

Abstract
Background
Primary sensory neurons express several types of ion channels including transient receptor potential vanilloid 1 (TRPV1) and voltage-gated Na+ channels. Our previous studies showed an increased excitability of bladder primary sensory and spinal neurons triggered by inflammation in the distal colon as a result of pelvic organ cross-sensitization. The goal of this work was to determine the effects of TRPV1 receptor activation by potent agonists and/or colonic inflammation on voltage-gated Na+ channels expressed in bladder sensory neurons.
Methods
Sprague-Dawley rats were treated with intracolonic saline (control), resiniferatoxin (RTX, 10−7 M), TNBS (colonic irritant) or double treatment (RTX followed by TNBS).
Key Results
TNBS-induced colitis increased the amplitude of total Na+ current by 2-fold and of tetrodotoxin resistant (TTX-R) Na+ current by 78% (p≤0.05 to control) in lumbosacral bladder neurons during acute phase (3 days post-TNBS). Instillation of RTX in the distal colon caused an enhancement in the amplitude of total Na+ current at −20 mV from −112.1±18.7 pA/pF (control) to −183.6±27.8 pA/pF (3 days post-RTX, p≤0.05) without changes in TTX resistant component. The amplitude of net Na+ current was also increased by 119% at day 3 in the group with double treatment (RTX followed by TNBS, p≤0.05 to control) which was significantly higher than in either group with a single treatment.
Conclusions & Inferences
These results provide evidence that colonic inflammation activates TRPV1 receptors at the peripheral sensory terminals leading to an up-regulation of voltage gated Na+ channels on the cell soma of bladder sensory neurons. This mechanism may underlie the occurrence of peripheral cross-sensitization in the pelvis and functional chronic pelvic pain.
Keywords: pelvic organ cross-sensitization, colonic inflammation, bladder DRG neurons, resiniferatoxin
INTRODUCTION
Transient receptor potential vanilloid subtype 1 (TRPV1) and voltage-gated Na+ channels (VGSC) are well known transducers of nociceptive processing in pain pathways 1-5. Both types of ion channels are expressed on nerve terminals and somata of dorsal root ganglion (DRG) neurons receiving primary sensory input from the pelvic organs 6, 7. Experiments on TRPV1 knockout mice revealed deficient responses of DRG neurons to noxious stimuli, absence of vanilloid-evoked pain behavior, and reduced thermal hypersensitivity upon somatic inflammation. Despite the aforementioned defects, DRG neurons from TRPV1−/− mice exhibited normal resting membrane potential and unchanged expression of VGSC 8.
TRPV1 agonists trigger membrane depolarization of sensory neurons due to activation of a non-selective cation current leading to an increase in calcium influx, increased action potential firing, and the release of modulatory neurotransmitters and neuropeptides from sensory axons 1, 9. This primary response is followed by a prolonged refractory period of receptor desensitization making the neurons less sensitive to subsequent noxious stimulation 10-12. The feature of TRPV1 receptors to internalize after agonist application and cause prolonged insensitivity to subsequent activation was tested in multiple clinical trials with the best results observed for the treatment of disorders with neuropathic pain (reviewed in 13).
Several lines of evidence suggest that TRPV1 receptors are involved in the development of cross-sensitization and neurogenic inflammation in the pelvis 14, 15. Pelvic organ cross-sensitization is one of the potential mechanisms underlying chronic pelvic pain of undefined etiology and functional pelvic pain syndromes 16-18. It develops as a result of complex neural interactions in the ascending pain pathways mainly due to convergence of sensory inputs in the autonomic and central nervous system 15, 16, 19-21. Our previous studies established a correlation between excitability of bladder projecting and colon/bladder convergent neurons in the lumbosacral spinal cord and DRG, neurogenic inflammation in the pelvis and colon/bladder cross-sensitization 20, 22, 23.
Under in vitro conditions, direct application of TRPV1 agonists in sensory neurons expressing both VGSC and TRPV1 channels significantly inhibits neuronal excitability due to blockade of VGSC 11, 24. The magnitude of the VGSC block is proportional to the amplitude of capsaicin-induced current and, in part, is dependent on activation of second messengers, such as cAMP. No studies were carried out so far to test if activation of TRPV1 receptors at the peripheral terminals of DRG neurons in vivo has the same effects on the expression and kinetics of VGSC. We aimed to determine the effects of peripheral TRPV1 receptor activation by RTX on the expression and kinetics of voltage-gated Na+ channels in bladder sensory neurons in the presence and absence of colonic inflammation. We also clarified the role of TRPV1-VGSC “cross-talk” in the development of pelvic organ cross-sensitization and the role of colonic inflammation in modulation of TRPV1-VGSC nociceptive pathways.
MATERIALS AND METHODS
Animal model and experimental groups
Adult male Sprague-Dawley rats (N=55, 250-300 g, Charles River Laboratories, Malvern, PA) were used in this study. Animals were housed two per cage, maintained on a 12 hour light/dark cycle with ad libitum access to water and food. Animals were divided into four experimental groups: 1-control (intracolonic saline enema); 2-colonic inflammation induced by TNBS (2,4,6-trinitrobenzene sulfonic acid, 12 mg/kg in 50% ethanol); 3 - resiniferatoxin (RTX, TRPV1 agonist, 10−7 M in saline) instillations in the colon; and 4 - RTX treatment (colon) followed by the induction of colonic inflammation with TNBS 2 days later. An additional group of rats (N=6) was treated with 50% ethanol (distal colon) to evaluate the effects of ethanol on the structure of the colonic wall as well as VGSC in bladder sensory neurons. Intracolonic instillations were performed via a flexible catheter connected to a 1 ml syringe under isoflurane anesthesia (2 min in duration). Animals received 0.7-0.8 ml of either saline, RTX in saline, ethanol or TNBS in ethanol. To avoid significant loss of instilled liquid, the rats were kept elevated by the tail until they regained consciousness and then were returned to their cages. Animals from each group were euthanized at 3 and 30 days after the last treatment. These time points correspond to the acute and resolved phases of TNBS-induced colitis, respectively.23 To evaluate the level of inflammation induced by TNBS, a segment of the distal colon was removed during tissue collections and sectioned in half. One part of the colon was fixed in 4% paraformaldehyde for histological evaluation and another part was snap-frozen to measure concentration of myeloperoxidase (MPO, enzyme released by neutrophils at the site of inflammation) using MPO ELISA kit (Alpco, Salem, NH). All protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and adhered to the guidelines of the Committee for Research and Ethical Issues published by the International Association for the Study of Pain.
Surgical procedure for labeling colonic and urinary bladder DRG neurons
Rats were anaesthetized with 2% isoflurane and held on a warming pad inside the designated hood to minimize the investigator’s exposure to the anesthetic. A midline laparotomy was performed under sterile conditions to gain access to the pelvic organs. The distal colon was exposed and DiI (1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate; Molecular Probes, Eugene, OR, USA; 1.5% w/v in methanol) was injected into the colonic wall using a Hamilton syringe with 26 gauge needle at 6-10 sites. The colon was placed back into the abdominal cavity and the urinary bladder was exposed for injections. Fast Blue (Polysciences Inc., Warrington, PA, USA; 1.5% w/v in water) was injected into the urinary bladder wall (detrusor) using the same approach as described for the colon. We intentionally performed double labeling to exclude from the experiments cells receiving dual afferent input from the distal colon and urinary bladder as these convergent neurons would be directly affected by colonic treatments 22. The total volume of dye injected into each organ was 20-25 μl. Adjacent pelvic organs were isolated with gauze to soak up any spills and prevent the labeling of adjacent structures during dye injections. Additionally, the needle was kept in place for 30s after each injection. Any leaked dye was removed with a cotton swab before placing the organ into the pelvic cavity. Incisions were sutured in layers under the sterile conditions followed by subcutaneous injection of buprenorphine (0.5 mg/kg). Animals were allowed to recover on a warm blanket until they gained full consciousness and then were returned to their cages. Rats underwent subsequent treatments with RTX or TNBS 5-7 days after the labeling of DRG neurons.
Isolation of DRG neurons for patch-clamp experiments
Animals were euthanized by overdose of sodium pentobarbital (130 mg/kg) at 3 and 30 days after the last treatment. Dorsal root ganglia were dissected and removed bilaterally at L6-S2 levels. The ganglia were treated with collagenase (Worthington, type 2, Biochemical Corp., Lakewood, NJ, USA) in F-12 medium (Invitrogen, Carlsbad, CA, USA) for 90 min in an incubator with 95% O2 and 5% CO2 at 37°C. Isolated ganglia were then rinsed in phosphate-buffered saline (PBS) and incubated for 15 min in the presence of trypsin (Sigma, St Louis, MO, USA; 1 mg/ml) at room temperature. The enzymatic reaction was terminated in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% of fetal bovine serum (FBS). Single neurons were obtained by gentle trituration using fire polished Pasteur pipettes in DMEM with trypsin inhibitor (Sigma; 2 mg/ml) and deoxyribonuclease (Sigma; DNase 1 mg/ml). The cell suspension was centrifuged for 10 min at 700 rpm (4°C), and supernatant was discarded. The pellet, containing sensory neurons, was resuspended in 2 ml of DMEM containing 10% FBS. Neurons were plated on poly-L-ornithine coated 35 mm Petri dishes. Isolated cells were maintained overnight in an incubator at 37°C with 95% O2/5% CO2 and were used for electrophysiological experiments within 24 hours.
Electrophysiological recordings of Na+ currents from isolated bladder sensory neurons
Bladder labeled neurons were identified using specific filters for DiI (UV-1A, Nikon, Lewisville, TX, USA) and Fast Blue (UV-2A, Nikon) under an inverted fluorescent microscope (Ti E2000–5, Nikon). Only neurons exhibiting bright blue fluorescence (Fast Blue labeled) were used for Na+ current recordings using the whole-cell patch clamp technique. For voltage clamp experiments the external solution contained (in mM): NaCl 45, TEA Chloride 30, Choline Chloride 60, KCl 5.4, MgCl2 1, CaCl2 1, HEPES 5, D-glucose 5.5, adjusted with NaOH to pH 7.4. Pipette solution for these experiments consisted of (in mM): L-aspartic acid 100, CsCl 30, MgCl2 2, Na-ATP 5, EGTA 5, HEPES 5 adjusted with CsOH to pH 7.2. CdCl (100 μM) was added to the external solution in order to block voltage gated calcium currents. Freshly made Amphotericin B (0.24 mg/ml) was added to the pipette solution for perforated whole cell recordings. Tetrodotoxin (TTX, 10−6 M) was used in the bath solution to block TTX-sensitive (TTX-S) Na+ currents. Microelectrodes were fabricated from borosilicate capillary glass (Sutter instruments, Novato, CA) and had resistances of 2-5 MΩ when filled with internal solution. Recordings commenced 5 min after the establishment of whole cell access. Series resistance was compensated ≥80-85%, and the calculated junction potential was around 5 mV. Cells were excluded from analysis if uncompensated series resistance resulted in a maximum voltage error >5 mV or if the seal or access resistance were unstable.
The steady-state activation of VGSC was assessed by using a three-pulse protocol with a negative pre-pulse to −110 mV and a series of following short pulses of 10 ms duration from −110 mV to +70 mV to activate Na+ currents. The amplitude of steady-state activation was measured at the peak of tail current at −70 mV, normalized and plotted as I/Imax against the voltage. Activation curves were fit with a Boltzmann function in the form of G = Gmax/{1 +exp((V½,act − V)/k)}, where Gmax is the maximal sodium conductance; V½,act is the potential at which activation is half-maximal; V is the test potential, and k is the slope factor. The amplitude of steady-state inactivation was measured at 0 mV after 150 ms depolarizing pulses ranging from −100 mV to 70 mV. Steady-state inactivation was normalized with the maximal peak current (Imax) and fit with a Boltzmann function, I/Imax = A + (1 − A)/{1 + exp((V − V½,inact)/k)}, where V represents the inactivating prepulse potential, V½,inact represents the midpoint of inactivation curve, and A is the offset. Voltages of half activation and inactivation were obtained from the individual neurons. The cells were held at −70 mV between the recordings. All experiments were performed at room temperature (23°C) and recorded using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA). All neurons were tested for response to RTX (10−7 M) in vitro at the end of each experiment. This was done in order to evaluate the level of TRVP1 receptor desensitization upon initial in vivo application of RTX. In case of receptor desensitization, responses of bladder sensory neurons to the second application of RTX in vitro should be significantly reduced or absent (see Table 1). Recordings were made from only one neuron in each coverslip due to exposure of all neurons to TTX and RTX during the bath application of the drugs.
Table 1.
Relationship between the presence of TTX-R Na+ channels and responses to TRPV1 agonist in isolated bladder sensory neurons.
| Experimental groups | Number of rats (N) |
Number of cells recorded (n) |
Cell capacitance (pF) |
Number of cells responsive to RTX (n) |
Number of cells expressing TTX-R Na+ current (n) |
Number of cells expressing TTX-R current and responsive to RTX (n) |
|---|---|---|---|---|---|---|
| Control | 8 | 11 | 55.2±5.5 | 10 | 6 | 3 |
| TNBS (3 days) | 6 | 14 | 41.1±4.1 | 11 | 5 | 3 |
| TNBS (30 days) | 5 | 13 | 43.8±6.5 | 11 | 8 | 5 |
| RTX (3 days) | 5 | 10 | 47.2±4.6 | 2 | 3 | 2 |
| RTX (30 days) | 5 | 14 | 49.2±5.2 | 3 | 8 | 3 |
| RTX+TNBS (3 days) | 5 | 15 | 46.8±5.4 | 6 | 10 | 3 |
| RTX+TNBS (30 days) | 9 | 14 | 42.6±4.6 | 4 | 5 | 1 |
Chemicals and drugs
All chemicals were obtained from Sigma with the exception of DiI (Molecular Probes, Invitrogen, Eugene, Oregon, USA) and Fast Blue (Polysciences, Warrington, PA, USA). Tetrodotoxin citrate (TTX) was purchased from Tocris Bioscience (Ellisville, Missouri, USA) and Amphotericin B from PCROS (New Jersey, USA).
Statistical analysis
pCLAMP software (Axon Instruments, CA) was used for data acquisition and analysis. All data are expressed as means ± SE. A positive response to RTX was defined as an inward current recorded in gap-free mode with the amplitude over 100 pA. Statistical significance between the groups was assessed by one-way ANOVA followed by Bonferroni’s test. Data with p≤0.05 were considered statistically significant.
RESULTS
TNBS induced colitis increases Na+ currents in bladder sensory neurons
Severity of colonic inflammation induced by TNBS was assessed by histological and biochemical methods as previously described 23. Analysis of H&E-stained sections of the distal colon revealed that neither saline (Fig. 1A) nor ethanol (Fig. 1B) caused detectable structural changes in the colonic wall. However, visible changes in the colonic histology including crypt segmentation, local infiltration and thickening of the muscle layer were detected at day 3 after TNBS treatment (Fig. 1C). By day 30 post-TNBS, the cytoarchitecture of the colon did not significantly differ from the control group (Fig. 1D). The MPO assay is a validated biochemical method of grading inflammatory changes in the viscera and represents the amount of MPO enzyme at the site of inflammation. The MPO data showed 4-fold increase in the colon on day 3 post-TNBS instillation, which was back to normal values at day 30 post-TNBS (Fig. 1E). Taken together, histological and MPO data demonstrated substantial inflammatory changes in the distal colon at day 3 corresponding to the acute phase of experimental colitis which was followed by recovery from inflammation at day 30 post-TNBS.
Figure 1. Histological and biochemical evaluation of the severity of colonic inflammation induced by TNBS.
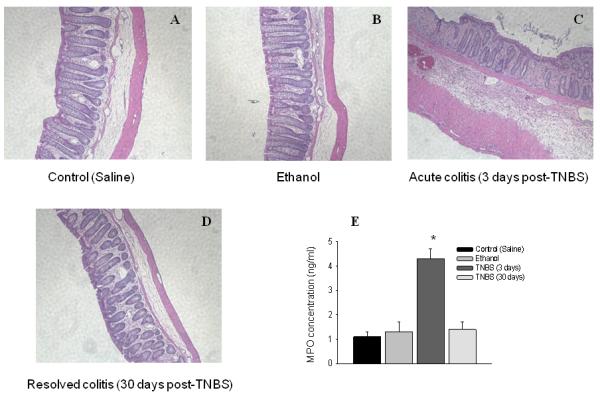
Hematoxylin and eosin (H&E) staining of the distal colon from the following groups of animals: control (A, saline), ethanol (B, 3 days), acute colitis (C, TNBS - 3 days) and resolved colitis (D, TNBS - 30 days). Please note that only group with acute colitis (3 days post-TNBS) shows significant tissue damage including sites of infiltration, thickening of the muscle layer, and disruption of the colonic crypts. E, Concentration of myeloperoxidase (MPO) enzyme in the distal colon from control and experimental groups correlates with the results of histological evaluation. *P≤ 0.05 to control group.
Retrograde labeling of lumbosacral sensory neurons revealed three populations of isolated DRG neurons: colon-projecting, bladder-projecting and colon-bladder convergent neurons. We intentionally performed double labeling to exclude from the experiments neurons receiving dual afferent input from the distal colon and urinary bladder as these neurons would be directly affected by colonic treatments. The number of double labeled cells was around 10% as previously reported. 22 Only neurons labeled with Fast Blue (bladder projecting) underwent electrophysiological evaluation and were used for data analysis.
Experimental colitis induced by TNBS triggered an increase in the amplitude of total Na+ current recorded from bladder afferent neurons. Raw traces of total Na+ current obtained from the control groups (saline and ethanol), acute (3 days post-TNBS), and resolved (30 days post-TNBS) colonic inflammation are presented in Fig. 2A. The current-voltage (I-V) relationship of total Na+ current normalized to the cell size shows that these neurons produced a large amplitude Na+ current upon membrane depolarization reaching maximal amplitude at −30 mV (Fig. 2B). Intracolonic instillation of ethanol caused some increase in total Na+ current, however, the difference did not reach statistical significance in comparison to the group with intracolonic saline (Fig. 2B). Acute colonic inflammation increased the peak amplitude of total Na+ current in bladder DRG neurons by 2-fold at −30 mV (from −106.4±19.4 pA/pF (n=11) in the control group to −209.6±20.1 pA/pF (n=14) in the acute colitis group (p≤0.05). At 30 days post-TNBS, when colonic inflammation resolved, the peak amplitude of total Na+ current was still enhanced and reached −202.2±27.5 pA/pF at the same voltage (n=13, p≤0.05). Bath application of TTX showed an increase in TTX-R component of Na+ current upon acute colonic inflammation from −28.4±8.3 pA/pF to −50.1±10.5 pA/pF at −10 mV (Fig. 1C, p≤0.05 to control). However, by day 30 the density of TTX-R Na+ current was back to control levels (Fig. 2C).
Figure 2. Effects of colonic inflammation on the amplitude of voltage-gated Na+ currents in bladder sensory neurons.
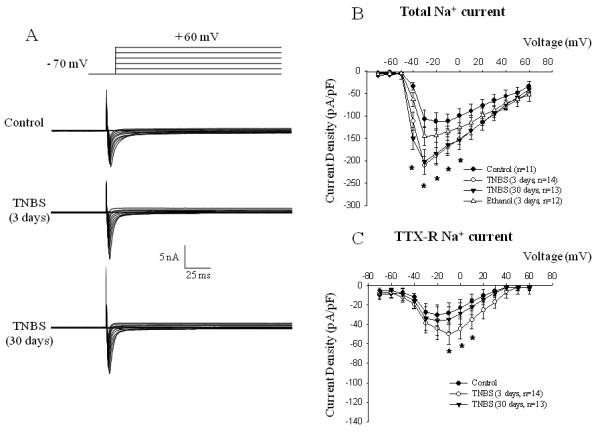
A, Top panel presents the scheme of a typical voltage protocol for recordings of Na+ currents including depolarizing pulses from −70 mV to +60 mV (10 mV increments) from the holding potential of −70 mV. Lower panel shows representative raw traces recorded from the control group and after acute (TNBS 3 days) or resolved (TNBS 30 days) colonic inflammation. B, Current-voltage (I-V) relationship of the total Na+ current recorded in bladder DRG after experimental colitis and application of intracolonic ethanol as a positive control. C, I-V relationship of TTX-resistant Na+ current in bladder projecting cell bodies. * - p≤0.05 when compared to control group.
We next assessed the kinetic parameters of Na+ currents after the induction of experimental colitis. The steady-state activation was studied by using a three-pulse protocol with a negative pre-pulse to −110 mV and a series of short pulses (10 ms duration) to activate Na+ currents (Fig. 3A). The amplitude of steady-state activation was measured at the peak of tail current upon the voltage step to −70 mV, normalized and plotted as I/Imax against the voltage (Fig 3D). Acute experimental colitis did not affect the activation of total Na+ current compared to the control group. However, resolved colitis led to the leftward shift in the steady-state activation of total Na+ current by 11 mV (V½=-34.9± 2.5 mV in the control group vs −45.6±1.7 mV at day 30 post-TNBS (Fig. 3D, p≤0.05). The amplitude of steady-state inactivation was measured at a series of membrane depolarizing steps ranging from −100 mV to +70 mV (Fig. 3B). Neither acute nor resolved colonic inflammation affected the parameters of steady-state inactivation of total Na+ current in bladder sensory neurons (Fig 3E).
Figure 3. Kinetics of Na+ channels recorded from bladder DRG neurons in rats with TNBS-induced colitis.
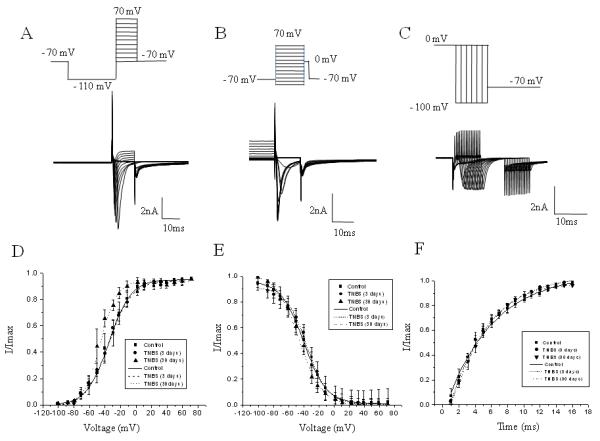
A, The protocol (top panel) of steady-state activation and raw traces (bottom panel) of total Na+ current. The steady-state activation of VGSC was assessed by using a three-pulse protocol with a negative pre-pulse to −110 mV and a series of short pulses of 10 ms duration from −110 mV to +70 mV to activate Na+ currents. B, The protocol of steady-state inactivation and raw traces of total Na+ current. The amplitude of steady-state inactivation was measured at 0 mV after 150 ms depolarizing pulses ranging from −100 mV to 70 mV. C, The protocol of recovery from inactivation and raw traces of total Na+ current. The time course of recovery from inactivation was assessed using a 100 ms pre-pulse to −100 mV from the membrane holding potential of −70 mV. Na+ channels were then inactivated by a 100 ms conditioning pulse to 0 mV, and the membrane was returned to a potential of −100 mV for a variable time period (from 1 to 20 ms, 1 ms increments) before eliciting a second current with a test pulse to 0 mV in order to assess recovery of channels from inactivation after a 5 s pulse-free period at the holding potential. D, Voltage dependence of steady-state activation in bladder neurons from control and TNBS treated animals. Please note a leftward shift in the group with acute colitis. E, Voltage dependence of steady-state inactivation in lumbosacral DRG neurons. F, Time dependence of recovery from inactivation in control and experimental groups. * - p≤0.05 indicates statistical significance when compared to the control group.
The time course of recovery from inactivation was assessed using a 100 ms pre-pulse to −100 mV from the membrane holding potential of −70 mV. Na+ channels were then inactivated by a 100 ms conditioning pulse to 0 mV, and the membrane was returned to a potential of −100 mV for a variable time period (from 1 to 20 ms, 1 ms increments) before eliciting a second current with a test pulse to 0 mV in order to assay recovery of channels from inactivation after a 5 s pulse-free period at the holding potential. Peak currents during the test pulse were normalized to peak current during conditioning pulse plotted against the recovery interval. As shown in Fig. 3F, there was no significant difference between the control and colitis groups in the kinetics of recovery of total Na+ current from inactivation state.
Intracolonic application of RTX enhances the amplitude of Na+ currents in bladder projecting neurons
Series of recordings were performed to clarify the role of desensitization of intracolonic TRPV1 receptors on the current density of VGSC in L6-S2 bladder sensory neurons. Instillation of RTX in the distal colon caused an enhancement in the amplitude of total Na+ current at −20 mV from −112.1±18.7 pA/pF (n=11, control) to −183.6±27.8 pA/pF (3 days post-RTX, n=10, p≤0.05, Fig. 4). The same effect was observed at day 30 after RTX instillation with an increase in total Na+ current from −106.4±19.3 pA/pF to −162.9±26.5 pA/pF (−30 mV, Fig. 4A and B, n=14, p≤0.05 to control). Analysis of TTX-R component of Na+ current in both experimental groups (3 and 30 days) did not reveal significant changes (data not shown) suggesting that RTX treatment did not influence TTX-R Na+ channels. RTX application also led to a leftward shift in steady-state activation from −34.9±2.5 mV in the control group to −43.9±2.0 mV in the experimental group (day 3 post-RTX, Fig. 4C). This change did not reach statistical significance (p>0.05) and returned to control levels at day 30 post-RTX application. No substantial alterations in inactivation parameters of total Na+ current were noted at either time point (Fig. 4D and E).
Figure 4. Activation of colonic TRPV1 receptors in vivo increases the amplitude of voltage-gated Na+ channels in bladder sensory neurons.
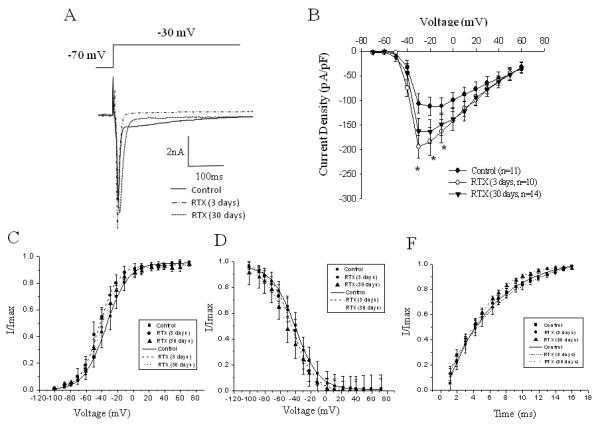
A, Representative raw traces of total Na+ current recorded at −30 mV in control neurons and after RTX treatment. B, Current-voltage (I-V) relationship of the total Na+ current recorded in bladder DRG after intracolonic RTX application. The peak amplitude of the current was normalized to the cell size and plotted as I/Imax against the voltage. C, Voltage dependence of steady-state activation in bladder neurons from control and RTX groups. D, Voltage dependence of steady-state inactivation of total Na+ current in lumbosacral DRG neurons. E, Time dependence of recovery from inactivation in control and experimental groups. * - p≤0.05 indicates statistical significance when compared to the control group.
Desensitization of colonic TRPV1 receptors in vivo followed by colonic inflammation up-regulate VGSC in bladder DRG neurons
To establish whether desensitization of TRPV1 receptors in the distal colon prior to colonic inflammation affected VGSC in bladder sensory neurons, animals were treated with intracolonic RTX followed by TNBS enema 2 days later. This interval between the treatments was selected to allow sufficient time for TRPV1 receptor internalization at the peripheral terminals and transmission of the signal to the cell body located within DRG. RTX pre-treatment did not prevent an increase in the amplitude of total Na+ current, previously shown to be up-regulated in both groups with experimental colitis. In fact, the effect was opposite - the peak amplitude of total Na+ current was increased by 119% at day 3 after the final treatment (n=15, p≤0.05 to control, Fig. 5A and B). An enhancement of total Na+ current was observed at all positive voltages (Fig. 5B). One month after the last treatment, current density of expressed Na+ channels was reduced by 40-44% of day 3 values (depending on the voltage) but was still significantly higher than in the control group (by 50% of control value at day 30, n=14, p≤0.05 to control from −30 mV to +10 mV).
Figure 5. Comparison of the expression and kinetic parameters of voltage dependent Na+ currents in bladder DRG neurons between control group and groups with double (RTX followed by TNBS) treatment.
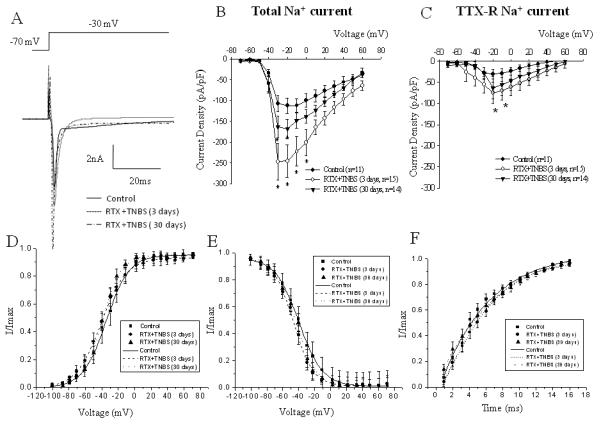
A, Representative traces of the peak of total Na+ current in control neurons and after RTX-TNBS treatment. B, Current-voltage (I-V) relationship of the total Na+ current recorded in bladder DRG after intracolonic RTX application. The peak amplitude of the current was normalized to the cell size and plotted as I/Imax against the voltage. C, I-V relationship of TTX-resistant Na+ current in bladder projecting sensory neuons. D, Voltage dependence of steady-state activation in bladder neurons from control and RTX-TNBS groups. E, Voltage dependence of steady-state inactivation of total Na+ current in lumbosacral DRG neurons. F, Time dependence of recovery from inactivation in control and experimental groups. * - p≤0.05 indicates statistical significance when compared to the control group.
These changes were accompanied by a slight leftward shift in steady-state activation from −34.9±2.5 mV (control) to −40.7±1.9 mV in the double treatment group (3 days, p≥0.05 to control). This parameter was close to one previously reported for the RTX group (also at day 3) without any statistically significant difference between RTX and RTX-TNBS groups. Neither steady-state inactivation nor recovery from inactivation of total Na+ current were affected by double treatment (Fig. 5E and F). Bath application of TTX revealed an up-regulation of TTX-R component of Na+ current at day 3 in bladder neurons in the group with double treatment. At −20 mV, an increase of TTX-R Na+ current was from −30.3±8.7 pA/pF (control) to −74.9±14.9 pA/pF (3 days, n=14, p≤0.05 to control, Fig. 5C). At day 30, the amplitude of TTX-R Na+ current was not statistically different from the control values. The steady-state activation and inactivation kinetics of TTX-R Na+ current were not affected by dual treatment (data not shown).
Desensitization of peripheral TRPV1 receptors in vivo reduced the sensitivity of isolated bladder nociceptors to subsequent RTX application in vitro
In order to determine if intracolonic RTX desensitized bladder afferent neurons and to define the level of desensitization, RTX (10−7 M) was applied in the bath solution at the end of each recording. The cell was considered to be responsive to RTX if the amplitude of the evoked current (recorded in “gap-free” mode) was larger than 100 pA. The peak amplitude of the response was normalized to the cell size and is presented in Fig. 6B along with the representative raw traces (Fig. 6A). We recorded Na+ currents and TRPV1 activated currents from both populations of RTX-responsive and non-responsive neurons as we could not predict if intracolonic RTX would affect bladder-projecting afferents. The results of our study showed that the amplitude of RTX response during both acute (3 days post-TNBS) and recovery (30 days post-TNBS) colonic inflammation was within the range of control values. However, there was more than 2-fold decrease in the response to RTX in all groups after either intracolonic RTX or double treatment (Fig. 6A and B).
Figure 6. Responses of isolated bladder sensory neurons to in vitro application of RTX.
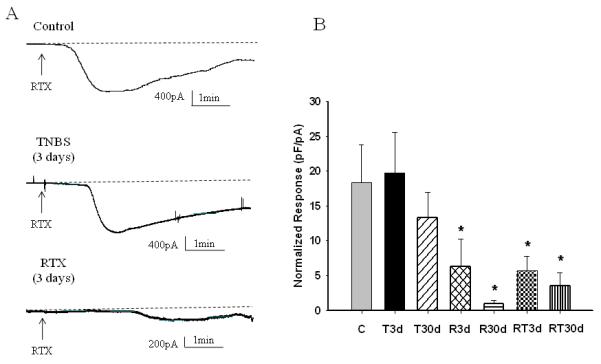
A, Raw traces of inward current recorded in response to 10−7 M of RTX in control and experimental groups. B, Amplitude of the RTX response normalized to the cell size in all groups. C- Control, T3d – 3 days after TNBS application, T30d – 30 days after TNBS treatment, R3d – 3 days after intracolonic RTX instillation, R30d – 30 days after intracolonic RTX instillation, RT3d - 3 days after TNBS treatment following intracolonic RTX, RT30d - 30 days after TNBS treatment following intracolonic RTX. * p≤0.05 indicates statistical significance when compared to the control group.
Previous studies reported a correlation between TTX-R Na+ channels and responses to TRPV1 agonists in isolated sensory neurons in vitro 11, 24. We also performed the quantitative analysis of association between TTX-R Na+ channels and responses to RTX in all experimental groups. These results are presented in Table 1 and show that only 20-21% of all bladder neurons responded to RTX after peripheral desensitization of colonic TRPV1 receptors. In both groups with double treatment (RTX+TNBS) the percentage of neurons responded to RTX in vitro was 30-40% in comparison to 91% in the control group and 79-85% in both TNBS groups (3 and 30 days). No correlation between the cell size and response to TRPV1 agonist was noted (Table 1).
DISCUSSION
In the present study we characterized the function of voltage-gated Na+ channels (VGSC) in bladder projecting lumbosacral DRG neurons upon noxious stimulation of the distal colon and activation of intracolonic TRPV1 receptors. Our findings confirmed the previously reported results that acute colonic inflammation increases the amplitude of net Na+ current in bladder sensory neurons mostly via an up-regulation of TTX-R Na+ currents 25. The novel findings of this study include electrophysiological analysis of VGSC upon intracolonic applications of RTX and resolution of colonic inflammation. To our knowledge, this is the first report which provides direct evidence that activation of peripheral TRPV1 receptors in the colon either by application of a TRPV1 agonist or by inflammatory insult triggers an increase in VGSC in sensory neurons receiving input from the urinary bladder. Our results also clarify the role of TRPV1/VGSC cross-talk in the development of pelvic organ cross-sensitization. Dorsal root ganglion neurons receiving input from the pelvic viscera play a key role in the development of peripheral cross-sensitization 22, 26. Modulation of VGSC in bladder sensory neurons is one of the peripheral mechanisms underlying cross-sensitization between the distal colon and urinary bladder. Abdominal discomfort, visceral hypersensitivity and chronic pelvic pain are associated with colon/bladder cross-sensitization which, in turn, contributes to etiology of co-morbid gastrointestinal and urologic disorders.
Excitability and action potential propagation in primary afferents are defined by the expression and function of VGSC on axons and neuronal somata of extrinsic sensory neurons 4, 5. We detected an up-regulation of VGSC in bladder sensory neurons during both acute phase and after the recovery from TNBS-induced colitis. The peak amplitude of total Na+ current in bladder projecting neurons was also enhanced after activation of intracolonic TRPV1 receptors with RTX. However, TTX-R Na+ currents were significantly increased in neurons isolated from TNBS-treated animals but not from RTX-treated rats. We speculate that effects of colonic inflammation are mediated predominantly via up-regulation of TTX-R Na+ current whereas RTX application affected mostly TTX-S Na+ current in bladder DRG neurons. Up-regulation of TTX-S (Nav1.6 and Nav1.7) and/or TTX-R (Nav1.8 and Nav1.9) Na+ channels by noxious stimulation is linked to the occurrence of visceral hyperalgesia and activation of pain pathways 5-7, 27. Studies utilizing animal models of acute and chronic inflammation in different organs of gastrointestinal 28-32 and genitourinary 33 systems showed an increased excitability of DRG neurons predominantly due to an increase in density of TTX-R Na+ channels. Activation of TRPV1 receptors may also modulate the function of VGSC in sensory ganglia. Studies on capsaicin-responsive DRG neurons in vitro showed that the majority of these neurons express TTX-R Na+ channels 11, 24. Recent study by Miranda-Morales et al 34 established that activation of TRPV1 receptors on peripheral axons of extrinsic sensory neurons can induce the release of neurotransmitters at remote sites from the initial stimulus via “axon reflex” mechanisms. The “axon reflex” was resistant to TTX but was blocked by low Na+ solution suggesting that TTX-R Na+ channels may play a major role in the observed effects 34.
The results of our study did not support correlation between the expression of TTX-R Na+ channels on sensory neurons and responses to peripheral RTX application. This discrepancy could be explained, in part, by the facts that a) we targeted TRPV1 receptors in vivo, not in vitro, and b) we stimulated colonic TRPV1 receptors, not the bladder ones. Nevertheless, secondary application of RTX in vitro in neurons isolated from animals treated previously with intracolonic RTX showed significant reduction in RTX response. At present, it is still unclear how activation of TRPV1 signaling pathways in the distal colon can lead to an up-regulation of VGSC on extrinsic sensory neurons innervating the adjacent organ - urinary bladder. The mechanisms underlying these “cross-organ” and “cross-channel” interactions are quite complex and could be organized into the following categories: 1) mechanisms of colon/bladder cross-sensitization (cross-organ); 2) mechanisms of TRPV1/VGSC cross-sensitization (cross-channel); and 3) mechanisms of “peripheral-somal” channel cross-activation.
The mechanisms of peripheral (excluding the spinal cord and brain) colon/bladder cross-sensitization have been previously discussed in several papers by our group 14, 17, 22 and by others 16, 18, 26, 35. Peripheral cross-sensitization includes convergence of sensory inputs from the viscera at the level of sensory ganglia whereas central mechanisms involve the levels of the spinal cord and the brain. Development of peripheral cross-sensitization between the distal colon and urinary bladder may occur due to: 1 – existence of convergent DRG neurons with dichotomized peripheral axons which may release neurotransmitters and neuropeptides via “axon-reflex” mechanism from their peripheral terminals; 2 – cross-depolarization between sensory neurons within the same dorsal root ganglion receiving single input from either the colon or urinary bladder; 3 – activation of bladder sensory neurons by the input from colon/bladder convergent spinal cord neurons via dorsal root reflexes (reviewed in 16, 17).
Development of TRPV1/VGSC “cross-channel” activation may depend on some of the mechanisms discussed above for the “cross-organ” sensitization. For instance, cross-depolarization between neighboring sensory neurons and/or glial cells within the same dorsal root ganglion could underlie an increase in Na+ current. Several studies on excised lumbar dorsal root ganglia revealed that neurons within a single ganglion are electrically 36-38 and chemically 39 coupled. Electrical “cross-depolarization” includes predominantly cross-excitation of a silent small diameter neuron (with unmyelinated C-type axon) by a stimulation of a neighboring neuron with myelinated axon (Aβ- or Aδ-type) 36. Chemical coupling is usually a result of intraganglionic release of neurotransmitters and neuropeptides from sensory neurons activated by a peripheral insult 39. It was determined that an increase in calcitonine gene-related peptide (CGRP) and substance P within a sensory ganglion may cause an enhancement in TTX-R current 40-42. Axonal activation of colonic TRPV1 receptors is also associated with the synthesis and release of substance P and CGRP in the sensory ganglia and urinary bladder 23. Alternatively, cross-sensitization can occur between afferent axons themselves. Both TRPV1 12 and Na+43 channels are expressed along the branches of afferent axons. The role of axonal Na+ and TRPV1 channels may change under pathophysiological conditions and trigger translocation of channel proteins from the soma to peripheral terminals with subsequent accumulation at the site of injury. These alterations create local ectopic discharges at peripheral terminals of sensory neurons and contribute to visceral hyperalgesia 38, 44. Experiments on Aδ- and C-fiber conduction in afferent fibers showed that TTX-S Na+ channels dominated conduction velocity and action potential propagation along both the peripheral and central branches of primary afferents despite the fact that the central branch of the afferent axon had a significantly higher percentage of TTX-R Na+ channels than its peripheral branch 43.
Activation of intracolonic TRPV1 receptors in vivo led to an up-regulation of VGSC on neuronal somata of bladder sensory neurons in our experiments. These results are different from the ones observed under in vitro conditions which determined that direct application of capsaicin (or RTX) on isolated DRG neurons responsive to TRPV1 stimulation blocks neuronal excitability due to fast inhibition of VGSC (11, 24 and our unpublished observations). A number of reasons could underlie the difference between in vivo and in vitro observed effects of TRPV1 agonists on Na+ channel function. First, there is a possibility that some of the studied bladder projecting neurons could belong to a subpopulation of convergent cells receiving afferent input from both the colon and urinary bladder and, thus, be directly activated by intracolonic RTX 22. Second, it is unclear if there might be any specific interaction between TRPV1 and VGSC channels at the membrane level. The functional interconnection between these channels in sensory neurons occurs either indirectly via recruitment of second messengers (e.g. cAMP or intracellular Ca2+) or directly, involving interactions within a complex as it was proven for other channels 45-47. Additionally, extracellular Na+ may surround proton-sensitive sites on the extracellular pore-forming loop of the TRPV1 channels and supress the channel activity under physiological conditions 48. Activation of TRPV1 channels by nitro-oleic acid simultaneously inhibits Na+ currents in isolated DRG neurons 49. Third, due to a 3 day time lapse between the RTX treatment and recordings of Na+ channels in our experiments, the expression of both TRPV1 and Na+ channels could have altered on the cell soma. It is established that the turnover half-life of Na+ channels synthesis and expression lies within 1-3 days 5, 7. Fourth, there may be some intercrossing of TRPV1 and VGSC signaling pathways via modulation of the same transcriptional or translational factors. It also would be of interest to determine whether there is some kind of capsaicin receptor on one of the Na+ channels expressed in DRG neurons and/or what the role of modulatory subunits of both channels is in TRPV1-VGSC “cross-talk”.
In summary, the results of our study demonstrate for the first time that activation of peripheral TRPV1 receptors in the colon either by application of a TRPV1 agonist or by inflammatory insult triggers the expression of VGSC in sensory neurons receiving input from the urinary bladder. The underlying mechanisms of these “cross-organ” and “cross-channel” interactions require further studiess and could provide important information for the development of new pharmacological approaches and therapeutic strategies for the treatment of co-morbid conditions characterized by functional pelvic pain arising from gastrointestinal or genitourinary systems.
ACKNOWLEDGEMENTS AND DISCLOSURES
We would like to thank Thomas Mathai and Shaohua Chang for excellent technical assistance. Preliminary results of this work were presented in an abstract form at the AUA 2010 meeting. Dr. Lei was a Scholar of the Seventh Annual Young Investigators Forum on Functional GI Disorders (April 2010, Arizona) sponsored by the Functional Brain-Gut Research Group. The study was supported by the NIH/NIDDK grants DK077699 (A.P.M.) and DK 077699-S2 (A.P.M).
Footnotes
AUTHOR CONTRIBUTION
QL performed the experiments, analyzed the data and drafted the manuscript; APM designed the study, performed the experiments, discussed the data and finalized the manuscript.
COMPETING INTERESTS
The authors have no competing interests.
REFERENCES
- 1.Caterina MJ, Schumacher MA, Tominaga M, et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389(6653):816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 2.Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- 3.Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 2009;10(7):1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- 4.Dib-Hajj SD, Cummins TR, Black JA, et al. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 5.Waxman SG. Channel, neuronal and clinical function in sodium channelopathies: from genotype to phenotype. Nat Neurosci. 2007;10(4):405–409. doi: 10.1038/nn1857. [DOI] [PubMed] [Google Scholar]
- 6.Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52(1):77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006;7(1 Suppl 1):S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Caterina MJ, Leffler A, Malmberg AB, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288(5464):306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 9.Holzer P, Maggi CA. Stimulation and propagation of the ascending enteric reflex contraction: role of tachykinins and acetylcholine. Regul Pept. 1993;46(1-2):383–385. doi: 10.1016/0167-0115(93)90094-o. [DOI] [PubMed] [Google Scholar]
- 10.Rigoni M, Trevisani M, Gazzieri D, et al. Neurogenic responses mediated by vanilloid receptor-1 (TRPV1) are blocked by the high affinity antagonist, iodo-resiniferatoxin. Br J Pharmacol. 2003;138(5):977–985. doi: 10.1038/sj.bjp.0705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su X, Wachtel RE, Gebhart GF. Capsaicin sensitivity and voltage-gated sodium currents in colon sensory neurons from rat dorsal root ganglia. Am J Physiol. 1999;277(6 Pt 1):G1180–G1188. doi: 10.1152/ajpgi.1999.277.6.G1180. [DOI] [PubMed] [Google Scholar]
- 12.Szallasi A, Nilsson S, Farkas-Szallasi T, et al. Vanilloid (capsaicin) receptors in the rat: distribution in the brain, regional differences in the spinal cord, axonal transport to the periphery, and depletion by systemic vanilloid treatment. Brain Res. 1995;703(1-2):175–183. doi: 10.1016/0006-8993(95)01094-7. [DOI] [PubMed] [Google Scholar]
- 13.Wong GY, Gavva NR. Therapeutic potential of vanilloid receptor TRPV1 agonists and antagonists as analgesics: Recent advances and setbacks. Brain Res Rev. 2009;60(1):267–277. doi: 10.1016/j.brainresrev.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Asfaw TS, Hypolite J, Northington GM, et al. Acute colonic inflammation triggers detrusor instability via activation of TRPV1 receptors in a rat model of pelvic organ cross-sensitization. Am J Physiol Regul Integr Comp Physiol. 2011;300(6):R1392–R1400. doi: 10.1152/ajpregu.00804.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ustinova EE, Fraser MO, Pezzone MA. Colonic irritation in the rat sensitizes urinary bladder afferents to mechanical and chemical stimuli: an afferent origin of pelvic organ cross-sensitization. Am J Physiol Renal Physiol. 2006;290(6):F1478–F1487. doi: 10.1152/ajprenal.00395.2005. [DOI] [PubMed] [Google Scholar]
- 16.Brumovsky PR, Gebhart GF. Visceral organ cross-sensitization - an integrated perspective. Auton Neurosci. 2010;153(1-2):106–115. doi: 10.1016/j.autneu.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malykhina AP. Neural mechanisms of pelvic organ cross-sensitization. Neuroscience. 2007;149(3):660–672. doi: 10.1016/j.neuroscience.2007.07.053. [DOI] [PubMed] [Google Scholar]
- 18.Ustinova EE, Fraser MO, Pezzone MA. Cross-talk and sensitization of bladder afferent nerves. Neurourol Urodyn. 2010;29(1):77–81. doi: 10.1002/nau.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pezzone MA, Liang R, Fraser MO. A model of neural cross-talk and irritation in the pelvis: implications for the overlap of chronic pelvic pain disorders. Gastroenterology. 2005;128(7):1953–1964. doi: 10.1053/j.gastro.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Qin C, Malykhina AP, Akbarali HI, et al. Cross-organ sensitization of lumbosacral spinal neurons receiving urinary bladder input in rats with inflamed colon. Gastroenterology. 2005;129(6):1967–1978. doi: 10.1053/j.gastro.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Ustinova EE, Gutkin DW, Pezzone MA. Sensitization of pelvic nerve afferents and mast cell infiltration in the urinary bladder following chronic colonic irritation is mediated by neuropeptides. Am J Physiol Renal Physiol. 2007;292(1):F123–F130. doi: 10.1152/ajprenal.00162.2006. [DOI] [PubMed] [Google Scholar]
- 22.Malykhina AP, Qin C, Greenwood-Van MB, et al. Hyperexcitability of convergent colon and bladder dorsal root ganglion neurons after colonic inflammation: mechanism for pelvic organ cross talk. Neurogastroenterol Motil. 2006;18(10):936–948. doi: 10.1111/j.1365-2982.2006.00807.x. [DOI] [PubMed] [Google Scholar]
- 23.Pan XQ, Gonzalez JA, Chang S, et al. Experimental colitis triggers the release of substance P and calcitonin gene-related peptide in the urinary bladder via TRPV1 signaling pathways. Exp Neurol. 2010;225(2):262–273. doi: 10.1016/j.expneurol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Oortgiesen M, Li L, et al. Capsaicin inhibits activation of voltage-gated sodium currents in capsaicin-sensitive trigeminal ganglion neurons. J Neurophysiol. 2001;85(2):745–758. doi: 10.1152/jn.2001.85.2.745. [DOI] [PubMed] [Google Scholar]
- 25.Malykhina AP, Qin C, Foreman RD, et al. Colonic inflammation increases Na+ currents in bladder sensory neurons. Neuroreport. 2004;15(17):2601–2605. doi: 10.1097/00001756-200412030-00008. [DOI] [PubMed] [Google Scholar]
- 26.Christianson JA, Liang R, Ustinova EE, et al. Convergence of bladder and colon sensory innervation occurs at the primary afferent level. Pain. 2007;128(3):235–243. doi: 10.1016/j.pain.2006.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cregg R, Momin A, Rugiero F, et al. Pain channelopathies. J Physiol. 2010;588(Pt 11):1897–1904. doi: 10.1113/jphysiol.2010.187807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beyak MJ, Ramji N, Krol KM, et al. Two TTX-resistant Na+ currents in mouse colonic dorsal root ganglia neurons and their role in colitis-induced hyperexcitability. Am J Physiol Gastrointest Liver Physiol. 2004;287(4):G845–G855. doi: 10.1152/ajpgi.00154.2004. [DOI] [PubMed] [Google Scholar]
- 29.Beyak MJ, Vanner S. Inflammation-induced hyperexcitability of nociceptive gastrointestinal DRG neurones: the role of voltage-gated ion channels. Neurogastroenterol Motil. 2005;17(2):175–186. doi: 10.1111/j.1365-2982.2004.00596.x. [DOI] [PubMed] [Google Scholar]
- 30.Bielefeldt K, Ozaki N, Gebhart GF. Mild gastritis alters voltage-sensitive sodium currents in gastric sensory neurons in rats. Gastroenterology. 2002;122(3):752–761. doi: 10.1053/gast.2002.31901. [DOI] [PubMed] [Google Scholar]
- 31.Bielefeldt K, Ozaki N, Gebhart GF. Experimental ulcers alter voltage-sensitive sodium currents in rat gastric sensory neurons. Gastroenterology. 2002;122(2):394–405. doi: 10.1053/gast.2002.31026. [DOI] [PubMed] [Google Scholar]
- 32.King DE, Macleod RJ, Vanner SJ. Trinitrobenzenesulphonic acid colitis alters Na 1.8 channel expression in mouse dorsal root ganglia neurons. Neurogastroenterol Motil. 2009;21(8):880–e64. doi: 10.1111/j.1365-2982.2009.01279.x. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Gebhart GF. Differential purinergic signaling in bladder sensory neurons of naive and bladder-inflamed mice. Pain. 2010;148(3):462–472. doi: 10.1016/j.pain.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miranda-Morales M, Ochoa-Cortes F, Stern E, et al. Axon reflexes evoked by transient receptor potential vanilloid 1 activation are mediated by tetrodotoxin-resistant voltage-gated Na+ channels in intestinal afferent nerves. J Pharmacol Exp Ther. 2010;334(2):566–575. doi: 10.1124/jpet.110.165969. [DOI] [PubMed] [Google Scholar]
- 35.Berkley KJ. A life of pelvic pain. Physiol Behav. 2005;86(3):272–280. doi: 10.1016/j.physbeh.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Amir R, Devor M. Functional cross-excitation between afferent A- and C-neurons in dorsal root ganglia. Neuroscience. 2000;95(1):189–195. doi: 10.1016/s0306-4522(99)00388-7. [DOI] [PubMed] [Google Scholar]
- 37.Amir R, Michaelis M, Devor M. Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J Neurosci. 2002;22(3):1187–1198. doi: 10.1523/JNEUROSCI.22-03-01187.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amir R, Kocsis JD, Devor M. Multiple interacting sites of ectopic spike electrogenesis in primary sensory neurons. J Neurosci. 2005;25(10):2576–2585. doi: 10.1523/JNEUROSCI.4118-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amir R, Devor M. Chemically mediated cross-excitation in rat dorsal root ganglia. J Neurosci. 1996;16(15):4733–4741. doi: 10.1523/JNEUROSCI.16-15-04733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cang CL, Zhang H, Zhang YQ, et al. PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol Pain. 2009;5:33. doi: 10.1186/1744-8069-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma K, Zhou QH, Chen J, et al. TTX-R Na+ current-reduction by celecoxib correlates with changes in PGE(2) and CGRP within rat DRG neurons during acute incisional pain. Brain Res. 2008;1209:57–64. doi: 10.1016/j.brainres.2008.02.096. [DOI] [PubMed] [Google Scholar]
- 42.Natura G, von Banchet GS, Schaible HG. Calcitonin gene-related peptide enhances TTX-resistant sodium currents in cultured dorsal root ganglion neurons from adult rats. Pain. 2005;116(3):194–204. doi: 10.1016/j.pain.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 43.Pinto V, Derkach VA, Safronov BV. Role of TTX-sensitive and TTX-resistant sodium channels in Adelta- and C-fiber conduction and synaptic transmission. J Neurophysiol. 2008;99(2):617–628. doi: 10.1152/jn.00944.2007. [DOI] [PubMed] [Google Scholar]
- 44.Bernardini N, Neuhuber W, Reeh PW, et al. Morphological evidence for functional capsaicin receptor expression and calcitonin gene-related peptide exocytosis in isolated peripheral nerve axons of the mouse. Neuroscience. 2004;126(3):585–590. doi: 10.1016/j.neuroscience.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 45.Bautista DM, Jordt SE, Nikai T, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124(6):1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 46.Salas MM, Hargreaves KM, Akopian AN. TRPA1-mediated responses in trigeminal sensory neurons: interaction between TRPA1 and TRPV1. Eur J Neurosci. 2009;29(8):1568–1578. doi: 10.1111/j.1460-9568.2009.06702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem. 2010;285(20):15167–15177. doi: 10.1074/jbc.M110.106153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohta T, Imagawa T, Ito S. Novel gating and sensitizing mechanism of capsaicin receptor (TRPV1): tonic inhibitory regulation of extracellular sodium through the external protonation sites on TRPV1. J Biol Chem. 2008;283(14):9377–9387. doi: 10.1074/jbc.M709377200. [DOI] [PubMed] [Google Scholar]
- 49.Sculptoreanu A, Kullmann FA, Artim DE, et al. Nitro-oleic acid inhibits firing and activates TRPV1- and TRPA1-mediated inward currents in dorsal root ganglion neurons from adult male rats. J Pharmacol Exp Ther. 2010;333(3):883–895. doi: 10.1124/jpet.109.163154. [DOI] [PMC free article] [PubMed] [Google Scholar]