

Abstract
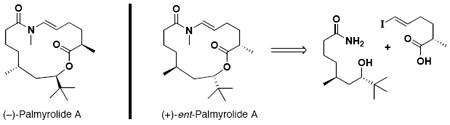
The first asymmetric total synthesis and determination of the absolute configuration for the neuroactive marine macrolide Palmyrolide A is described. The highlight of the synthesis is macrocyclization via trans-enamide formation catalyzed by copper (I) iodide and cesium carbonate. Comparison with the authentic spectral data confirms the synthesis of (+)-ent-Palmyrolide A.
The study of marine organisms continues to provide natural products with intricate molecular architecture, unique biological activity, and great potential for use as modern chemotherapeutics. Recently, Gerwick and coworkers reported the isolation and structural elucidation of palmyrolide A (1), a neuroactive macrolide from a cyanobacterial assemblage comprised of Leptolyngbya and Oscillatoria species collected at Palmyra Atoll, south of Hawaii.1 Initial biological studies revealed 1 to be a potent inhibitor of calcium ion oscillations in murine cerebrocortical neurons and to possess sodium ion channel blocking ability in neuroblastoma cells.1 When screened against human lung adenocarcinoma cells, the authors note no appreciable cytotoxicity.1
The overall connectivity of (−)-palmyrolide A was determined by detailed NMR studies.1 However, due to the resistance of the lactone to hydrolyze under a variety of reaction conditions, the authors were unable to degrade the macrolide into acyclic fragments that would prove useful in determining the absolute stereochemistry.1 As a result, the authors performed the Murata J-based configurational analysis2 on the macrocycle itself to determine the relative stereochemistry between the C(5) methyl and the C(7) t-butyl centers. This data, in conjunction with NOE correlations, suggested the relationship between C(5) and C(7) was syn.1
We became interested in (−)-palmyrolide A as a synthetic target not only because of its interesting biological profile, but also due to the presence of two unique structural elements − the rare t-butyl moiety and the trans-N-methyl enamide. A search of the literature reveals few examples of isolated natural products that contain a sterically-encumbered t-butyl group α to the lactone ester,3 with (−)-apratoxin A being the sole example confirmed by total synthesis.4 It should be noted that for apratoxin, the relative stereochemistry between its C(37) methyl and C(39) t-butyl is anti.5 In the case of palmyrolide A, Gerwick speculates that the t-butyl structural unit may be evolutionary, developed as a way to prevent hydrolysis of the bio-active lactone moiety under natural conditions.1
N-Methyl enamide macrocycles are exceedingly rare in the natural product literature, with few reported examples;3b-c none have been confirmed by total synthesis. Interestingly, these compounds all contain a t-butyl group α to the lactone ester within their molecular framework, and are likely derived from the same genus of cyanobacteria.1 A related family of macrolides contains an N-H enamide,6 although with cis olefin geometry. Several other compounds have sidechains decorated with N-H enamides,7 however to the best of our knowledge, there is only one that features an N-methyl enamide subunit.7f
Due to uncertainty regarding the absolute configuration of palmyrolide A,1 at the outset of our synthetic campaign we decided to target all possible diastereomeric combinations. While Gerwick identified the relative configuration between the C(5) methyl and the C(7) t-butyl to be syn,1 based on the apratoxin A literature,3a, 4 we believed that the relationship between these two groups could also be anti. In order to most efficiently address the unknown absolute stereochemistry, we decided to exploit a synthetic route that would allow us to synthesize all diastereomers concurrently. Herein, we report our work on the C(5)-C(7) anti series.
In the isolation report, the absolute stereochemistry of the C(10) methyl group was unequivocally assigned to be in the R configuration.1 Rather than design our synthetic plan around the known C(10) center, we thought that a more economical approach would be to target only one of the two possible C(5)-C(7) anti combinations, and then vary the stereochemistry at C(10). In this way, we would utilize a common fragment (cf. 2) to gain access to both anti diastereomeric combinations of palmyrolide A (1a and 1b, Scheme 1) representing one compound from each enantiomeric set. We chose the C(5)-S, C(7)-S arrangement, based on guidance from Gerwick,8 to coincide with the absolute stereochemistry found in apratoxin A.3a
Scheme 1.
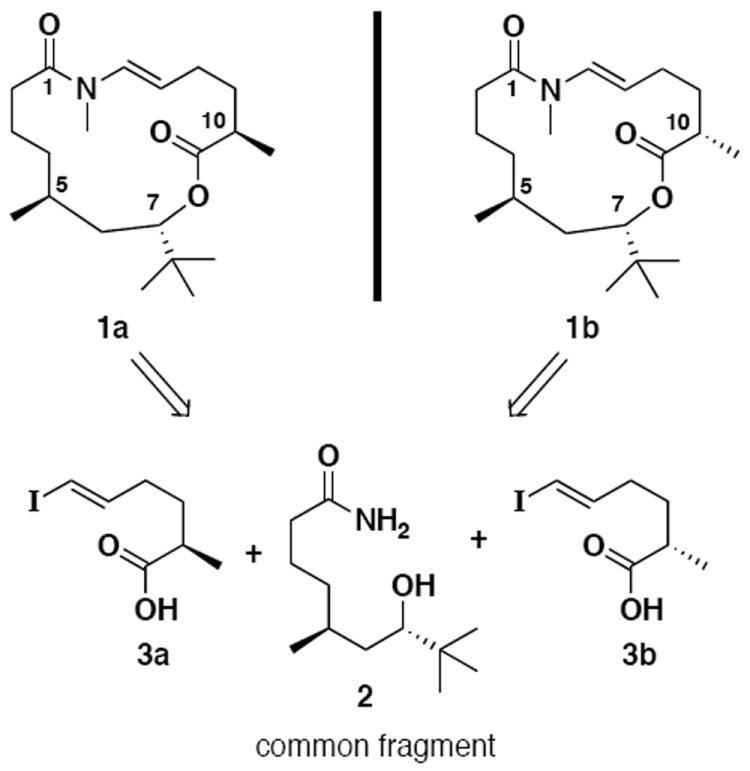
Retrosynthetic Analysis
Retrosynthetically, we believed that macrocyclization exploiting a method to unite a primary amide with a vinyl iodide would prove facile.9-10 A related macrocyclization has been documented,9j however to the best of our knowledge, there has been no use of this strategy for the construction of a 15-membered macrocycle, or for the formation of a trans enamide. Further simplification reveals amide alcohol 2, which is common to both targets, and vinyl iodides 3a and 3b (Scheme 1). These fragments could be combined in the forward direction under mild reaction conditions via formation of a mixed-anhydride.
To establish the key anti-stereorelationship between C(5) and C(7), we relied on elegant chemistry developed by Cavelier and coworkers during their recent synthesis of oxoapratoxin,11 an oxazoline analogue of apratoxin A. The synthesis of fragment 2 commenced with a D-proline catalyzed asymmetric aldol union between pivaldehyde and acetone to furnish β-hydroxy ketone (−)-4, following the known literature account11 (Scheme 2). In the Cavelier studies, stereoselective syn-reduction of (−)-4 was affected using diethylmethoxyborane/NaBH4,12 which provided an acceptable mixture of syn and anti diastereomers (95:5). Unfortunately, in our hands this reduction strategy did not yield a synthetically workable mixture of isomers. Chromatographic separation also proved difficult. Pleasingly, recourse to a stereoselective reduction using 2.5 equivalents of DIBAl-H13 provided the requisite diol in excellent yield and high diastereoselectivity (Scheme 2). This modification also obviated the need for a challenging silica gel purification step. Next, in two synthetic operations involving: 1) treatment with thionyl chloride in pyridine, and 2) oxidation using RuO4, the syn-diol was easily converted into cyclic sulfate (−)-5 in good overall yield.14
Scheme 2.
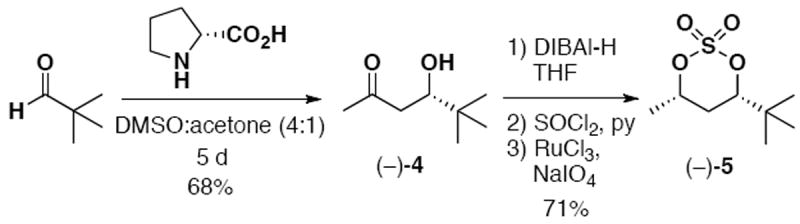
Synthesis of Cyclic Sulfate 5
Nucleophilic ring opening of (−)-515 using a mixed organometallic species derived from allylmagnesium bromide and copper iodide, following the procedure of Cavelier,11 and alcohol protection with TESCl provided alkene (−)-6, possessing the anti-C(5)-S, C(7)-S stereochemical arrangement we targeted (Scheme 3).16 Of note, the triethylsilyl group in (−)-6 is the only protecting group employed during our synthesis of palmyrolide A.
Scheme 3.
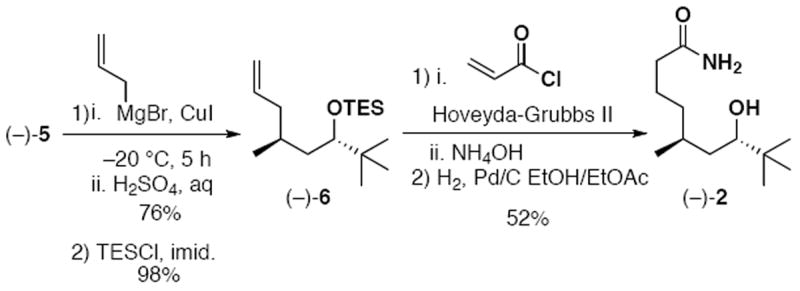
Synthesis of Fragment 2
Union of (−)-6 with freshly distilled acryloyl chloride17a employing the Hoveyda-Grubbs II precatalyst, followed by in situ addition of ammonium hydroxide allowed access to the primary amide. The analogous cross metathesis using acrylamide and the Grubbs II precatalyst17b was also tried, however this process gave good but somewhat inferior results compared to acryloyl chloride (36% vs. 58%, respectively). In the next step, hydrogenation of the α,β-unsaturation occurred with concomitant loss of the labile triethylsilyl group,18 and provided fragment (−)-2 in good overall yield (Scheme 3).
It should be noted that during the cross metathesis step, trace HCl from the acryloyl chloride is sufficient to cleave some of the silyl ether protecting group. As a result, the balance in chemical yield is byproduct arising from free alcohol esterification with acryloyl chloride. Unfortunately, attempts to hydrolyze the ester were met with no success. We believe this is due to the steric constraints of the neighboring t-butyl group preventing nucleophilic attack on the ester carbonyl. This result is consistent with the reported unsuccessful attempts to hydrolyze the lactone of palmyrolide A.1
With amide alcohol 2 in hand, we turned our attention to the fabrication of vinyl iodides 3a and 3b. The syntheses of these fragments began employing known alcohol 7, produced in three literature operations from commercially available 3-butyn-1-ol.19 Alcohol to iodide interconversion proceeded in high yield, and the resultant diiodide (8) was used in the Myers alkylation20 with (S,S)-propionamide 9a and separately with (R,R)- propionamide 9b (Scheme 4). Pleasingly, both alkylations proceeded in good yield and in high diastereoselectivity (>20:1 by 1H NMR). These yields are consistent with the Myers studies which noted limited reactivity with substrates bearing β-alkyl branching or alkoxy groups.20 We believe the presence of the β-vinyl iodide of 8 may be responsible for the diminished yields we observed relative to unhindered literature examples.20 Subsequent hydrolysis with NaOH provides acids (−)-3a and (+)-3b,21 with negligible loss of stereopurity, and complete preservation of the trans vinyl iodide unit.22
Scheme 4.
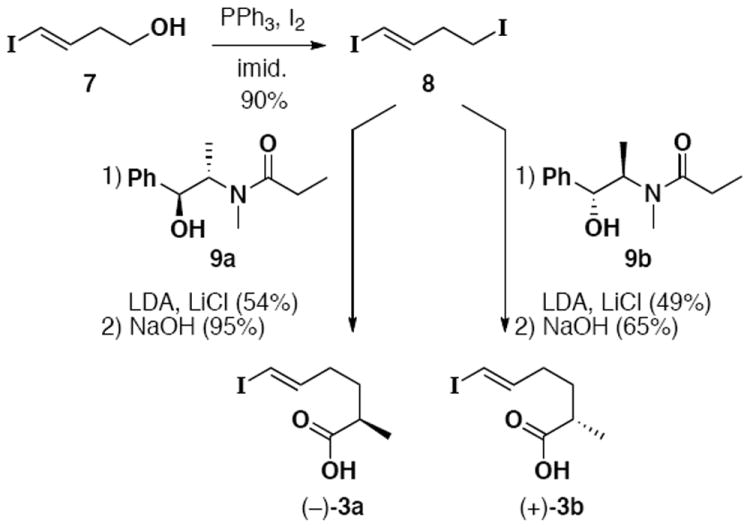
Assembly of Vinyl Iodides 3a and 3b
Union of the common amide alcohol fragment (2) with either (−)-3a or (+)-3b, exploiting 2,4,6-trichlorobenzoyl chloride as coupling agent,23 allowed access to macrocyclization precursors (−)-10a and (−)-10b, respectively (Scheme 5). Formation of the 15-membered ring, using a high-dilution modification (0.01 M) to the copper (I) iodide / cesium carbonate conditions developed by Buchwald9d afforded the trans-N-H enamide macrocycles in modest yield. To the best of our knowledge, this is the first use of the copper-promoted reaction conditions for the formation of trans-enamide macrocycles. In the final step, treatment of each macrocycle separately with sodium hydride followed by iodomethane24 provided the requisite trans-N-methyl enamides (+)-1a and (+)-1b (Scheme 5) in excellent yield.
Scheme 5.
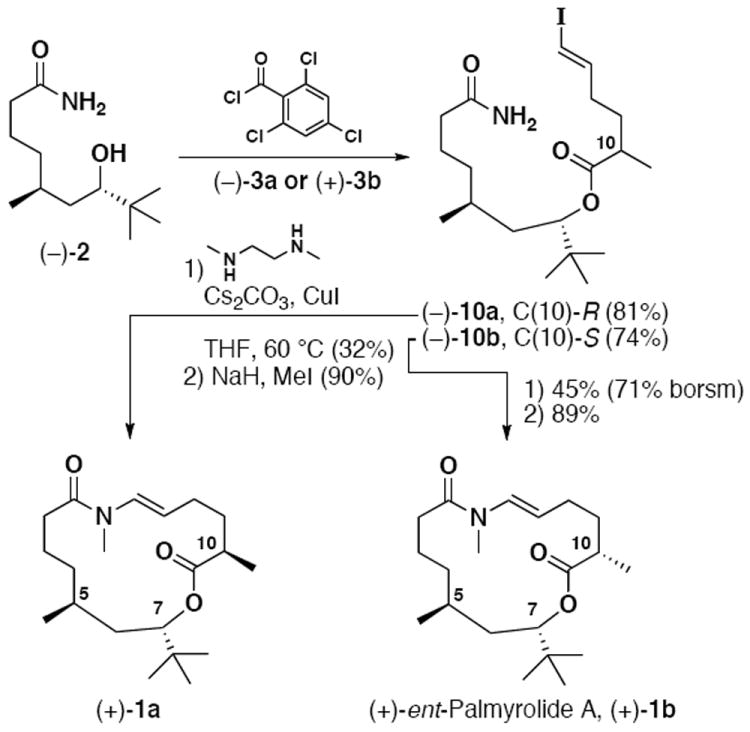
Synthesis of Macrocycles 1a and 1b
At this stage, a comparison with the reported spectra of palmyrolide A1 was made. Compound (+)-1a, featuring the natural C(10)-R stereochemistry, did not match the literature values reported by Gerwick. Pleasingly, there was a complete 1H and 13C NMR match with macrolide (+)-1b, where the C(10) methyl group is inverted relative to the natural macrolide. Optical rotation comparison confirms that we have synthesized the enantiomer of (−)-palmyrolide A, (+)-ent-palmyrolide A {[α]D = + 23 (c = 0.65, CHCl3), lit. [α]D = − 29 (c = 0.9, CHCl3)}.1 The longest linear sequence is 11 steps from commercially available starting materials, or 6 steps from known silyl ether (−)-6. Importantly, we have confirmed that the stereochemical relationship between the C(5) methyl and the C(7) t-butyl is anti, and not syn, as originally proposed in the isolation report.1 Also of interest, the absolute stereochemistry at C(5) and C(7) found in (−)-palmyrolide A is opposite that found in apratoxin A.3a
A full account of this work, including syntheses of the complementary C(5) methyl, C(7) t-butyl syn series, and the synthesis of natural (−)-palmyrolide A, will be reported in due course.
Supplementary Material
Acknowledgments
We would like to thank New Mexico State University for providing financial assistance in terms of a startup package, as well as the NMSU-MBRS Research Initiative for Scientific Enhancement (NIH R25GM061222) and NMSU-Howard Hughes Medical Institute (Grant # 52006932) programs for providing salary support (CKV and EMJ, respectively). Finally, we would like to thank Professor William Gerwick (UCSD) and Dr. Alban Pereira (UCSD) for helpful discussions, and for providing FIDs of their palmyrolide A work.
Footnotes
This manuscript is dedicated to Professor Amos B. Smith, III, with admiration and respect.
Supporting Information Available Detailed experimental procedures, as well as scans of 1H and 13C NMR spectra are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pereira AR, Cao Z, Engene N, Soria-Mercado IE, Murray TF, Gerwick WH. Org Lett. 2010;12:4490–4493. doi: 10.1021/ol101752n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. J Org Chem. 1999;64:866–876. doi: 10.1021/jo981810k.. (b) This method was successfully applied to the structure elucidation of the (−)-apratoxin A macrolide. See reference 3a.
- 3.(a) Luesch H, Yoshida WY, Moore RE, Paul VJ, Corbett TH. J Am Chem Soc. 2001;123:5418–5423. doi: 10.1021/ja010453j. [DOI] [PubMed] [Google Scholar]; (b) Klein D, Braekman JC, Daloze D, Hoffmann L, Castillo G, Demoulin V. J Nat Prod. 1999;62:934–936. doi: 10.1021/np9900324. [DOI] [PubMed] [Google Scholar]; (c) Matthew S, Salvador LA, Schupp PJ, Paul VJ, Luesch H. J Nat Prod. 2010;73:1544–1552. doi: 10.1021/np1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For the most recent total synthesis, see: Numajiri Y, Takahashi T, Doi T. Chem Asian J. 2009;4:111–125. doi: 10.1002/asia.200800365.. For the first total synthesis, see: Chen J, Forsyth CJ. J Am Chem Soc. 2003;125:8734–8735. doi: 10.1021/ja036050w.
- 5.For the apratoxin numbering convention, see reference 3a.
- 6.For a review article showcasing several examples, see: Gournelis DC, Laskaris GG, Verpoorte R. Nat Prod Rep. 1997;14:75–82. doi: 10.1039/np9971400075.. See also: Lin H-Y, Chen C-H, You B-J, Liu KCSC, Lee S-S. J Nat Prod. 2000;63:1338–1343. doi: 10.1021/np000136a.
- 7.For several examples, see: Erickson KL, Beutler JA, Cardellina JH, II, Boyd MR. J Org Chem. 1997;62:8188–8192. doi: 10.1021/jo971556g.Galinis DL, McKee TC, Pannell LK, Cardellina JH, II, Boyd MR. J Org Chem. 1997;62:8968–8969.McKee TC, Galinis DL, Pannell LK, Cardellina JH, II, Laakso J, Ireland CM, Murray L, Capon RJ, Boyd MR. J Org Chem. 1998;63:7805–7810.Kim JW, Shin-ya K, Furihata K, Hayakawa Y, Seto H. J Org Chem. 1999;64:153–155. doi: 10.1021/jo9814997.Kohno J, Koguchi Y, Nishio M, Nakao K, Kuroda M, Shimizu R, Ohnuki T, Komatsubara S. J Org Chem. 2000;65:990–995. doi: 10.1021/jo991375+.Nogle LM, Gerwick WH. Org Lett. 2002;4:1095–1098. doi: 10.1021/ol017275j.
- 8.Personal communication. 2011 [Google Scholar]
- 9.For copper-catalyzed vinyl halide / amide unions, see: Ogawa T, Kiji T, Hayami K, Suzuki H. Chem Lett. 1991:1443–1446.Shen R, Porco JA., Jr Org Lett. 2000;2:1333–1336. doi: 10.1021/ol005800t.Shen R, Lin CT, Porco JA., Jr J Am Chem Soc. 2002;124:5650–5651. doi: 10.1021/ja026025a.Jiang L, Job GE, Klapars A, Buchwald SL. Org Lett. 2003;5:3667–3669. doi: 10.1021/ol035355c.Nakamura R, Tanino K, Miyashita M. Org Lett. 2003;5:3583–3586. doi: 10.1021/ol0352299.Han C, Shen R, Su S, Porco JA., Jr Org Lett. 2004;6:27–30. doi: 10.1021/ol0360041.Coleman RS, Liu P-H. Org Lett. 2004;6:577–580. doi: 10.1021/ol036381d.Pan X, Cai Q, Ma D. Org Lett. 2004;6:1809–1812. doi: 10.1021/ol049464i.Shen R, Inoue T, Forgac M, Proco JA., Jr J Org Chem. 2005;70:3686–3692. doi: 10.1021/jo0477751.Toumi M, County F, Evano G. Angew Chem Int Ed. 2007;46:572–575. doi: 10.1002/anie.200602865.Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054–3131. doi: 10.1021/cr8002505.
- 10.For a good review of the enamide literature pre-2000, see 10b and references cited therein. For select methods post-2000, see: Fürstner A, Brehm C, Cancho-Grande Y. Org Lett. 2001;3:3955–3957. doi: 10.1021/ol016848p.Wallace DJ, Klauber DJ, Chen C-y, Volante RP. Org Lett. 2003;5:4749–4752. doi: 10.1021/ol035959g.Brice JL, Meerdink JE, Stahl SS. Org Lett. 2004;6:1845–1848. doi: 10.1021/ol0494360.Yudha SS, Kuninobu Y, Takai K. Org Lett. 2007;9:5609–5611. doi: 10.1021/ol702564e.Bolshan Y, Batey RA. Angew Chem. 2008;120:2139–2142. doi: 10.1002/anie.200704711.Arndt M, Salih KSM, Fromm A, Goossen LJ, Menges F, Niedner-Schatteburg G. J Am Chem Soc. 2011;133:7428–7449. doi: 10.1021/ja111389r.Nicolaou KC, Jiang X, Lindsay-Scott PJ, Corbu A, Yamashiro S, Bacconi A, Fowler VM. Angew Chem Int Ed. 2011;50:1139–1144. doi: 10.1002/anie.201006780.
- 11.Gilles A, Martinez J, Cavelier F. J Org Chem. 2009;74:4298–4304. doi: 10.1021/jo900583j. [DOI] [PubMed] [Google Scholar]
- 12.Chen KM, Gunderson KG, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Chem Lett. 1987:1923–1926. [Google Scholar]
- 13.Kiyooka S-i, Kuroda H, Shimasaki Y. Tetrahedron Lett. 1986;27:3009–3012. [Google Scholar]
- 14.Gao Y, Sharpless KB. J Am Chem Soc. 1988;110:7538–7539.. Also see reference 11.
- 15.For a review article documenting the reactivity of cyclic sulfates see: Byun H-S, He L, Bittman R. Tetrahedron. 2000;56:7051–7091.
- 16.Silyl ether (−)-6 is a known compound. See reference 11.
- 17.(a) Ferrié L, Bouzbouz S, Cossy J. Org Lett. 2009;11:5446–5448. doi: 10.1021/ol9021386. [DOI] [PubMed] [Google Scholar]; (b) Choi T-L, Chatterjee AK, Grubbs RH. Angew Chem Int Ed. 2001;40:1277–1279. doi: 10.1002/1521-3773(20010401)40:7<1277::aid-anie1277>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 18.Sajiki H, Ikawa T, Hattori K, Hirota K. Chem Commun. 2003:654–655. [PubMed] [Google Scholar]
- 19.Munakata R, Ueki T, Katakai H, Takau K, Tadano K-i. Org Lett. 2002;3:3029–3033. doi: 10.1021/ol016449u.Trend RM, Ramtohul YK, Stoltz BM. J Am Chem Soc. 2005;127:17778–17788. doi: 10.1021/ja055534k.. Alcohol 7 can also be prepared in one synthetic step from 3-butyn-1-ol. See: Huang Z, Negishi E-i. Org Lett. 2006;8:3675–3678. doi: 10.1021/ol061202o.
- 20.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J Am Chem Soc. 1997;119:6496–6511. [Google Scholar]
- 21.The yield of (+)-3b is unoptimized.
- 22.Elimination of Z-vinyl iodides readily occurs upon treatment with NaOMe. For example, see reference 19a
- 23.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 24.Gooβen LJ, Blanchot M, Arndt M, Salih KSM. Synlett. 2010:1685–1687. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.