

Abstract
Based on the anti-hepatitis C activity of 2′-C-methyl-adenosine and 2′-C-methyl-guanosine, a series of new modified purine 2′-C-methyl nucleosides was prepared as potential anti-hepatitis C virus agents. Herein, we report the synthesis of both 6-modified and 2-modified purine 2′-C-methyl-nucleosides along with their anti-HCV replication activity and cytotoxicity in different cells.
Hepatitis C virus (HCV) infects more than 200 million individuals worldwide according to the World Health Organization. In addition, an estimated 3 to 4 million people contract HCV each year. Long-term infection can lead to chronic liver disease, such as cirrhosis of the liver or hepatocellular carcinoma. Until recently the standard of care therapy employed interferon-α (IFN) often in combination with the nucleoside analog ribavirin. The impact on standard of care by approval of the two HCV protease inhibitors Incivek and Victrelis remains unclear as both drugs require response-guided therapy regimens that can shorten the duration of IFN therapy in infected persons with an early viral response from 48 weeks to as few as 24 weeks. In addition, serious side effects and limited efficacy emphasize the urgent need for improved therapeutic agents.1 Moreover, there is no established vaccine for HCV. As a result, there is an urgent need for safe and effective therapeutic agents that combat HCV infection and that have high genetic barrier to resistance.
Among the most successful preclinical and clinical nucleosides with potent HCV NS5B polymerase inhibition are 2′-C-methyl analogs. 2 2′-C-Methyl adenosine, 1 and 2′-C-methyl guanosine, 2 (Fig. 1) display selective in vitro anti-HCV activity as non-obligatory chain terminators toward RNA elongation. However, in vivo these analogs display poor bioavailability due to deamination of the adenine base and purine nucleoside phosphorylase (PNP) promoted glycosidic bond cleavage in the case of 1 and poor cellular uptake and/or inefficient phosphorylation for 2.3
Figure 1.
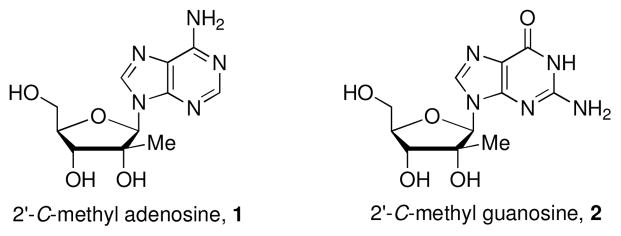
Important 2′-C-methyl purine analogs.
Base-modification of nucleoside analogs has been successful in the development of potent antiviral agents.4 Within the 2′-C-methyl nucleoside series, the 7-deaza modification of the adenosine analog results in a significant improvement of in vivo anti-HCV activity.5 In this study, novel purine modified 2′-C-methyl nucleoside analogs were prepared which explored the steric, electronic, and hydrogen bonding effects on anti-HCV activity as measured by a cell-based replication assay.
The synthesis of 2′-C-methyl-2-amino-6-methyl purine analogs is outlined in Scheme 1. Compound 3, as a single isomer, was readily prepared in three sugar modification steps5 from commercially available starting materials. Condensation of 3 with 2-amino-6-chloropurine in the presence of DBU and TMSOTf at 80 °C for 5 h gave the 6-chloro nucleoside 4 as the pure β-anomer in 92% yield. A modification to the standard Vorbrüggen conditions involving the addition of TMSOTf at −78 °C improved the reaction yield from around 60%3a to over 90%. Cross-coupling reactions have been used successfully for the introduction of carbon-linked substituents into purine moieties.6 Hence compound 4 was allowed to react with trimethylaluminum under Pd(PPh3)4 catalyzed conditions in THF to give the corresponding 6-methyl-purine derivative 5 in 54% yield. Subsequently, the benzoyl groups were removed to afford the 6-methyl purine nucleoside analog 6. 7 Monophosphate prodrug, (aryloxy)phosporamidate 7, was prepared by following Uchiyama’s procedure in the presence of tert-butyl magnesium chloride in THF.8,9
Scheme 1.

Reagents and conditions: a) 2-amino-6-chloropurine, DBU, TMSOTf, CH3CN, 80 °C, 5 h, 92%; b) Pd(Ph3P)4, Al(CH3)3, THF, 75 °C, 8 h, 54%; c) NH3/CH3OH, rt, 2 d, 62%; d) t-BuMgCl, phenyl ethoxyalaninyl phosphorochloridate, THF, rt, 18 h, 9 %.
The 6-phosphonate substituted purine is an attractive target because the phosphonate group may mimic the hydrogen bonding characteristics of a 6-amino or 6-hydroxyl group of endogenous nucleoside bases and thus may result in nucleoside analogs that are inhibitors of adenosine deaminase (ADA).10 Application of the Arbuzov reaction to 6-chloro purine 4 by treatment with triethyl phosphite afforded 6-phosphonate 8 in 78% yield (Scheme 2).11 Removal of the benzoyl groups with saturated NH3 in ethanol or NaOEt in ethanol provided 912 and 10 respectively.
Scheme 2.

Reagents and conditions: a) P(OEt)3, 130°C, 18 h; 78%; b) NH3/EtOH, rt, 4 d, 44%; c) NaOEt/EtOH, rt, 2 d, then 50°C for 30 min, 19%.
A series of 1,2,4-triazolo [5,1-i] adenine derivatives were identified as potent adenosine A2a receptor antagonists.13 Application of these interesting base modifications to a 2′-Me sugar nucleoside provides potential HCV inhibitors. Among many synthetic approaches for preparation of these purine base modifications,14 an efficient cyclization method utilizing N, O-bis-(trimethylsilyl)acetamide (BSA) to undergo a dehydrative rearrangement was chosen as shown in Scheme 3.
Scheme 3.

Reagents and conditions: a) H2NNHCOR, 110 °C, 2 d; b) BSA, 130 °C, 5 h, 12a: 39%; 12b: 72%; c) NaOMe/MeOH, rt, 18 h, 13a: 50%; 13b: 90%.
The ipso displacement of 6-chloro group of compound 4 by acetohydrazide or 2-furoic hydrazide afforded compounds 11a and 11b, which are used for the next step without purification. Heating with BSA for 5 h furnished the desired tricycles 12 via a dehydrative rearrangement process.13 After removal of the benzoyl protecting groups, 13a and 13b were isolated in 50% and 90% yield, respectively.
The 6-H nucleoside 15 was isolated in 21% yield during the preparation of 12a (Fig. 2). Formation of 15 can be explained by a base promoted Wolff-Kishner reduction.15 This postulate is supported by the detection of the 6-hydrazo nucleoside 14 by LC/MS during the preparation of 11a. During the BSA promoted dehydrative rearrangement reaction, intermediate 14 was converted to 15 while the intermediate 11a proceeded to 12a. In a confirmation reaction, 6 equivalents of hydrazine were allowed to react with compound 4 at 110 °C for 2 days. The reduced nucleoside 15 was cleanly formed without removal of the benzoyl groups.
Figure 2.
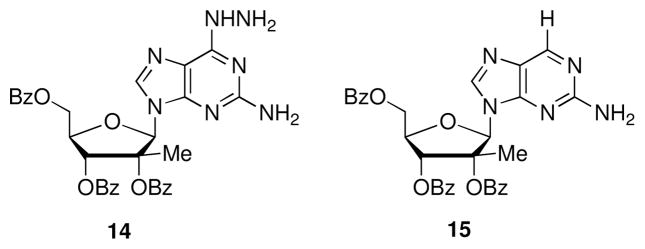
Side products of 12a formation.
Previous studies have found 2-position modified purines with potent antiviral activity and/or a reduction of toxicity.16 The 2-hydrazine-, 2-azido- and 2-triazole-purine nucleosides were targeted as they potentially offer a variety of steric, electronic, and hydrogen bonding interactions which may enhance recognition by HCV NS5B polymerase. Treatment of the 2,6-dichloro purine nucleoside 16 with methanolic ammonia removed the benzoyl protecting groups with concomitant amination and methoxylation to afford 6-aminopurine 17 and 6-methoxypurine 18 respectively (Scheme 4). Nucleophilic substitution of the 2-chloro group of 17 with hydrazine hydrate gave the 2-hydrazine substituted purine nucleoside 19.17 Treatment of 19 with sodium nitrite in acetic acid provided a 63% yield of the 2′-azidopurine 20. Based on analysis of the 1H-NMR, compound 20 exists in both an azido and an N1 tetrazole tautomeric form. 18 1,3-Dipolar cycloaddition of azide 20 with ethynyltrimethylsilane through a Cu (I)-catalyzed 1,3-cycloaddition reaction generated the triazole analogs 2119 and compound 22.20 Compound 21 is formed by desilylation during the cycloaddition reaction. Compound 21 may also be prepared from 22 by treatment with aqueous HF in THF and formation of 21 during the cycloaddition step may be suppressed by addition of BSA to the reaction mixture.21
Scheme 4.

Reagents and conditions: a) NH3/CH3OH, THF, rt, 3 d, 17: 46%, 18: 21%; b) hydrazine, EtOH, rt, 16 h, 92%; c) NaNO2, HOAc, rt, 1 h, 78%. e) sodium L-ascorbate, CuSO4•5 H2O, TMSCCH, t-BuOH/H2O (v/v, 1:1), rt, 18h, 21: 12%, 22: 30%.
The 2-hydroxylamino substitution was selected for evaluation due to its structural and electronic similarity to hydrazine. As shown in Scheme 5 the 6-chloropurine 23 was nitrated with tetrabutylammonium nitrate (TBAN) and trifluoroacetic anhydride (TFAA) to afford the 2-nitro analog 24 in 79% yield.22 Nucleophilic substitution with sodium azide produced compound 25 in 99% yield. Side reactions and lower yields occurred when more than 1 equivalent of sodium azide was utilized in this reaction. Careful hydrogenation under mild heating afforded 2-hydroxypurines 26 and 27 in modest yields.23 However, removal of the benzoyl groups of 26 or 27 with NH3/CH3OH or sodium methoxide under a variety of conditions failed to give the desired compounds, but instead lead to complex mixtures.
Scheme 5.

Reagents and conditions: a) TBAN, TFAA, CH2Cl2, 85°C, 3 h, 79%; b) NaN3, DMF, −18 °C, 1 h, then 0 °C, 2 h, 99%; c) H2 (1 atm), Pd/C, EtOAc, EtOH, 45–50 °C, 26: 1.5 h, 43%; 27: 3 h, 41%.
Based on the biological activity of the 4-amino-1H-imidazo[4,5-d]pyridazin-7(6H)-one ring system as a purine isostere, 24 4-amino-1-(2-methyl-β-D-ribofuranosyl)-3H-imidazo[4,5-d] pyriazin-7-(6H)-one, 32 and 7-amino-1-(2-methyl-δ -D-ribofuranosyl)-1H-imidazo [4,5-d] pyridazin-4-(5H)-one, 35 were synthesized (Scheme 6). The Vorbrüggen glycosylation reaction of 4 with ethyl 5(4)-cyano-1H-imidazole 4(5)carboxylate 28 gave two regioisomeric nucleosides 29 and 30 in 64% and 36%, respectively. The regioisomeric structures were assigned based on analysis of their 1H-NMR data and comparison to their ribofuranosyl analogs.25 Compounds 29 and 30 were independently treated with excess t-butylamine in ethanol to obtain the deprotected and transesterified 31 and 34 in 91% and 78% yield, respectively. The two compounds were separately reacted with hydrazine, followed by heating with sodium ethoxide in anhydrous ethanol at reflux to yield the respective target nucleoside analogs 32 and 35 in 71% and 62% yield. The unexpected bis-adduct products 33 and 36 were also obtained albeit in low yields of 5% and 12%, respectively.
Scheme 6.

Reagents and conditions: a) DBU, TMSOTf, CH3CN, 0°C, 30 min, 50 °C, overnight, 29: 64%, 30: 36%; b) t-butylamine, MeOH, rt, 2 days, for 31: 91%, for 34: 78%; c) i) hydrazine monohydrate, MeOH, 0 °C; ii) NaOEt (cat), EtOH, reflux, 32: 71%, 33: 5%, 35: 62%, 36: 12%.
All of the modified purine 2′-C-methyl nucleosides were evaluated for inhibition of HCV RNA replication at 10 μM in Huh7 cells using a subgenomic HCV replicon system.26 Cytotoxicity in Huh7 cells was determined simultaneously with anti-HCV activity by extraction and amplification of both HCV RNA and ribosomal RNA (rRNA).27 To determine the spectrum of activity of the compounds, anti-HIV activity was evaluated versus HIV-1LAI in primary human peripheral blood mononuclear (PBM) cells and AZT was used as a positive standard. Cytotoxicity was determined in PBM, human lymphoblastoid CEM, and African Green monkey Vero cells.28 A subset of the compounds were tested for their ability to inhibit and/or act as substrates of adenosine deaminase.29 The antiviral and cytoxicity results are summarized in Table 1.
Table 1.
Anti-HCV activity, anti-HIV activity, and cytotoxicity of synthesized compounds in cellular assays.
| Cmpd | Anti-HCV activity: % inhib @ 10 μM in Huh7 cells | Anti-HIV activity, μM | Cytotoxicity in: CC50, μM | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| HCV | rRNA | EC50 | EC90 | PBM | CEM | Vero | |
| 6 | 0 | 0 | 70.1 | >100 | >100 | 12.9 | >100 |
| 7 | 0 | 0 | 68.2 | >100 | >100 | >100 | >100 |
| 9 | 18.7 | 14.8 | >100 | >100 | >100 | >100 | >100 |
| 10 | 19.3 | 14.9 | >100 | >100 | >100 | >100 | >100 |
| 13a | 0 | 0 | >100 | >100 | >100 | >100 | >100 |
| 13b | 0 | 0 | >100 | >100 | >100 | >100 | >100 |
| 17 | 0 | 5.8 | >100 | >100 | >100 | >100 | >100 |
| 18 | 14.3 | 9.8 | >100 | >100 | >100 | >100 | >100 |
| 19 | 85.2 | 7.0 | >100 | >100 | >100 | >100 | >100 |
| 20 | 5.2 | 0 | >100 | >100 | >100 | >100 | >100 |
| 21 | 0 | 0 | >100 | >100 | >100 | >100 | >100 |
| 22 | 0 | 0 | >100 | >100 | >100 | >100 | >100 |
| 26 | 65.9 | 76.1 | 8.7 | 42.1 | >100 | 27.4 | 41.5 |
| 27 | 99.5 | 99.9 | 16.6 | 65.4 | 43.2 | 7.3 | >100 |
| 32 | 0 | 0 | >100 | >100 | >100 | >100 | >100 |
| 33 | 0 | 0 | >100 | >100 | >100 | >100 | >100 |
| 35 | 0 | 0 | >100 | >100 | >100 | >100 | >100 |
| 36 | 0 | 9.0 | >100 | >100 | >100 | >100 | >100 |
In general, nucleosides with the 6-position modifications of the purine base did not display any marked anti-HCV activity (up to 10 μM) or toxicity to Huh7 PBM, CEM, or Vero cell lines. The lack of a nti-HCV activity or cytotoxicity of these nucleosides may be due to inefficient uptake and/or their inability to be intracellularly metabolized to the corresponding nucleoside triphosphates. However, the monophosphate prodrug 7, which would bypass the initial phosphorylation step, also did not display any inhibition of HCV replicon RNA replication.
Modification in the 2-position of the purine base proved to be somewhat interesting. The substituted purines 2-Cl,6-NH2, 17, 2-Cl,6-OMe, 18, 2-azido,6-NH2, 20, 2-(1H-triazole),6-NH2, 21, 2-(4-(trimethylsilyl)-1H-triazole),6-NH2, 22 along with the purine isosteres 32–36 all displayed no antiviral activity or cytotoxicity. In contrast, hydrazine 19 possessed anti-HCV activity (85% inhibition at 10 μM; EC50 = 6 μM) with no observed cytotoxicity which encouraged us to prepare its (2S)-ethyl(phenoxy)phosphorylamino)–propanoate prodrug (Fig. 3). This phosphoramidate prodrug was more active versus HCV replication with an EC50 of 0.9 μM. However stability studies of 19 in pH 7.4 phosphate buffer at 23 °C showed complete conversion of 19 to a complex mixture of products after 3 days. One of decomposition products was identified as 2′-C-methyladenosine, a known inhibitor of HCV NS5B polymerase.30
Figure 3.
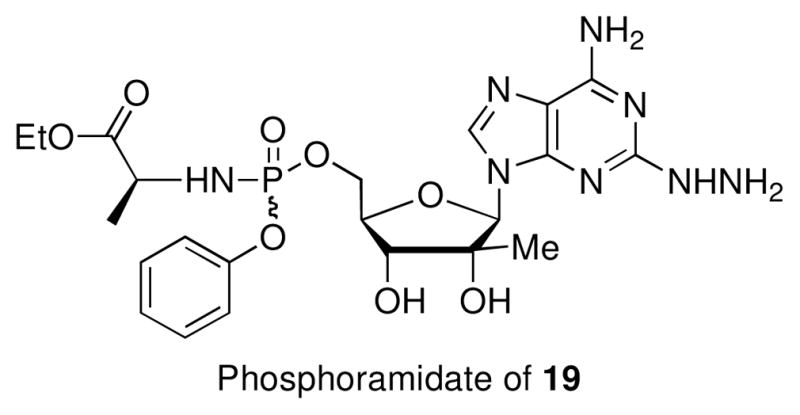
(2S)-ethyl(phenoxy)phosphorylamino)–propanoate prodrug.
The two 2-hydroxyamino nucleosides 26 and 27 displayed broad toxicity against most cell lines tested. Compounds 9, 10, 13b, 21, 32, and 35 were tested for their ability to inhibit the ADA catalyzed conversion of adenosine to inosine. These compounds were chosen due to their structural similarity to known ADA inhibitors.10,13 Only compound 21 was found effective with an average IC50 = 3.7 ± 0.4 μM. Compounds 9, 10, 21, and 32 were evaluated as potential substrates of adenosine deaminase; however, none underwent a nitrogen to oxygen transformation.
In conclusion, a variety of purine-modified 2′-C-methyl nucleosides were synthesized and evaluated as potential anti-HCV agents. Among these synthesized nucleoside analogs, none displayed potent and selective anti-HCV or anti-HIV activity. Interestingly, some of these purine analogs warrant further study as potential inhibitors of adenosine deaminase.31
Acknowledgments
This work is supported in part by NIH grant 2P30-AI-050409 (CFAR), 5R37-AI-041980, and by the Department of Veterans Affairs. Dr. R. F. Schinazi is the principal founder of RFS Pharma, LLC. His laboratory received no funding for this work from the company and vice versa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Zeuzem S, Feinman SV, Rasenack J, Heathcote EJ, Lai MY, Gane E, O’Grady J, Reichen J, Diago M, Lin A, Hoffman J, Brunda M. N Engl J Med. 2000;343:1666. doi: 10.1056/NEJM200012073432301. [DOI] [PubMed] [Google Scholar]
- 2.(a) Carroll SS, Tomassini JE, Bossernan M, Getty K, Stahlhut MW, Eldrup AB, Bhat B, Hall D, Simcoe AL, Lafemina R, Rutkowski CA, Wolanski B, Yang Z, Migliaccio G, Francesco RD, Kuo LC, MacCoss M, Olsen DB. J Biol Chem. 2003;278:11979. doi: 10.1074/jbc.M210914200. [DOI] [PubMed] [Google Scholar]; (b) Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. J Med Chem. 2005;48:5504. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 3.(a) Eldrup AB, Allerson CR, Bennett CF, Bera S, Bhat B, Bhat N, Bosserman MR, Brooks J, Burlein C, Carroll SS, Cook PD, Getty KL, MacCoss M, McMasters DR, Olsen DB, Prakash TP, Prhavc M, Song Q-L, Tomassini JE, Xia J. J Med Chem. 2004;47:2283. doi: 10.1021/jm030424e. [DOI] [PubMed] [Google Scholar]; (b) Eldrup AB, Prhavc M, Brooks J, Bhat B, Prakash TP, Song QL, Bera S, Bhat N, Dande P, Cook PD, Bennett CF, Carroll SS, Ball RG, Bosserman M, Burlein C, Colwell LF, Fay JF, Flores OA, Getty K, LaFemina RL, Leone J, MacCoss M, McMasters DR, Tomassini JE, Langen DV, Wolanski B, Olsen DB. J Med Chem. 2004;47:5284. doi: 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]
- 4.Kumar R, Sharma N, Nath M, Saffran HA, Tyrrell DL. J Med Chem. 2001;44:4225. doi: 10.1021/jm010227k. [DOI] [PubMed] [Google Scholar]
- 5.(a) Harry-O’kuru RE, Smith JM, Wolfe MS. J Org Chem. 1997;62:1754. [Google Scholar]; (b) Tamerlani G, Salsini L, Lombardi I, Bartalucci D, Cipolletti G. 2004158059. US Pat Appl Publ. 2004:12.; (c) Storer R, Moussa A, Chaudhuri N, Waligora F. WO 2004052899. PCT Int Appl. 2004:90.
- 6.Hocek M, Pohl R, Cisarova I. Eur J Org Chem. 2005;14:3026. [Google Scholar]; (b) Hocek M, Dvorakova H. J Org Chem. 2003;68:5773. doi: 10.1021/jo034351i. [DOI] [PubMed] [Google Scholar]
- 7.Spectral data for compound 6: 1H-NMR (CD3OD, 400 MHz): δ 8.46 (s, 1 H), 5.98 (s, 1 H), 4.17 (d, J = 9.2 Hz, 1 H), 4.00 (m, 1 H), 3.97 (d, J = 2.0 Hz, 1 H), 3.81 (dd, J = 2.8 Hz, J = 12.4 Hz, 1 H), 2.56 (s, 3 H), 0.92 (s, 3 H). LC/MS (m/z), calcd for C12H18N5O4 (M+ + H): 296.13; found, 296.19.
- 8.Uchiyama M, Aso Y, Noyori R, Hayakawa Y. J Org Chem. 1993;58:373. [Google Scholar]
- 9.Bobeck DR, Schinazi RF, Coats SJ. Antiviral Therapy. 2010;15:935. doi: 10.3851/IMP1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Gillerman I, Fischer B. J Med Chem. 2011;54:107. doi: 10.1021/jm101286g. [DOI] [PubMed] [Google Scholar]; (b) Šilhár P, Pohl R, Votruba I, Hocek M. Org Lett. 2004;6:3225. doi: 10.1021/ol049059r. [DOI] [PubMed] [Google Scholar]; (c) Šilhár P, Pohl R, Votruba I, Hocek M. Org Biomol Chem. 2005;3:3001. doi: 10.1039/b508122j. [DOI] [PubMed] [Google Scholar]
- 11.Qu G-R, Xia R, Yang X-N, Li J-G, Wang D-C, Guo H-M. J Org Chem. 2008;73:2416. doi: 10.1021/jo702680p. [DOI] [PubMed] [Google Scholar]
- 12.Spectral data for compound 9: 1H-NMR (CD3OD, 400 MHz): δ 8.63 (s, 1 H), 6.04 (s, 1 H), 4.27 (m, 4 H), 4.18 (d, J = 9.2 Hz, 1 H), 3.97 (m, 2 H), 3.81 (dd, J = 3.2 Hz, J = 12.8 Hz, 1 H), 1.33 (t, J = 7.2 Hz, 6 H), 0.94 (s, 3 H). 13C-NMR (CD3OD, 400 MHz): δ 158.97, 152.61, 149.80, 147.58, 141.32, 89.56, 81.47, 77.54, 70.59, 62.46, 58.15, 17.45, 13.91. LC/MS (m/z), calcd for C15H25N5O7P (M+ + H), 418.14; found, 418.33.
- 13.Silverman LS, Caldwell JP, Greenlee WJ, Kiselgof E, Matasi JJ, Tulshian DB, Arik L, Foster C, Bertorelli R, Monopoli A, Ongini E. Bioorg & Med Chem Lett. 2007;17:1659. doi: 10.1016/j.bmcl.2006.12.104. [DOI] [PubMed] [Google Scholar]
- 14.Baraldi PG, Tabrizi MA, Gessi S, Borea PA. Chem Rev. 2008;108:238. doi: 10.1021/cr0682195. [DOI] [PubMed] [Google Scholar]
- 15.(a) Unciti-Broceta A, Infantas MJ, Gallo MA, Espinosa A. Chem Eur J. 2007;13:1754. doi: 10.1002/chem.200600282. [DOI] [PubMed] [Google Scholar]; (b) Kos NJ, Jongejan H, Van der Plas HC. Gazz Chim Ital. 1987;117:369. [Google Scholar]
- 16.(a) Gupta M, Nair V. Tetrahedron Lett. 2005;46:1165. doi: 10.1016/j.tetlet.2005.04.143. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohgo S, Ohrui H, Kodama E, Matsuoka M, Mitsuya H. 20090234110. US Pat Appl Publ. 2009:25.
- 17.(a) Xu Y, Ikeda R, Sugiyama H. J Am Chem Soc. 2003;125:13519. doi: 10.1021/ja036233i. [DOI] [PubMed] [Google Scholar]; (b) Schaeffer HJ. 19804199574. US Pat. 1980:18.
- 18.(a) Lioux T, Gosselin G, Mathé C. Eur J Org Chem. 2003:3997. [Google Scholar]; (b) Sodum RS, Fiala ES. Chem Res Toxicol. 1998;11:1453. doi: 10.1021/tx980160c. [DOI] [PubMed] [Google Scholar]; (c) Elzein E, Kalla R, Li X-f, Perry T, Marquart T, Micklatcher M, Li Y, Wu Y-Z, Zeng D, Zablocki J. Bioorg Med Chem Lett. 2007;17:161. doi: 10.1016/j.bmcl.2006.09.065. [DOI] [PubMed] [Google Scholar]
- 19.Spectral data for compound 21: 1H-NMR (CD3OD, 400 MHz): δ 8.79 (d, J = 1.2 Hz, 1 H), 8.59 (s, 1 H), 7.87 (d, J = 1.2 Hz, 1 H), 6.16 (s, 1 H), 4.18 (d, J = 9.2 Hz, 1 H), 4.00 (m, 2 H), 3.86 (dd, J = 3.2 Hz, J = 12.8 Hz, 1 H), 0.96 (s, 3 H). 13C-NMR (CD3OD, 400 MHz): δ 156.96, 149.80, 149.56, 140.32, 133.57, 123.63, 118.39, 91.87, 83.21, 79.03, 72.28, 59.85, 19.07. LC/MS (m/z), calcd for C13H17N8O4 (M+ + H), 349.13; found, 349.16.
- 20.Cosyn L, Palaniappan KK, Kim SK, Duong HT, Gao ZG, Jacobson KA, Calenbergh SV. J Med Chem. 2006;49:7373. doi: 10.1021/jm0608208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coats SJ, Link JS, Gauthier D, Hlasta D. Org Lett. 2005;7:1469. doi: 10.1021/ol047637y. [DOI] [PubMed] [Google Scholar]
- 22.Wanner MJ, KuÈnzel JK, Jzerman IAP, Koomen G-J. Bioorg Med Chem Lett. 2000;10:2141. doi: 10.1016/s0960-894x(00)00415-7. [DOI] [PubMed] [Google Scholar]
- 23.Wanner MJ, Koomen G-J. J Chem Soc, Perkin Trans. 2001;11908 [Google Scholar]
- 24.Ujjinamatada RK, Paulman RL, Ptak RG, Hosmane RS. Bioorg Med Chem. 2006;14:6359. doi: 10.1016/j.bmc.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 25.(a) Ujjinamatada RK, Phatak P, Burger AM, Hosmane RS. J Med Chem. 2008;51:694. doi: 10.1021/jm700931t. [DOI] [PubMed] [Google Scholar]; (b) Berry DA, Wotring LL, Drach JC, Townsend LB. Nucleosides Nucleotides. 1994;13:2001. [Google Scholar]
- 26.Rondla R, Coats SJ, McBrayer TR, Grier J, Johns M, Tharnish PM, Whitaker T, Zhou L-H, Schinazi RF. Antivir Chem Chemother. 2009;20:99. doi: 10.3851/IMP1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuyver LJ, Whitaker T, McBrayer TR, Hernandez-Santiago BI, Lostia S, Tharnish PM, Ramesh M, Chu CK, Jordan R, Shi J, Rachakonda S, Watanabe KA, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2003;47:244. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie MW, Hart GC, Smith GA, Hahn EF. Antimicrob Agents Chemother. 1990;34:1061. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The assay was performed by combining 140 μM of adenosine (final concentration) and 0.01 units of adenosine deaminase (Sigma catalog # A5043) in 50 mM potassium phosphate (pH 7.4). Compounds were tested at 10 μM. Absorbance @ 265 nm was measured over four minutes and the slope was observed. Changes in the slope by incubation with compound versus no compound indicate inhibition as the slope approaches zero.
- 30.Sodum RS, Fiala SE. Chem Res Toxicol. 1998;11:1453. doi: 10.1021/tx980160c. [DOI] [PubMed] [Google Scholar]
- 31.(a) Koscsó B, Csóka B, Pacher P, Haskó G. Expert Opin Investig Drugs. 2011;20:757. doi: 10.1517/13543784.2011.573785. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Antonioli L, Fornai M, Colucci R, Tuccori M, Blandizzi C. Expert Opin Investig Drugs. 2011;20:717. doi: 10.1517/13543784.2011.579104. [DOI] [PubMed] [Google Scholar]