

Palladium-catalyzed cross-coupling reactions of aryl halides with aromatic C–H bonds have emerged as a powerful method for the preparation of biaryls.[1,2] Despite substantially increased attention to the field, typical reaction conditions still require high temperatures (> 120°C) for insertion into aromatic C–H bonds, which can be viewed as a major drawback to this chemistry. Such forcing conditions often appear to be critical to overcoming the low reactivity of aryl C–H bonds. A much milder C–H activation reaction at ambient temperatures would, in particular, likely be more dependent on activation by the catalyst.[3]
Although there are many ortho-directing groups for C–H activation reactions,[1] the amide residue in anilides is especially attractive as a coupling partner for the synthesis of valuable aniline derivatives. In 1984, Tremont and co-workers used acetanilides for C–H alkylation with alkyl iodides, albeit promoted by stoichiometric Pd(OAc)2.[4] Both the Daugulis[5] and Sanford[6] groups have demonstrated Pd-catalyzed ortho-arylations of anilides with aryl iodides or iodonium salts at temperatures above 100°C. Moreover, ortho-directed C–H activation can suffer from double arylations with respect to the directing group.[1,5] C–H arylations of reactive indoles have been reported at room temperature,[7] but to the best of our knowledge C–H arylation of anilide derivatives with aryl halides at ambient temperatures have not yet been achieved.[1,8] Herein, we describe the first room temperature mono-C–H activation of urea derivatives and their cross-couplings with aryl iodides in water (Scheme 1). This methodology provides a convenient route to various aniline derivatives by means of C–H activation under mild conditions.
Scheme 1.

C–H activation at room temperature in water.
Optimization studies employed the combination of anilides (1a–f) and 4-iodoanisole (2a, 2 equiv) in the presence of Pd(OAc)2 (10 mol%), AgOAc (2 equiv) and aqueous HBF4 (5 equiv) in 2 wt% surfactant/water solutions at room temperature (Table 1). The effectiveness of various directing groups was initially examined, and among a number of different anilide derivatives 1a–f explored, only the aromatic urea 1f smoothly underwent C–H arylation at room temperature (Table 1, runs 1–6). Recently, Lloyd-Jones and Booker-Milburn have also found aryl ureas to be more active coupling partners for C–H functionalizations than other anilides.[9] Acetanilide 1a reacted with 2a only upon heating to 50°C. Pivaloylanilide 1c has been reported as an effective directing group at 130°C,[1,2,5] but gave a low yield under these room temperature conditions (run 3). Generally, acetic acid or trifluoroacetic acid (TFA) is required to carry out C–H activation;[1,2] in this case, HBF4 was found to be critical for generation of biaryl 3 in good yield (run 6).
Table 1.
Optimization of C–H arylations at room temperature.[a]
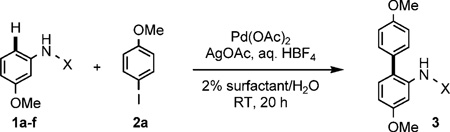 | ||||
|---|---|---|---|---|
| Run | X | Surfactant | Yield [%] | |
| 1 | COMe | 1a | PTS | trace |
| 2 | COiPr | 1b | PTS | trace |
| 3 | COtBu | 1c | PTS | 24 |
| 4 | CSNMe2 | 1d | PTS | 0 |
| 5 | COCF3 | 1e | PTS | trace |
| 6 | CONMe2 | 1f | PTS | 67(0,[b] 0,[c] 0,[d] 60[e]) |
| 7 | CONMe2 | 1f | Triton X-100 | 73 (47,[f] 20[g]) |
| 8 | CONMe2 | 1f | Solutol | 65 |
| 9 | CONMe2 | 1f | Brij 35 | 76 |
| 10 | CONMe2 | 1f | Brij 30 | 72 |
| 11 | CONMe2 | 1f | TPGS | 73 |
| 12 | CONMe2 | 1f | Cremophor EL | 40 |
| 13 | CONMe2 | 1f | none | 35 |
Conducted at room temperature for 20 h in 2 wt% surfactant/water with 10 mol% Pd(OAc)2, AgOAc (2 equiv), aq. HBF4 (5 equiv), 1 (0.25 mmol), and 2 (2 equiv).
AcOH (instead of HBF4).
HCl.
TFA.
TsOH.
1.2 equiv AgOAc.
3 mol% catalyst.
Although use of the surfactant PTS[10] gave good yields, comparable results were realized with several commercially available amphiphiles. Best yields were obtained using 2 wt% Brij35 in water (Table 1, runs 7–13). Reduced amounts of HBF4, silver salt, or palladium catalyst led to lower yields. A plausible rationale for these results involves generation of a highly active cationic palladium species (Scheme 2).[7c,11]
Scheme 2.

Generation of a cationic palladium(II) species.
As illustrated by several representative examples in Table 2, the scope of this transformation is broad, applying to aryl urea derivatives and aryl iodides bearing a variety of functional groups with yields in the 70–97% range, all done in water at room temperature. Under these mild conditions, only mono-arylated products of net substitution were typically obtained.
Table 2.
Products from reactions of aryl ureas with aryl iodides.[a]
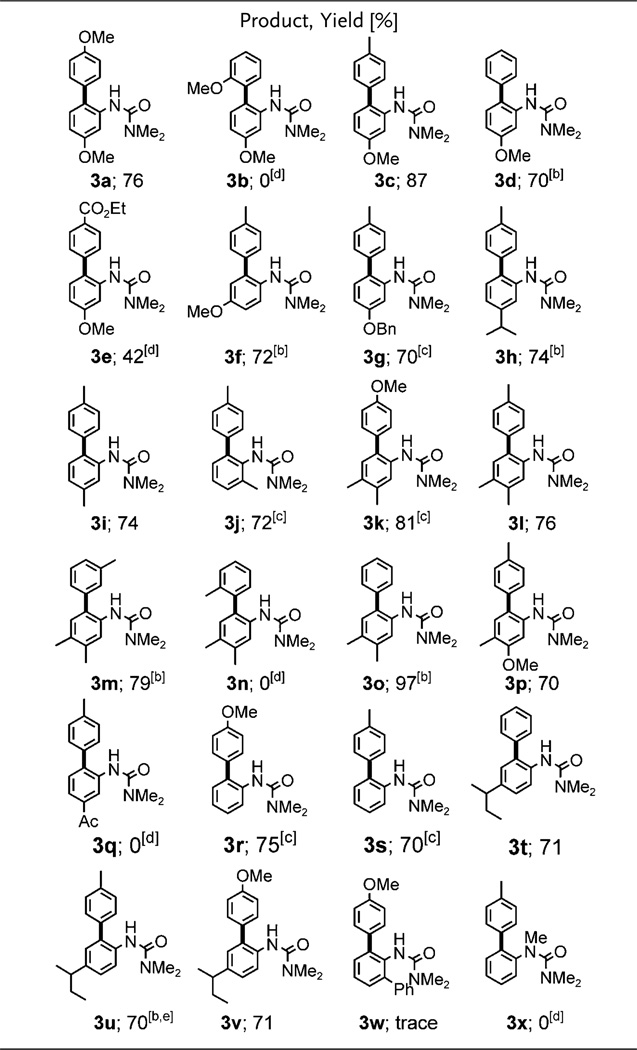 |
Conducted at room temperature for 20 h in 2 wt% Brij 35/water with 10 mol% Pd(OAc)2, AgOAc (2 equiv), aq. HBF4 (5 equiv), 1 (0.25 mmol), and 2 (2 equiv).
Run for 48 h.
Run for 72 h.
Run for 96 h.
1.2 equiv of ArI.
Especially noteworthy are aniline derivatives lacking ortho- or meta-substitution, which have previously been shown to be prone to double arylation. Under these conditions, couplings are selective for singly arylated products (3f, 3r, 3s, 3v). Only reactions of the aryl urea bearing a 4-sec-butyl group with phenyliodide and 4-tolyl iodide produced small amounts (10–15% yields) of doubly arylated products. Reduced aryl iodide loading or reaction time, however, suppressed double arylation to less than 3% (3t, 3u).
While current reaction conditions were effective for a variety of substrates, products resulting from sterically hindered aryl iodides having ortho-substituents, such as 2-anisyl iodide and 2-tolyl iodide, were not formed (3b, 3n). The reactivity of N-methyl substituted ureas (e.g., 3x) appears to be much lower than that of their non-N-methyl-substituted analogues, possibly due to palladium coordination in the initial C–H activation step. Electron-deficient ureas (e.g., 3q) were also inert, suggesting that electrophilic attack of cationic palladium may be critical for activating aromatic C–H bonds. The reactivity of aryl iodides possessing electron-rich groups is also much higher than that of more electron-withdrawing aryl iodides (3a vs. 3e).
Further advantage can also be taken of the reaction conditions associated with these cross-couplings to allow for tandem processes. Thus, in the presence of silver nitrate, arylation afforded a product of type 3 exclusively, following standard treatment with hydrogen carbonate (Scheme 3). Without exposure to this aqueous workup, nitrated biaryl 4 was isolated. Since use of silver acetate gave only arylated product 3a regardless of quenching conditions, the potential for carrying out secondary electrophilic aromatic substitution could readily be demonstrated. Simple introduction of bromine prior to workup afforded the C-arylated, regiospecifically brominated adduct 5 in good overall isolated yield (70%). The identities of products 3a and 4 were confirmed by X-ray analyses (see Supporting Information).
Scheme 3.

Tandem C–H arylation–electrophilic trapping.
While the exact reaction mechanism is currently unclear, one possibility involves a cationic PdII complex-catalyzed electrophilic C–H activation step.[7,11] Nevertheless, the reaction of 1f and 2a (see Table 2) in the presence of [Pd(MeCN)4](BF4)2, a commercially available cationic palladium(II) complex, did not result in the formation of product (Scheme 4, top). It was found, however, that adding 40 mol% MeCN under the standard, optimized, and otherwise successful conditions (cf. Table 2, product 3a; 76% yield), only traces of product formation was observed (Scheme 4, bottom). This suggests that the low reactivity of the pre-formed cationic palladium complex may actually be due to suppression of the reaction by MeCN coordination to the Lewis acidic PdII.
Scheme 4.

Effect of cationic palladium species.
With the goal of generating a highly active cationic PdII complex without the aid of strong acid, and in the absence of coordinating ligands, the combination of Pd(OAc)2 and AgBF4 was examined (Scheme 5). As expected, these conditions led to C–H activation. Unlike the reaction with AgOAc, the reaction with AgBF4 produced the corresponding C–H arylated product 3a without assistance of external acid at room temperature. This result, under such mild conditions, is indicative of the potential for highly active cationic palladium species to serve as especially effective catalysts for C–H arylation reactions. The silver salt may not only weaken the C–I bond and/or function as halogen scavenger, but may also play an important role in the generation of cationic palladium(II) species.
Scheme 5.

C–H activation without acids at room temperature in water.
In summary, the first room temperature C–H arylation of anilides with aryl iodides to give biaryl derivatives in good yields is described. These are accomplished using aryl urea derivatives, and are all done in water in the absence of phosphine ligands. Further studies of metal-catalyzed C–H activation reactions at room temperature, including both Heck couplings and mechanistic studies, are currently under investigation.
Experimental Section
General procedure
Aryl urea 1 (0.25 mmol), aryl iodide 2 (0.5 mmol), AgOAc (0.5 mmol, 83 mg), and Pd(OAc)2 (0.025 mmol, 5.6 mg) were sequentially added in air to a reaction tube equipped with a stir bar and a septum. The aqueous solution containing the surfactant (1.0 mL, 2 wt%), and 48 wt% HBF4 solution (1.25 mmol, 0.16 mL) were added by syringe and vigorously stirred for 20 h. After completion, the contents of the flask were quenched with NaHCO3 and extracted with EtOAc. The solution obtained was filtered through a plug of silica gel and anhydrous MgSO4, and concentrated by rotary evaporation. The residue was purified by flash chromatography eluting with hexane/EtOAc to afford the product.
Supplementary Material
Footnotes
Financial support provided by the NIH (GM 86485) is gratefully acknowledged.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200905967.
References
- 1.Reviews: Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem. 2009;121:5196. doi: 10.1002/anie.200806273. Angew. Chem. Int. Ed.2009, 48, 5094. Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n. Kakiuchi F, Kochi T. Synthesis. 2008:3013. Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173. doi: 10.1039/b606984n. Ishiyama T, Miyaura N. Pure Appl. Chem. 2006;78:1369. Nevado C, Echavarren AM. Synthesis. 2005:167. and references therein.
- 2.Recent reports on C–H activation/arylation with aryl halides: Join B, Yamamoto T, Itami K. Angew. Chem. 2009;121:3698. doi: 10.1002/anie.200806358. Angew. Chem. Int. Ed.2009, 48, 3644. Campeau L-C, Stuart DR, Leclerc J-P, Bertrand-Laperle M, Villemure E, Sun H-Y, Lasserre S, Guimond N, Lecavallier M, Fagnou K. J. Am. Chem. Soc. 2009;131:3291. doi: 10.1021/ja808332k. Yang F, Wu Y, Li Y, Wang B, Zhang J. Tetrahedron. 2009;65:914. Scarborough CC, McDonald RI, Hartmann C, Sazama GT, Bergant A, Stahl SS. J. Org. Chem. 2009;74:2613. doi: 10.1021/jo802632v. Kim J, Jo M, So W, No Z. Tetrahedron Lett. 2009;50:1229. Gorelsky SI, Lapointe D, Fagnou K. J. Am. Chem. Soc. 2008;130:10848. doi: 10.1021/ja802533u.
- 3.C–H activation at room temperature is very rare: a) Reaction of an arene with benzoquinone: Zhang H-B, Liu L, Chen Y-J, Wang D, Li C-J. Adv. Synth. Catal. 2006;348:229.; b) Fujiwara–Moritani reaction in TFA: Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:1586. doi: 10.1021/ja0176907.; c) C–H borylation: Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew. Chem. 2002;114:3182. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. Angew. Chem. Int. Ed.2002, 41, 3056.; d) reaction of an arene forming multiple bonds: Jia C, Piao D, Oyamada J, Lu W, Kitamura T, Fujiwara Y. Science. 2000;287:1992. doi: 10.1126/science.287.5460.1992.; e) methoxycarbonylation: see ref. [9]
- 4.Tremont SJ, ur Rahman H. J. Am. Chem. Soc. 1984;106:5759. [Google Scholar]
- 5.a) Shabashov D, Daugulis O. J. Org. Chem. 2007;72:7720. doi: 10.1021/jo701387m. [DOI] [PubMed] [Google Scholar]; b) Daugulis O, Zaitsev VG. Angew. Chem. 2005;117:4114. doi: 10.1002/anie.200500589. Angew. Chem. Int. Ed.2005, 44, 4046. [DOI] [PubMed] [Google Scholar]
- 6.Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]
- 7.a) Deprez NR, Kalyani D, Krause A, Sanford MS. J. Am. Chem. Soc. 2006;128:4972. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]; b) Campeau L-C, Bertrand-Laperle M, Leclerc J-P, Villemure E, Gorelsky S, Fagnou K. J. Am. Chem. Soc. 2008;130:3276. doi: 10.1021/ja7107068. [DOI] [PubMed] [Google Scholar]; c) Lebrasseur N, Larrosa I. J. Am. Chem. Soc. 2008;130:2926. doi: 10.1021/ja710731a. [DOI] [PubMed] [Google Scholar]; d) Zhao J, Zhang Y, Cheng K. J. Org. Chem. 2008;73:7428. doi: 10.1021/jo801371w. [DOI] [PubMed] [Google Scholar]
- 8.a) Ohnmacht SA, Mamone P, Culshaw AJ, Greaney MF. Chem. Commun. 2008:1241. doi: 10.1039/b719466h. [DOI] [PubMed] [Google Scholar]; b) Flegeau EF, Popkin ME, Greaney MF. Org. Lett. 2008;10:2717. doi: 10.1021/ol800869g. [DOI] [PubMed] [Google Scholar]; c) Turner GL, Morris JA, Cheng K. Angew. Chem. 2007;119:8142. Angew. Chem. Int. Ed.2007, 46, 7996. [Google Scholar]; d) Herrerías CI, Yao X, Li Z, Li C-J. Chem. Rev. 2007;107:2546. doi: 10.1021/cr050980b. [DOI] [PubMed] [Google Scholar]
- 9.Houlden CE, Hutchby M, Bailey CD, Ford JG, Tyler SNG, Gagné MR, Lloyd-Jones GC, Booker-Milburn KI. Angew. Chem. 2009;121:1862. doi: 10.1002/anie.200805842. Angew. Chem. Int. Ed.2009, 48, 1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For other reactions at room temperature in water, see: Nishikata T, Lipshutz BH. J. Am. Chem. Soc. 2009;131:12103. doi: 10.1021/ja905082c. Krasovskiy A, Duplais C, Lipshutz BH. J. Am. Chem. Soc. 2009;131:15592–15593. doi: 10.1021/ja906803t. Lipshutz BH, Ghorai S. Aldrichimica Acta. 2008;41:58.
- 11.The mechanism of C–H activation has been well studied: Racowski JM, Dick AR, Sanford MS. J. Am. Chem. Soc. 2009;131:10974. doi: 10.1021/ja9014474. Desai LV, Stowers KJ, Sanford MS. J. Am. Chem. Soc. 2008;130:13285. doi: 10.1021/ja8045519. Lane BS, Brown MA, Sames D. J. Am. Chem. Soc. 2008;130:8050. doi: 10.1021/ja043273t. García-Cuadrado D, de Mendoza P, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2007;129:6880. doi: 10.1021/ja071034a. Lafrance M, Fagnou K. J. Am. Chem. Soc. 2006;128:16496. doi: 10.1021/ja067144j. Lafrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754. doi: 10.1021/ja062509l. García-Cuadrado D, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2006;128:1066. doi: 10.1021/ja056165v. Davies DL, Donald SMA, Macgregor SA. J. Am. Chem. Soc. 2005;127:13754. doi: 10.1021/ja052047w.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.