of years in Californian Raphanus (Hegde et al. 2006; Ridley et al. 2008) and British Rhododendron (Milne and Abbott 2000). However, the fate of individual parts of the parental genomes – and associated characteristics – after a hybridization event is as yet less obvious (Baack et al. 2008; Rose et al. 2009). Therefore, predicting the likelihood of introgression of specific genes in a hybrid genome is a tough task, which should start with adequate quantitative estimations of the likelihood of different genomic regions to introgress into wild species (Stewart et al. 2003; Martin et al. 2005). Our study is aiming at providing the first steps in exactly doing that, employing nontransgenic crop-wild relative hybrids in lettuce as model system.
Hooftman et al. (2006) hypothesized that crop-wild hybridization could have contributed to the recent invasiveness of prickly lettuce (Lactuca serriola) in Europe. Experimentally, an advantage in survival capacity was identified for at least four generations of synthetic hybrids between lettuce (L. sativa) and wild L. serriola over wild-type plants (Hooftman et al. 2005, 2007). However, increased survival could not readily be related to specific morphological or reproductive traits, indicating complex trait interactions (Hooftman et al. 2005). We suggest that a sorting of genotypes by mortality, favoring distinct genomic segments from one of the parents, under field conditions could be responsible (Hooftman et al. 2009). Under experimentally applied selection targeted at leaf length and flowering time, phenotypes have been shown to change rapidly in Raphanus (Campbell et al. 2009). Therefore, in this contribution to the series of papers into hybrid fitness in Lettuce, we will study the underlying genetic mechanisms of these fitness differences in more depth.
We will test whether selection attributable to natural field conditions leads to preferred combinations of chromosomal elements originating from both species instead of one preferential parent. Putatively, such mosaic could provide the suggested new additive interactions. We employ progeny derived from a cross between a lettuce butterhead cultivar (L. sativa var. capitata) and two Dutch/German wild L. serriola accessions. Three generations of hybrids (S1, BC1, and BC1S1) were grown under natural field conditions, providing nonartificial and a likely wide variety of different selective forces. A control generation was grown in the greenhouse. Markers generated by three different molecular genetic methods (AFLP, SSAP, and NBS profiling) were used, following the association map of Lactuca made for this identical cross (Syed et al. 2006). Our study is based upon two analyses, which in combination could provide tentative indications of genomic hotspots and cold areas for crop allele introgression under natural field conditions.
Changes in allele frequencies. Following ‘conventional wisdom’, alleles originating from crop taxa in hybrids are less favorable under natural conditions (Hails and Morley 2005), which would lead to selective retention of alleles from the wild taxon under natural conditions. We estimate changes in allele frequencies among surviving hybrids after field exposure relative to frequencies observed in the absence of such exposure (‘distortion’).
Linkage disequilibrium. Introgression likelihood of individual genes does not only depend on their own fitness associations but also on the strength of their linkage with other loci (Stewart et al. 2003). Here, we test for consistent postzygotic preservation and disruption of LD.
Materials and methods
Study organisms
For extensive descriptions of the two parental taxa, we refer to Tutin et al. (1976), as well as Hooftman et al. (2005, 2006). In short, Lactuca serriola L. (Asteraceae, 2n = 18) is a common annual weed found in anthropogenically disturbed habitats, such as roadsides, railways, and ruderal sites in urban areas. The species is native to North Africa, Western Asia, and Europe (Carter and Prince 1985). Nowadays, it has been reported as well in South Africa and the Americas (Lebeda et al. 2001). Plants are predominantly autogamous with outcrossing rates of approx. 1% via insect vectors. Seeds of L. serriola are suited for wind dispersal having a parachute-like pappus (D ‘Andrea 2006).
Lactuca sativa L. (Asteraceae, lettuce, 2n = 18) is a common annual crop, which flowers frequently in allotments, largely simultaneously and often sympatrically with L. serriola. It is considered conspecific with, and derived from, L. serriola. Both taxa are fully interfertile, with no known pre- or postzygotic barriers (Koopman et al. 2001). The outcrossing rate of L. sativa is similar to that reported for L. serriola (0.5%; Giannino et al. 2008). Seeds of L. sativa have similar parachute-like pappus as L. serriola, but through cultivated erect involucral bracts are less able to be released.
Plant material and field experiments
The employed hybrid generations and their performances are described in Hooftman et al. (2005, 2007); their pedigree is depicted in Fig. 1. Explaining the hybridization procedure in short: F1 hybrids were created using L. serriola as mother plants and butterhead L. sativa cultivar (var. capitata cv. Dynamite) as pollen donors. The crossing technique followed the protocol by Nagata (1992) with minor alterations. We let F1 hybrids self-pollinate to obtain a generation of autogamous hybrid progeny (hereafter called S1). Backcrossing (BC1) was performed using F1 hybrids maternally and L. serriola as pollen donors; doing so we mimic the most likely scenario of gene escape into a wild population. Here, a single hybrid would be surrounded by wild plants leading to combined backcrossing and selfing.
Figure 1.
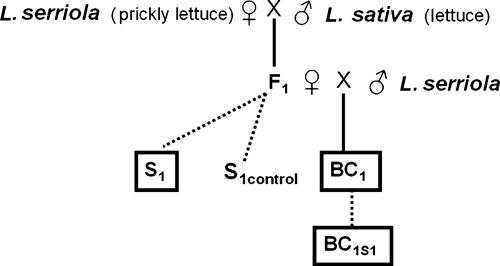
Pedigree of hybrid generations used. Solid lines indicate (back)crossing, and dotted lines indicate selfing. Boxed generations were exposed to nonartificial natural field conditions. The S1 and BC1 generations underwent one growing season of field exposure and the BC1S1 generation two growing seasons. The control generation (S1control) underwent no field exposure.
We employed the genetic map of Syed et al. (2006) based on an S1 generation from one of these interspecific crosses to obtain hybrid populations for field testing. This map consists of nine linkage groups (LGs), which are assumed to correspond to the nine chromosomes of lettuce. Based on preliminary analysis by both SSRs and AFLPs (Keygene N. V., unpublished data), the L. serriola paternal material employed here consisted of four genotypes, with possible heterozygosity identified for 40 loci. Those genotypes were present in the wild populations from which L. serriola seeds for F1 creation were derived (The Netherlands, N50°49′, E05°55′; Germany, N53º32′ E10°54′).
The experiments were split over three locations: Amsterdam (Netherlands, N 52°21′, E 04°58′), Sijbekarspel (Netherlands, N 52°42′, E 05°00′), and Aachen (Germany, N 50°46′, E 06°03′). At each location, two 100 m2 plots were demarcated and ploughed, mimicking anthropogenic soil disturbance. In April 2003, seeds of L. serriola, L. sativa, and S1- and BC1 hybrids were sown individually with 30-cm spacing. The grid positions of seeds within plots were fully randomized. A pioneer vegetation was allowed to emerge spontaneously next to Lactuca, exerting conditions similar to many natural habitats of L. serriola. The resulting mortality rate of Lactuca plants was approximately 90%, offering a window of opportunity for postzygotic selection. In August 2003, autogamous progeny (BC1S1) of these BC1 plants was collected and resown in April 2004 at the site of production using a similar experimental design. Through this setup, field exposure comprising two growing seasons was applied to this BC1S1 generation.
Sampling, molecular marker methods, and loci used
Leaf material was sampled in July 2003 (BC1 and S1) and July 2004 (BC1S1) over the three experimental locations (92 plants generation−1). Sampled plants included almost all surviving individuals with seed-set for these generations. Hooftman et al. (2005, 2007), using these very same plants, did not observe significant interactions between site and fitness, including survival and germination rates as well as fecundity. Therefore, all sites were pooled to obtain sufficient statistical power for the main analysis (but see Table S4 for an analysis per field).
The control S1 generation (91 plants) was grown under fully controlled greenhouse conditions; individual plants were the same as used for the map of Syed et al. (2006), utilizing the same DNA extracts. In further contrast to the field sampling, young plants were collected, which could include genotypes with low or no reproductive fitness. Consequently, the control generation represents only very limited postzygotic selection and hence can be used as baseline for segregation patterns.
DNA was extracted using a modified CTAB extraction method (Syed et al. 2005). All subsequent procedures were identical as used in Syed et al. (2006), performed by the same laboratories and technical staff – i.e., Keygene N.V. for AFLP loci (scored codominantly), SCRI, Dundee for SSAP loci, and Plant Breeding, Wageningen for SSAP and NBS profiling loci (SSAP and NBS scored dominantly). NBS and SSAP are the more recently developed marker types. NBS loci linked to pathogen resistance (R) genes actively used in breeding (Van der Linden et al. 2004). SSAPs are retrotransposon-based markers, expected to behave as alleged neutral markers like AFLP (e.g., Cornman and Arnold 2009; Du et al. 2009). In AFLPs, differentiating between heterozygotes and dominant homozygotes was performed with Xcelerator v.1.3.1. software package (proprietary software, Keygene N.V.). This program differentiates between those two classes depending on the intensity of the bands. To eliminate possible selfings that occurred during the BC1 crossing process (see Hooftman et al. 2005), we conducted a SAHN (Sequential Agglomerative Hierarchical Nested) cluster analysis using UPGMA parameters (Unweighted Pair-Group Method, arithmetic Average). One BC1 plant and one BC1S1 plant were excluded because this analysis suggested an autogamous origin instead of backcrossing for these plants.
We preselected 272 loci from the map in Syed et al. (2006) for our analysis, aiming at an optimal coverage of the genome and the nine linkage groups. Afterward, 40 loci with an alleged L. serriola band presence state were discarded because of the possible presence of wild parental alleles other than the one used for the map. We checked this by detecting false negatives –i.e., unexpected and theoretically impossible homozygotes for the alleged crop allele (band absence) – in the BC1 generation.
The cleaned data set consisted of 232 informative loci containing 89 AFLP loci, 115 SSAP loci, and 28 NBS loci; 186 of those loci were directly located on one of the nine linkage groups, with an average distance of 5.9 cm. We included an additional anonymous set of 46 loci yet to be assigned to one of the LGs. This latter group included AFLP loci from both parentage and SSAP and NBS loci originating from L. sativa. All analyses were performed separately for each of the three hybrid generations. Codominant loci failing to differentiate among hetero- and homozygotes were regarded as not interpretable. Loci for which less than half of the plants per generation had interpretable banding patterns were regarded as missing data.
Changes in allele frequencies in selected progeny
Significance levels for changes in allele frequencies were tested by estimating the bias in the segregation of individual loci in progeny being exposed to field conditions, conditions that provided a variety of nonartificial selective forces. This was tested against patterns observed in the control generation, which were not subjected to postzygotic selection. For this greenhouse-grown control (S1) generation, the distortion of loci was established through comparing to expected neutral segregation. The observed (baseline) distortion was subsequently extrapolated per individual locus to hypothetical BC1 and BC1S1 expected marker distributions (Supporting information: Tables S1 and S2). We will further refer to this significant bias as ‘segregation distortion’. We created confidence intervals using Monte Carlo simulations adjusting the significance threshold based on the number of tests. This analysis and the subsequent LD calculations were written in Matlab; code is available as Supporting information (Data S1). The two-step analysis proceeded as follows:
To take into account the random loss of alleles with low frequencies because of the sampling of all 92 available plants in the field, a Monte Carlo approach was implemented. In each simulation, 92 virtual diploid plants were created. In those plants, per locus, the state – homozygous for one of the parents or heterozygous – was drawn from the expected distribution of these states (Supporting information: Table S2). The probabilities of obtaining states per locus were based on the allele frequencies extrapolated from the control generation, i.e., not from Mendelian except for the control itself (Supporting information: Table S1). For 17 loci with missing data in the control generation, we assumed neutral segregation. Subsequently, the obtained frequencies per locus of this sample of 92 plants were calculated. This procedure was repeated 50 000 times.
Confidence intervals for a given significance level (α) were obtained from the above obtained sampling distribution of state frequencies per locus. To correct for the number of tests (=loci) performed, we allowed for a type I error of one single locus, consequently α = (1/# loci) per generation. To give an example, for 211 loci, the actual α would be 0.0047, and the confidence interval would represent the 49 764th value in the sorted array per locus from the above bootstrap procedure. The resulting α values are the following: control: 0.0047 (for N = 213); S1: 0.0045 (N = 220); BC1: 0.0063 (N = 158); and BC1S1: 0.0047 (N = 211). The corresponding confidence limits per locus per generation are provided in Supporting information (Table S3). A locus with an observed state frequency above the confidence interval is labeled as significantly distorted. The P-value distributions for all tests are provided in the Supporting information. These distributions show a bias in the direction of either high or low p-values of field-exposed generations, indicating a strong distinction between loci with segregation distortion and loci without distortion (Fig. S1).
Increased and decreased LD
We tested for nonrandom associations among loci, using their correlation coefficients (r; Flint-Garcia et al. 2003), and we will subsequently use the term linkage disequilibrium (LD) when referring to this estimate (Eqn 1).
| (1) |
where X11 as the frequency of homozygous L. sativa alleles for adjacent locus gap i; p1 and p2 the frequency of L. sativa and L. serriola alleles on the first haploid, respectively; q1 and q2 the frequency of L. sativa and L. serriola alleles on the second haploid, respectively.
Pairwise codominant/dominant combinations were integrated through transforming codominant scoring into dominant by assuming the band presence state being dominant in heterozygotes. To test for alterations in LD among generations, the observed levels of LD were compared with an expected level of LD, expressed as percentage. This comparison was achieved through calculating the ratio of LD per LG of control and field-tested generation, averaging over the adjacent pairwise combinations. We use the marginal LD (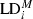 ) values per generation as our estimate, i.e., the value for each pairwise combination adjusted for the level of LD in the control generation according to Eqn 2. In this way, we are able to compare per adjacent pairwise combination the relative decrease or increase in the control generation. A Chi-square test was included to test for statistical significance (df = 1; Eqn 3). We employed a correction for multiple testing following Benjamini and Hochberg (1995), with α = 0.05 as base value.
) values per generation as our estimate, i.e., the value for each pairwise combination adjusted for the level of LD in the control generation according to Eqn 2. In this way, we are able to compare per adjacent pairwise combination the relative decrease or increase in the control generation. A Chi-square test was included to test for statistical significance (df = 1; Eqn 3). We employed a correction for multiple testing following Benjamini and Hochberg (1995), with α = 0.05 as base value.
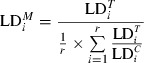 |
(2) |
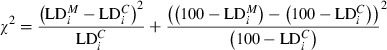 |
(3) |
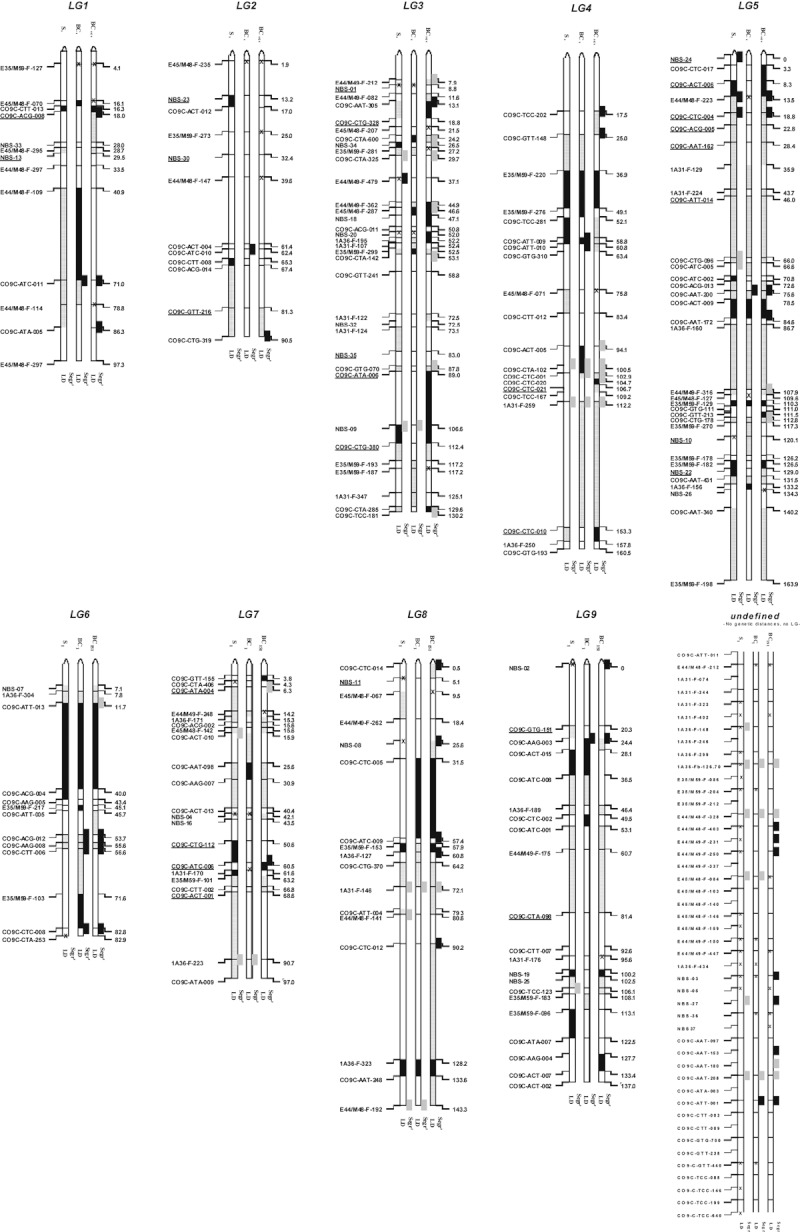
where 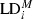 = marginal LD for adjacent locus gap i;
= marginal LD for adjacent locus gap i; 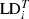 = observed LD in the tested generation for adjacent locus gap i;
= observed LD in the tested generation for adjacent locus gap i; 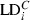 = observed LD in the control generation for adjacent locus gap i, and r = number of adjacent locus gaps in the Linkage Group.
= observed LD in the control generation for adjacent locus gap i, and r = number of adjacent locus gaps in the Linkage Group.
Genomic hotspots
We will in our discussion tentatively identify both genomic hotspots and cold areas for introgression of crop alleles into wild generations (Stewart et al. 2003). Such areas are defined by unidirectional distorted chromosomal parts of 2 loci or more, co-occurring with a higher than expected LD. We will, speculatively, indicate general genomic regions but refrain from being very specific. Further studies should narrow these regions.
Results
Single-locus allele frequency changes
Of the 232 informative loci, 66 showed allele frequencies significantly different from expected in one or more field-exposed generations, compared with the control generation (Table 1, Fig. 2). This included a large number of loci for which crop alleles were in excess of expectations. We attribute this distortion to the actual field exposure, as only few distorted loci were identified in the control series, in which the postzygotic possibility for selection was very limited (16 loci; Table 1).
Table 1.
Number of loci with significant segregation distortion after field exposure for either parental species compared with expected segregation based on an extrapolated control generation (Supporting information Table S1). Included are three marker types of which AFLP were codominantly scored and NBS and SSAP dominantly. Four hybrid generations are included with 92 surviving plants generation−1: the control generation underwent no field exposure; the S1 and BC1 generations both underwent one growing season of field exposure; and the BC1S1 generation two growing seasons
| Distortion with prevalence for | |||||
|---|---|---|---|---|---|
| Generation | Marker type/total no. of loci | L. serriola Allele | L. sativa allele | Hetero-zygotes | Directional† |
| AFLP | |||||
| Control | 73 | 1 | 2 | 0 | ns |
| S1 | 88 | 10 | 2 | 0 | * |
| BC1‡ | 80§ | 6 | 0 | ns | |
| BC1S1 | 72 | 6 | 6 | 0 | ns |
| NBS | |||||
| Control | 28 | 2 | 2 | ns | |
| S1 | 20§ | 2 | 1 | ns | |
| BC1¶ | 9 | 1 | 1 | ns | |
| BC1S1 | 24§ | 1 | 4 | ns | |
| SSAP | |||||
| Control | 112 | 0 | 9 | * | |
| S1 | 112§ | 5 | 2 | ns | |
| BC1¶ | 69 | 3 | 11 | * | |
| BC1S1 | 115 | 14 | 20 | ns | |
Significance (*P < 0.05) indicates a noneven, i.e., directional, distribution of distorted loci among parental species, Chi-square, AFLP: df = 2; NBS/SSAP: df = 1.
Codominantly scored BC1 loci (backcross with L. serriola) are either L. serriola homozygotes or heterozygotes.
Proportionally not more distorted than the control generation (P > 0.05; Chi-square, AFLP: df = 2; NBS/SSAP: df = 1).
N in the BC1 generation is lower, as dominant loci with a dominant L. serriola allele will show all a band being either heterozygous or homozygous L. serriola.
Figure 2.
Segregation distortion and direction of linkage disequilibrium (LD) change. Three hybrid generations of 92 plants each are included (S1, BC1, and BC1S1; columns 1–3, respectively), for each of nine linkage groups and an anonymous group of loci. Parentage, linkage group, and mapping distance in cm are according to Syed et al. (2006). Differences in gray-shading among loci within columns indicate LD alterations, based on marginal LD compared with the S1, greenhouse-grown, control generation (α = 0.05, corrected for multiple testing following Benjamini and Hochberg 1995): black segments indicate increased LD, and hatched segments indicate lowered LD. The allelic bias (‘distortion’) is indicated next to the loci per column (α = 1/loci per generation), compared with the segregation as observed in the (extrapolated) control generation. Dark-colored loci contain significantly more crop (L. sativa) alleles than expected over 92 plants; light-colored loci contain significantly more wild relative (L. serriola) alleles. SSAP loci are prefixed with ‘C0’; NBS with ‘NBS’; all other prefixes represent AFLP loci. ‘X’ represents missing data. Within the BC1 generation, loci either with a L. serriola band presence or dominantly scored provided no segregation and LD signal (underlined loci). For the ease of readability, distances smaller than 1.8 cm are not scaled.
We identified substantial differences among the hybrid generations, as well as differences among the three different types of markers employed. After 2 years of field exposure (BC1S1; 51 loci), the distortion was almost double compared with one year of field exposure (S1 and BC1, 22 and 22 loci, respectively). This increase is attributed to repeated selection in combination with an additional recombination possibility. For all but two SSAP loci, at LG5 and LG6 – CO9C-CTC-004 and CO9C-AAG-008 – this preference for one parental allele per locus was based on at least two fields being distorted in the same direction (Supporting information, Table S4). In general, there was no overall bias for one of the parental species for all the three marker types. Two exceptions were identified: in the S1 generation, distortion was significantly biased in the direction of the L. serriola alleles for AFLP markers. In contrast, for SSAP markers in the BC1 generation, we found a significant preference for L. sativa (Table 1). However, for none of the marker types, after 2 years of exposure, did we identify a significant overall preference for one of the parents. This suggests that elements of both parents are favored depending on the putative fitness association at the specific location. Also, no overall preferential distortion signal was found in the NBS loci (Table 1). For NBS markers, the amount of distorted loci was not larger than in the control generation, except for the BC1, but with low numbers (2). Strikingly, no tendency in favor of heterozygotes was identified for all generations (Table 1).
Pairwise linkage disequilibrium
For over half of the adjacent pairwise combinations, LD was significantly altered and putatively influenced by the field exposure (Table 2). In approximately half or more of the pairwise combinations, the level of LD was affected in field-exposed generations, compared with the extrapolated baseline from the control generation (Table 2). This proportion was different among the generations (S1: 48; BC1: 69%, and BC1S1: 58%). For both selfing, field exposed, generations (S1 and BC1S1), the ratio of alteration between lowered and elevated LD was about 2:1, significantly different from random (P < 0.001; Table 2). The direction of alterations from the baseline per pairwise combination is indicated in Fig. 2, marked by different gray shading.
Table 2.
Direction of alteration of adjacent pairwise linkage disequilibrium (LD) under field exposure compared with a S1 control (α = 0.05, corrected for multiple tests following Benjamini and Hochberg 1995). Comparisons are based on marginal, adjusted, values for the tested generations
| LD compared with (S1−)control | ||||
|---|---|---|---|---|
| Generation | N | Weakened (<) | Strengthened (>) | Unchanged |
| S1 | 157 | 53*** | 23 | 81 |
| BC1† | 77 | 31ns | 22 | 24 |
| BC1S1 | 145 | 57*** | 27 | 61 |
Significant (P < 0.001) directional effect deviating from 1:1 weakened/strengthened (Chi-square, df = 1).
N in the BC1 generation is lower, as dominant loci with a dominant L. serriola allele will show all a band being either heterozygous or homozygous L. serriola.
No directional effect deviating from 1:1 weakened/ strengthened (Chi-square, df = 1)
Many pairwise combinations are altered in the same direction (either lower or higher) for all generations (Table 3; Fig. 2). Very strongly significant, the two generations with an included selfing event (S1 and BC1S1) show that 44 combinations are altered in the same direction, whereas only six were not (P < 0.001; Table 3). Such strong bias makes it very unlikely that these patterns are caused by nondirectional, i.e., random, variation.
Table 3.
Among-generation comparison of the direction of adjacent pairwise Linkage disequilibrium alteration under field exposure compared with an S1 control (α = 0.05, corrected for multiple tests following Benjamini and Hochberg 1995). Comparisons are based on marginal, adjusted, values for the tested generations. Three categories are provided: equal direction (=), opposite directions (≠), and both unchanged. N represents the total viable pairwise combinations available for among-generation comparison
| S1 | BC1† | BC1S1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gen' | N | = | ≠ | Unchanged | N | = | ≠ | Unchanged | N | = | ≠ | Unchanged |
| S1 | 75*** | 22 | 4 | 17 | ||||||||
| BC1† | 63ns | 17 | 8 | 8 | ||||||||
| BC1S1 | 131*** | 44 | 6 | 36 | ||||||||
Significant (P < 0.001) directional effect deviating from 1:1 equal/ opposite (Chi-square, df = 1).
N in the BC1 generation is lower, as dominant loci with a dominant L. serriola allele will show all a band being either heterozygous or homozygous L. serriola.
No directional effect deviating from 1:1 equal/opposite (Chi-square, df = 1).
Throughout the Lactuca genome, there are clear areas in which LD is consistently strengthened even for all generations like in parts of LG4 (36.9–49.1 cM), LG5 (78.5–84.6 and 110.3–111.0), and LG6 (11.7–40). The locus starting this segment at LG6 is biased in the direction of L. serriola, making it a potential favorable spot for transgene insertion. However, the opposite, a consistent loosening of LD among all three generations, is found on various places throughout the genome. Putatively, this is caused by opposing forces, selecting toward different parental directions. Obvious examples can be found in LG5 (46.0–66.0) and LG8 (64.2–72.1).
Discussion
Our study together with its related work on hybrid fitness in natural field conditions (Hooftman et al. 2005, 2007, 2009) follows the fate of parental genomes through several generations after hybridization between wild and cultivated lettuce. This larger framework included the construction of a genetic map for the cross used in this study (Syed et al. 2006). Our study is among the first to empirically test the genomic structure in hybrids between crops and wild relatives as affected by natural, i.e., nonartificial, field conditions, others being Brassica and Helianthus in Rose et al. (2009) and Baack et al. (2008). Genomic hotspots and cool areas for introgression can be identified, as performed below, through the identification of distorted regions in which linkage disequilibrium (LD) is consistently strengthened through multiple generations. Such regions indicate that specific recombinants are favored under these field conditions. We consider such information very useful for plant breeding. Through marker-assisted breeding or related techniques (Varshney and Dubey 2009), plants could be bred having transgene insertion sites in genomic regions providing the highest likelihood of being purged after escape into wild relative populations (Stewart et al. 2003; Sweet 2009).
Allele frequency shifts
For a large number of loci, we identified a clear change in allele frequencies in favor of the retention of alleles specific to one or the other parental taxon in the natural field. This included many loci for which crop alleles were in excess of expectations for band present states of both L. sativa and L. serriola. The nonuniform distribution of P-values, skewed to contain many high values in field-exposed generations, indicates that the applied correction for multiple tests (Benjamini and Hochberg 1995) provided conservative results rather than speculative ones. Our results contrast to a view quite generally held until few years ago that alleles originating from the crop were considered deleterious for survival in the wild, assuming a general loss of adaptive capacity for crop genes in wild habitats (Hails and Morley 2005). It appears that at least part of the alleles, which have been (co−) selected by breeders, may confer advantages upon hybrid plants under field exposure, depending on the type of environment. This is a conclusion that we share with recent work in sunflower (Baack et al. 2008; Chapman et al. 2008), rice (Cao et al. 2009), Brassica (Rose et al. 2009), and Raphanus (Snow et al. 2010). In our study, the driving force seems to be directional selection. The approximately 90% mortality rate per growing season postzygotically removes many less-fit hybrid lineages (Whitney et al. 2006). Consequently, positively selected crop genes will promote subsequent introgression of many less advantageous crop alleles, including transgenes, via ‘genetic hitchhiking’ (Barton 2000; Stewart et al. 2003; Mackay and Powell 2007).
Differences between marker types
Distortion – i.e., changes in allele frequencies from those expected under neutrality – showed a tendency to the selection for many wild-type alleles after one growing season of field exposure in the S1 generation for AFLP loci as well as a preference for crop alleles for SSAP loci in the BC1 generation. No preferences were seen in the BC1S1 generation. Whether there is any biological explanation behind these opposite directions remains to be identified with further research. SSAPs have recently been successfully employed (e.g., Cornman and Arnold 2009; Du et al. 2009). However, the drawbacks and caveats of SSAPs are not yet completely known. Therefore, it is hard to tell whether there is any contribution from a marker-type bias in favor of a specific category of linked trait, which could explain the difference between AFLPs and SSAPs between the two generations.
Fitter Lactuca hybrids
In earlier work, we identified these identical hybrid plants to be substantially fitter than the wild parent (relative growth rate = 3). This difference is mostly because of a larger survival capacity at the seedling stage (Hooftman et al. 2007). The fitness advantage gradually declined through multiple generations but was still present in the BC3 and S4 generations as well as in the BC1S1 plants used in this study (relative growth rate = 1.7). However, as a group, hybrids were not morphologically distinguishable from the wild parent (Hooftman et al. 2005). Therefore, any crop genes connected to this fitness advantage seem not to be correlated with easily visible morphological characteristics. Furthermore, in light of the fitness advantage of these hybrid generations, it is remarkable that being heterozygous was hardly advantageous. Prior to this study, we expected to find a larger effect of heterosis after releasing these conspecific taxa from their inbreeding load by outcrossing (Johansen-Morris and Latta 2006). Accordingly, we now strongly suggest that additive interactions among genes rather than heterosis could be responsible for the increased fitness of these Lactuca hybrids compared with both parental species.
Fixation and selective strength
LD among loci could have led to coselection of large numbers of linked genes among which only one or few genes actually confer a fitness benefit, the rest being effectively neutral (Burke et al. 2005). Such segments could start functioning as independent units when experiencing opposing selective forces with respect to introgression. As a result, neighboring segments will become more and more separated and introgression will appear to occur in chromosomal ‘units’ surrounding an important locus associated with plant fitness (Stewart et al. 2003). Identification of such quantitative trait loci (QTLs) in this lettuce cross is currently underway (e.g., Uwinama et al. 2010). The various areas we identified with less than expected LD could be examples of areas that are more prone to tear apart in opposite parental directions under nonagricultural conditions. Rapid fixation of whole genomic areas could be the result, providing selection pressures do not vary much. The evidence that fixation of large segments is a distinct possibility is provided through the signature of domestication patterns by agriculture (Ehrenreich and Purugganan 2007; Wills and Burke 2007; Yamasaki et al. 2007). By applying repeated similar bottlenecks, fixation is rapid. After selecting for hundreds of generations with a constant and fairly similar environmental pressure provided by agricultural conditions, LD could be still significant up to 10 cm, such as reported in barley (Kraakman et al. 2004).
Hotspots and cold areas for introgression
We can tentatively identify genomic hotspots and cold areas for the introgression of crop alleles into wild generations (Stewart et al. 2003). The BC1S1 generation shows the clearest results for this having been exposed to 2 years of selection pressure. Being speculative, few suggestions for a judicious choice of insertion of a new (trans)gene to lettuce could be onto parts of LG3 (middle and bottom) or LG4 (lower middle), where manifold distortion to containing L. serriola segments combined with strengthened LD occurs, with a smaller similar suitable area on LG6 (top). When derived from L. sativa, those parts provide in general a higher likelihood of being purged from the hybrid genome after initial hybridization. In contrast, LG5 (top and middle) and LG8 (upper middle) seem to be better avoided, favoring L. sativa genomic segments. A combination with one or more of the proposed other biosafety containment constructs (e.g., Gressel and Valverde 2009; Moon et al. 2010) would decrease the likelihood of introgression even more.
Implications for future crop development and risk assessment
Our results are not only applicable to this single underlying association study of Syed et al. (2006), but to lettuce breeding in general. The majority of employed AFLP markers are integrated in the larger map of Truco et al. (2007). That map is based on an Iceberg lettuce cultivar and a Californian L. serriola accession and has been a prime outcome of the Compositae Genome Project (http://compgenomics.ucdavis.edu/). Currently, this database contains already over 42 000 lettuce unigenes (McHale et al. 2009).
Gene flow from crops to wild relatives has occurred and will continue to occur (Ellstrand 2003; Chandler and Dunwell 2008). Subsequently, LD induced by selection may cause genetic hitchhiking, which could be an important factor facilitating the introgression of specific genes (Stewart et al. 2003; Chapman and Burke 2006). Modeling studies have already shown that segments with only limited fitness advantage in the hybrid plants can provide rapid fixation of introduced genes (Morjan and Rieseberg 2004; Hooftman et al. 2008). Where recombination rates between a transgene and beneficial QTLs are low, introgression of such genes is either facilitated or mitigated, depending on the trait association (Stewart et al. 2003; Sweet 2009).
As a negative message from our study, we conclude that genes, should they be undesirable in natural settings (such as certain transgenes), could introgress into a wild background without conferring a direct fitness benefit themselves. More positively, a judicious choice of transgene insertion sites may inhibit transgene persistence because of selection against the segment in a nonagricultural, natural, habitat. Our observations of these mechanisms could have clear implications for developing transgene containment strategies based on LD. The first step is an adequate assessment of the likelihood of (trans) gene introgression over the whole genome (Stewart et al. 2003). The way forward could be using QTL approaches and related association studies (Mackay and Powell 2007; Heffner et al. 2009), employing the vastly expanding number of genetic maps (Collard and MacKill 2008). Such information could then be combined with developing appropriate predictive modeling techniques (Meirmans et al. 2009; Sweet 2009).
Acknowledgments
We thank Peter van Tienderen, Gerard Oostermeijer, Eric Schranz, Thure Hauser, Rene Smulders, Jeroen Rouppe van der Voort, Gerard van der Linden, as well as all members of ANGEL for discussions. Two anonymous reviewers and the associate editor are acknowledged for their constructive comments improving the manuscript. We also thank the technical staffs at the University of Amsterdam (Mirjam Jacobs, Maaike de Jong), SCRI, Keygene (Dick Lensink & Rudie Antonise), and PRI (Wendy van't Westende). This study was funded by EU-QLK3-2001-01657.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Histograms of P-value distributions
Table S1 Calculations for locus based expected segregation ratios
Table S2 Expected segregation per locus
Table S3 Confidence intervals for distortion
Table S4 P-values per field for theBC1S1 generation
Data S1 Data and Matlab code used
Literature cited
- Ashraf M. Inducing drought tolerances in plants: recent advances. Biotechnology Advances. 2010;28:169–183. doi: 10.1016/j.biotechadv.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Baack EJ, Sapir Y, Chapman MA, Burke JM, Rieseberg LH. Selection on domestication traits and quantitative trait loci in crop–wild sunflower hybrids. Molecular Ecology. 2008;17:666–677. doi: 10.1111/j.1365-294X.2007.03596.x. [DOI] [PubMed] [Google Scholar]
- Barton NH. Genetic Hitchhiking. Philosophical Transactions of the Royal Society B. 2000;355:1553–1562. doi: 10.1098/rstb.2000.0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beddington J. Food security: contributions from science to a new and greener revolution. Philosophical Transactions of the Royal Society B. 2010;365:61–71. doi: 10.1098/rstb.2009.0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B. 1995;57:289–300. [Google Scholar]
- Bhatnagar-Mathur P, Vadez V, Sharma KK. Transgenic approaches for abiotic tolerance in plants: retrospect and prospects. Plant Cell Reports. 2008;27:411–424. doi: 10.1007/s00299-007-0474-9. [DOI] [PubMed] [Google Scholar]
- Burke JM, Arnold ML. Genetics and fitness of hybrids. Annual Review in Genetics. 2001;35:31–52. doi: 10.1146/annurev.genet.35.102401.085719. [DOI] [PubMed] [Google Scholar]
- Burke JM, Knapp SJ, Rieseberg LH. Genetic consequences of selection during the evolution of cultivated sunflower. Genetics. 2005;171:1933–1940. doi: 10.1534/genetics.104.039057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LG, Snow AA, Sweeney PM, Ketner JM. Rapid evolution in crop-weed hybrids under artificial selection for divergent life histories. Evolutionary Applications. 2009;2:172–186. doi: 10.1111/j.1752-4571.2008.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao QJ, Xia H, Yang X, Lu BR. Performance of hybrids between weedy rice and insect resistant transgenic rice under field experiments: implication for environmental risk assessment. Journal of Integrative Plant Biology. 2009;51:1138–1148. doi: 10.1111/j.1744-7909.2009.00877.x. [DOI] [PubMed] [Google Scholar]
- Carter RN, Prince SD. The geographical distribution of Prickly Lettuce (Lactuca serriola): a general survey of its habitat and performance in Britain. Journal of Ecology. 1985;73:27–38. [Google Scholar]
- Chandler S, Dunwell JM. Gene flow, risk assessment and the environmental release of transgenic plants. Critical Reviews in Plant Sciences. 2008;27:25–49. [Google Scholar]
- Chapman MA, Burke JM. Letting the gene out of the bottle: the population genetics of genetically modified crops. New Phytologist. 2006;170:429–443. doi: 10.1111/j.1469-8137.2006.01710.x. [DOI] [PubMed] [Google Scholar]
- Chapman MA, Pashley CH, Wenzler J, Hvala J, Tang SX, Knapp SJ, Burke JM. A genomic scan reveals candidates for genes involved in the evolution of cultivated sunflower. Plant Cell. 2008;20:2931–2945. doi: 10.1105/tpc.108.059808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collard BCY, MacKill DJ. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philosophical Transactions of the Royal Society B. 2008;363:557–572. doi: 10.1098/rstb.2007.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornman RS, Arnold ML. Characterization and comparative analysis of sequence-specific amplified polymorphisms based on two subfamilies of IRRE retrotransposons in Iris missouriensis (Iridaceae) Genetica. 2009;135:25–38. doi: 10.1007/s10709-008-9248-8. [DOI] [PubMed] [Google Scholar]
- D ‘Andrea L. Historical and Molecular Biogeography of Prickly Lettuce (L. serriola L.) and Evaluation of Hybridisation with the Crop (L. sativa L.) Switzerland: University of Neuchâtel; 2006. PhD thesis. [Google Scholar]
- Du XY, Zhang ZR, Luo ZR. Comparison of four molecular markers for genetic analysis in Diospyros L. (Ebenaceae) Plant Systematics and Evolution. 2009;281:171–181. [Google Scholar]
- Ehrenreich IM, Purugganan MD. The molecular genetic basis of plant adaptation. American Journal of Botany. 2007;93:953–962. doi: 10.3732/ajb.93.7.953. [DOI] [PubMed] [Google Scholar]
- Ellstrand NC. Dangerous Liaisons? When Cultivated Plants Mate with their Wild Relatives. Baltimore, Maryland: John Hopkins University press; 2003. [Google Scholar]
- Flint-Garcia SA, Thornsberry JM, Buckler ES. Structure of Linkage Disequilibrium in plants. Annual Reviews of Plant Biology. 2003;54:357–374. doi: 10.1146/annurev.arplant.54.031902.134907. [DOI] [PubMed] [Google Scholar]
- Giannino D, Nicolodi C, Testone G, Di Giacomo E, Iannelli MA, Frugis G, Mariotti D. Pollen mediated transgene flow in lettuce (Lactuca sativa L.) Plant Breeding. 2008;127:308–314. [Google Scholar]
- Gressel J, Valverde BE. A strategy to provide long-term control of weedy rice while mitigating herbicide resistance transgene flow, and its potential use for other crops with related weeds. Pest Management Science. 2009;65:723–731. doi: 10.1002/ps.1754. [DOI] [PubMed] [Google Scholar]
- Hails RS, Morley K. Genes invading new generations: a risk assessment perspective. Trends in Ecology and Evolution. 2005;20:245–252. doi: 10.1016/j.tree.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Heffner EL, Sorrells ME, Jannink JL. Genomic selection for crop improvement. Crop Science. 2009;49:1–12. [Google Scholar]
- Hegde SG, Nason JD, Clegg JM, Ellstrand NC. The evolution of California's wild radish has resulted in the extinction of its progenitors. Evolution. 2006;60:1187–1197. [PubMed] [Google Scholar]
- Hooftman DAP, Oostermeijer JGB, Jacobs MMJ, den Nijs JCM. Vital rates determine the performance advantage of crop-wild hybrids in Lettuce. Journal of Applied Ecology. 2005;42:1086–1095. [Google Scholar]
- Hooftman DAP, Oostermeijer JGB, den Nijs JCM. Invasive behavior of Lactuca serriola (Asteraceae) in The Netherlands: spatial distribution and ecological amplitude. Basic and Applied Ecology. 2006;7:507–519. [Google Scholar]
- Hooftman DAP, de Jong MJ, Oostermeijer JGB, den Nijs JCM. Modelling the long-term consequences of crop-wild relative hybridization: a case study using four generations of hybrids. Journal of Applied Ecology. 2007;44:1035–1045. [Google Scholar]
- Hooftman DAP, Oostermeijer JGB, Marquard E, den Nijs JCM. Modelling the consequences of crop-wild relative gene flow: a sensitivity analysis of the effects of outcrossing rates and hybrid vigour breakdown in Lactuca. Journal of Applied Ecology. 2008;45:1094–1103. [Google Scholar]
- Hooftman DAP, Hartman Y, Oostermeijer JGB, den Nijs JCM. Existence of Vigorous Lineages of Crop-Wild Hybrids in Lettuce under Field Conditions. Environmental Biosafety Research. 2009;8:203–217. doi: 10.1051/ebr/2010001. [DOI] [PubMed] [Google Scholar]
- Johansen-Morris AD, Latta RG. Fitness consequences of hybridization between ecotypes of Avena barbata: hybrid breakdown, hybrid vigor, and transgressive segregation. Evolution. 2006;60:1585–1595. [PubMed] [Google Scholar]
- Koopman WJM, Zevenbergen MJ, van der Berg RG. Species relationships in Lactuca s.l. (Lactuceae, Asteraceae) inferred from AFLP fingerprints. American Journal of Botany. 2001;88:1881–1887. [PubMed] [Google Scholar]
- Kraakman ATW, Niks RE, van den Berg PMMM, Stam P, van Eeuwijk FA. Linkage disequilibrium of yield and yield stability in modern spring barley cultivars. Genetics. 2004;168:435–446. doi: 10.1534/genetics.104.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeda A, Doležalová I, Křístková E, Mieslerová P. Biodiversity and ecogeography of wild Lactuca spp. in some European countries. Genetic Resources and Crop Evolution. 2001;48:153–164. [Google Scholar]
- Mackay I, Powell W. Methods for linkage disequilibrium mapping in crops. Trends in Plant Science. 2007;12:57–63. doi: 10.1016/j.tplants.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Martin NH, Bouck AC, Arnold ML. Loci affecting long-term hybrid survivorship in Louisiana Irises: implication for reproductive isolation and introgression. Evolution. 2005;59:2116–2124. [PubMed] [Google Scholar]
- McHale LK, Truco M, Kozik A, Wroblewski T, Ochoa OE, Lahre KA, Knapp SJ, et al. The genomic architecture of disease resistance in lettuce. Theoretical and Applied Genetics. 2009;118:565–580. doi: 10.1007/s00122-008-0921-1. [DOI] [PubMed] [Google Scholar]
- Meirmans PG, Bousquet J, Isabel N. A metapopulation model for the introgression from genetically modified plants into their wild relatives. Evolutionary Applications. 2009;2:160–171. doi: 10.1111/j.1752-4571.2008.00050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RI, Abbott RJ. Origin and evolution of invasive naturalised material of Rhododendron ponticum L. in the British Isles. Molecular Ecology. 2000;9:541–556. doi: 10.1046/j.1365-294x.2000.00906.x. [DOI] [PubMed] [Google Scholar]
- Moon HS, Li Y, Stewart CN. Keeping the genie in the bottle: transgene containments by excision in pollen. Trends in Biotechnology. 2010;28:3–8. doi: 10.1016/j.tibtech.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Morjan CL, Rieseberg LH. How species evolve collectively: implications of gene flow and selection for the spread of advantageous alleles. Molecular Ecology. 2004;13:1341–1356. doi: 10.1111/j.1365-294X.2004.02164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata RT. Clip-and-wash method of emasculation for lettuce. Hortiscience. 1992;27:907–908. [Google Scholar]
- Ridley CE, Kim SC, Ellstrand NC. Bidirectional history of hybridization in California wild radish, Raphanus sativus (Brassicaceae), as revealed by chloroplast DNA. American Journal of Botany. 2008;95:1437–1442. doi: 10.3732/ajb.0800119. [DOI] [PubMed] [Google Scholar]
- Rose CW, Millwood RJ, Moon HS, Rao MR, Halfhill MD, Raymer PL, Warwick SI, et al. Genetic load and transgenic mitigating genes in transgenic Brassica rapa (field mustard) x Brassica napus (oilseed rape) hybrid populations. BMC Biotechnology. 2009;9:93. doi: 10.1186/1472-6750-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierenbeck KA, Ellstrand NC. Hybridization and the evolution of invasiveness in plants and other organisms. Biological Invasions. 2009;11:1093–1105. [Google Scholar]
- Snow AA, Andow DA, Gepts P, Hallerman EM, Power A, Tiedje JM, Wolfenbarger LL. Genetically engineered organisms and the environment: current status and recommendations. Ecological Applications. 2005;15:377–404. [Google Scholar]
- Snow AA, Culley TM, Campbell LG, Sweeney PM, Hegde SG, Ellstrand NC. Long-term persistence of crop alleles in weedy populations of wild radish (Raphanus raphanistrum. New Phytologist. 2010;186:537–548. doi: 10.1111/j.1469-8137.2009.03172.x. [DOI] [PubMed] [Google Scholar]
- Stewart CN, Halfhill MD, Warwick SI. Transgene introgression from genetically modified crops to their wild relatives. Nature Reviews Genetics. 2003;4:806–817. doi: 10.1038/nrg1179. [DOI] [PubMed] [Google Scholar]
- Sweet JB. The 10th International Symposium on the Biosafety of Genetically Modified Organisms (ISBGMO), Wellington, New Zealand, November 2008. Environmental Biosafety Research. 2009;8:161–181. doi: 10.1051/ebr/2009017. [DOI] [PubMed] [Google Scholar]
- Syed NH, Sureshsundar S, Wilkinson MJ, Bhau BS, Cavalcanti JJV, Flavell AJ. Ty1-copia retrotransposon-based SSAP marker development in Cashew (Anacardium occidentale L.) Theoretical and Applied Genetics. 2005;110:1195–1202. doi: 10.1007/s00122-005-1948-1. [DOI] [PubMed] [Google Scholar]
- Syed NH, Sørensen AP, Antonise R, van de Wiel C, van der Linden CG, van't Westende W, Hooftman DAP, et al. A detailed linkage map of Lettuce based on SSAP, AFLP and NBS markers. Theoretical and Applied Genetics. 2006;112:517–527. doi: 10.1007/s00122-005-0155-4. [DOI] [PubMed] [Google Scholar]
- Tanaka Y. Introgressive hybridization as the breakdown of postzygotic isolation: a theoretical perspective. Ecological Research. 2007;22:929–939. [Google Scholar]
- Truco MJ, Antonisse R, Lavelle D, Ochoa O, Kozik A, Witsenboer H, Fort SB, et al. A high-density, integrated genetic linkage map of Lettuce (Lactuca spp.) Theoretical and Applied Genetics. 2007;115:735–746. doi: 10.1007/s00122-007-0599-9. [DOI] [PubMed] [Google Scholar]
- Tutin TG, Heywood VH, Burges NA, Moore DM, Valentine DH, Walters SM, Webb DA. Flora Europaea. Vol. 4. Cambridge, UK: Cambridge University Press; 1976. [Google Scholar]
- Uwinama B, van der Wiel C, Smulders R, Visser R. 2010. A genetic analysis of the introgression process from crops to wild relatives under abiotic stress conditions: the case of Lettuce. Proceedings of the 11th ISBGMO conference. Buenos Aires, Argentina.
- Van der Linden CG, Outers DCAE, Mahaska V, Kochieva EZ, Smulders MJM, Vosman B. Efficient targeting of plant disease resistance loci using NBS profiling. Theoretical and Applied Genetics. 2004;109:384–393. doi: 10.1007/s00122-004-1642-8. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Dubey A. Novel genomic tools and modern genetic and breeding approaches for crop improvement. Journal of Biochemistry and Biotechnology. 2009;18:127–138. [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. Adaptive introgression of herbivore resistance traits in the weedy sunflower Helianthus annuus. The American Naturalist. 2006;167:794–807. doi: 10.1086/504606. [DOI] [PubMed] [Google Scholar]
- Wills DM, Burke JM. Quantitative Trait Locus analysis of the early domestication of sunflower. Genetics. 2007;176:2589–2599. doi: 10.1534/genetics.107.075333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Wright SI, McMullen MD. Genomic screening for artificial selection during domestication and improvement in Maize. Annals of Botany. 2007;100:967–973. doi: 10.1093/aob/mcm173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.