

Abstract
Botulinum neurotoxins (BoNTs) are the most toxic proteins known to man, exposure to which results in flaccid paralysis. Given their extreme potency, these proteins have become studied as possible weapons of bioterrorism; however, effective treatments that function after intoxication have not progressed to the clinic. Here, we have reexamined one of the most effective inhibitors, 2,4-dichlorocinnamyl hydroxamate, in the context of the known plasticity of the BoNT/A light chain metalloprotease. Our studies have shown that modifications of this compound are tolerated and result in improved inhibitors, with the best compound having an IC50 of 0.23 μM. Given the inconsistency of structure-activity relationship trends observed across similar compounds, this data argues for caution in extrapolating across structural series.
Keywords: botulinum neurotoxin, hydroxamic acids, bioterrorism, rational inhibitor design
Botulinum neurotoxins (BoNTs), the proteins that cause the disease botulism, have become heavily studied in recent years due to the dual nature of these proteins as both potent biopharmaceuticals as well as possible weapons of bioterrorism.1 These proteins are produced by the gram-positive bacteria Clostridium botulinum and consist of seven immunologically distinct serotypes (A-G) with varying tropism. Estimates have been made that approximately 1,000 people are afflicted with botulism each year, with the majority of cases being from BoNT/A, and a minority from BoNT/B and /E.2 With an estimated lethality of 1-5 ng/kg, BoNT/A is the most toxic protein known to man. Because of this risk, the CDC has classified BoNTs as ‘category A’ agents, and recent reevaluation by a U.S. Federal panel of scientists and security experts has recommended BoNT be designated a ‘Tier 1 select agent’, a category subject to the highest possible security standards.3
While vaccine-based therapeutics designed to counteract the extreme morbidity and mortality associated with BoNT intoxication have been reported, optimal efficacy is observed prior to toxin exposure, limiting their use primarily to prophylactic measures.4 The biochemical mechanism of action of BoNTs has been closely studied and three distinct stages of the intoxication process have been characterized: neuronal cell surface receptor binding and internalization, toxin translocation out of endosomes into the cytosol, and light chain (LC) metalloprotease recognition and cleavage of endogenous SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complexes essential for vesicle fusion and neurotransmitter exocytosis.5 The ultimate culmination of these molecular events is a blockage of neurotransmitter exocytosis, resulting in flaccid paralysis.
Most small molecule studies to date have focused on inhibition of the 50 kDa LC of BoNT/A,1 not only because it is this serotype that is most lethal to humans but also because it possesses an extremely long half-life in cell culture and in vivo.6 Indeed, the duration of action of BoNT/A when used therapeutically can be as long as 3 to 12 months.7 This serotype specifically recognizes and cleaves SNAP-25 (synaptosomal-associated protein of 25 kDa), a critical component of the SNARE complex anchored onto the intracellular face of motor neuron membrane. Small molecules have been espoused as particularly suitable therapeutics for botulism given that a successful therapeutic must be capable of treating the patient after exposure to the toxin, meaning after internalization of the toxin into peripheral motor neurons. While proteins and other biological therapeutics frequently suffer from poor cellular permeability, small molecules can be designed such that they have acceptable permeability profiles.
A number of small molecules have been reported to inhibit the BoNT/A LC through a variety of mechanisms.8-14 Among these compounds, cinnamyl hydroxamates have been particularly successful inhibitors of BoNT/A due to their tight binding to metal ions, and a variety of leads have been reported.8-10 One of the most potent compounds in vitro, 2,4-dichlorocinnamyl hydroxamic acid 1,8 displayed marginal in vivo activity in a mouse model of BoNT/A exposure and was the first to highlight the poor predictive value of common cell models of intoxication.9 More recently, we have reported a series of benzothiophene-2-yl hydroxamic acids that are among the most potent small molecule inhibitors discovered to date and also display more favorable pharmacologic properties.10
In contrast to rational design efforts, there also has been recent interest in the development of pharmacophore models for predicting BoNT inhibitors in silico. In particular, a “three zone pharmacophore” has been reported and validated in a screen of the National Cancer Institute Open Repository of compounds.12 Lead compounds identified in this screen have been further improved using this model as a guide.13 Interestingly, one of the other motivations for the development of an in silico screening model was the difficulty in optimizing early lead candidates into more efficacious inhibitors.14 Indeed, the authors reported an inability to obtain crystals suitable for crystallographic studies or improve lead candidates through synthetic studies guided by molecular docking experiments.
This difficulty in further optimizing lead compounds for BoNT/A inhibition is echoed in other studies where marginal improvement in inhibition has been achieved through rational design.15 As a result, this study was conducted to examine the flexibility of the active site of BoNT/A, particularly in the context of the plasticity present at the β-exosite that is adjacent to the active site.16 By designing compounds that reach further into the hydrophobic pockets of this region and provide a “handle” for the enzyme to recognize, we expected the potency of a given inhibitor would improve. Also, while not designed to uncover better therapeutic candidates per se, removal of the pharmacologically disfavored aryl halides without sacrificing inhibitor potency could provide a secondary benefit to this study.10
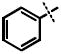
The available structural data indicates that the 2-chloro moiety of 1 makes contacts with the side chain of Arg363 in the BoNT/A LC, filling a void that is observed in the structures of complexes missing this group.16 We speculated that by fixing the 2-chloro substituent and varying the para substituent of 1, the flexibility of the β-exosite could be directly tested. Our initial studies commenced with the preparation of a common intermediate from which a number of analogs of 1 could be rapidly prepared via cross-coupling chemistry. Interestingly, this compound, 2-chloro-4-bromocinnamyl hydroxamate 2, was an equipotent inhibitor of BoNT/A LC as 1 (IC50 = 0.69 μM, Table 1). Suzuki coupling of protected 2 with phenylboronic acid followed by deprotection yielded biarylcinnamyl hydroxamate 4, which also was a comparable inhibitor of BoNT/A (IC50 = 1.23 μM, Table 1). This was a particularly surprising finding; the nonconservative change of a chloro substituent for a phenyl group had almost no effect on the inhibitory potency of the compound, despite the dramatic change in stereoelectronics. In contrast, 2-chlorocinnamyl hydroxamate 3 displays a greater than ten-fold loss in potency.
Table 1.
Inhibition of BoNT/A LC by 4-substituted 2-chlorocinnamyl hydroxamic acids.a
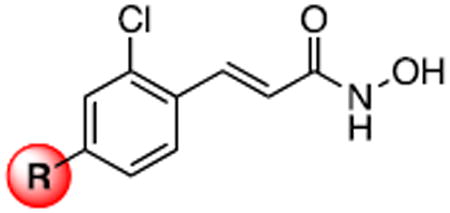
| ||
|---|---|---|
| Compound | R | IC50 (μM) |
| 1 |
|
0.67 ± 0.04 |
| 2 |
|
0.69 ± 0.05 |
| 3 |
|
13.4 ± 1.1 |
| 4 |
|
1.23 ± 0.57 |
| 5 |
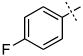
|
9.49 ± 4.11 |
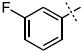
| 6 |
|
31.3 ± 4.6 |
| 7 |
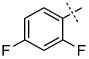
|
0.98 ± 0.52 |
| 8 |
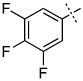
|
0.64 ± 0.17 |
| 9 |
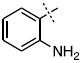
|
0.55 ± 0.10 |
| 10 |
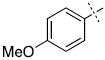
|
32.3 ± 2.6 |
| 11 |
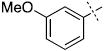
|
24.9 ± 3.0 |
| 12 |
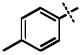
|
60.7 ± 3.8 |
| 13 |
|
>100 |
| 14 |
|
38.2 ± 7.0 |
| 15 |
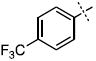
|
35.7 ± 3.4 |
| 16 |
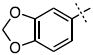
|
33.1 ± 5.4 |
| 17 |
|
15.4 ± 1.7 |
| 18 |
|
>100 |
| 19 |

|
68.4 ± 5.4 |
| 20 |
|
42.7 ± 2.8 |
| 21 |
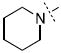
|
1.13 ± 0.29 |
| 22 |
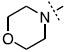
|
35 ± 6.3 |
| 23 |
|
>100 |
IC50 values were determined using the SNAPtide® assay.17
Assuming compounds such as 4 would bind to BoNT/A LC in a manner similar to the parent hydroxamate 1, one would anticipate a binding orientation with the cinnamyl moiety oriented towards the 370 loop that makes up the β-exosite of the light chain.16 In the case of 1, the sharp boundary created by the 370 loop creates a pocket that has been reported to be entirely filled by the 2,4-dichloro-substituted phenyl ring. Furthermore, it has been reported that bulky substituents such as in 4-tert-butylcinnamyl hydroxamate result in a dramatic loss of potency, arguing that changes in this position will result in poor inhibitor candidates.8 Yet, biaryl compound 4 is a comparable inhibitor to1, refuting this hypothesis as a general rule within this series.
With this data in hand, we further explored this series of biarylcinnamyl hydroxamates to determine the extent of substitution that would be tolerated without compromising inhibition. Using common intermediate 2, a series of biaryl cinnamyl hydroxamates were synthesized. All compounds were prepared using Suzuki or Heck cross-couplings to generate the desired hydroxamates as final products in good yield and excellent purity. This initial set of compounds was subsequently screened in a high-throughput SNAPtide® assay widely used by our laboratory as well as others as a primary triage for the discovery of BoNT/A inhibitors.17 This assay relies on the appearance of fluorescence upon cleavage of the FRET-quenched SNAPtide® substrate, allowing relative IC50 values to be calculated.
Surprisingly, many of the 4-aryl-cinnamyl hydroxamates were potent inhibitors of the BoNT/A LC, with five compounds (4, 7, 8, 9, and 21) being comparable or better inhibitors than the parent compound 1. More importantly, some SAR can be clearly seen from the data. First, while the biphenyl moiety is tolerated (1 vs. 4), meta substitution of the newly installed phenyl ring results in poor inhibition (i.e., 6, 11, 13, 14, 16). Also, the binding pocket seems not to tolerate additional steric bulk at the para position (10, 12, 15, 17, and 18), but does tolerate electronic modulation of the aromatic ring (4 vs. 7, 8). Interestingly, not only is the steric bulk of an aromatic ring tolerated, but also other groups such as the piperidyl moiety of 21. Additional SAR can be gleaned from compounds 21-23. In 21, the piperidine group is tolerated similar to the phenyl group of 4 (IC50 = 1.13 and 1.23 μM, respectively), yet changing the piperidine to a morpholine or piperazine group (22 and 23) results in very poor inhibitors, likely indicating that the lone pairs of the heteroatoms have disfavored interactions with the enzyme.
Given the structures of these compounds, a concern regarding their ability to interfere with the fluorescence assay and result in significant inner-filter effect quenching18 could be raised. To address this, a set of compounds were tested for intrinsic fluorescence, as well as their ability to quench the fluorescence of the product of SNAPtide® cleavage. Both potent inhibitors as well as compounds that showed poor to no inhibition were included in this testing. In all cases, no effect on SNAPtide® cleavage product fluorescence was observed, indicating that the observed inhibition is not an artifact of the assay.
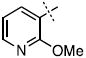
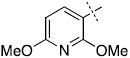
We next chose to replace the phenyl ring with various six-(24-38) or five-membered (39-44) heterocyclic analogs both in an attempt to improve on the interactions between the inhibitor and enzyme, as well as to test our SAR-derived hypotheses (Table 2). After screening this second-generation set, some interesting trends emerged. First, of the 21 compounds, 8 compounds had improved inhibition relative to the parent hydroxamate (Table 2). Indeed, 3-pyridyl substitution resulted in compound 24 which displayed an IC50 of 230 nM, a three-fold improvement in potency over 1.
Table 2.
Inhibition of BoNT/A LC by second generation pyridyl-cinnamyl hydroxamic acids.a
|
| ||
|---|---|---|
| Compound | R | IC50 (μM) |
| 24 |
|
0.23 ± 0.02 |
| 25 |
|
27.3 ± 7.1 |
| 26 |
|
35.3 ± 9.6 |
| 27 |
|
0.56 ± 0.15 |
| 28 |
|
0.75 ± 0.25 |
| 29 |
|
0.66 ± 0.19 |
| 30 |
|
8.01 ± 1.7 |
| 31 |
|
>100 |
| 32 |
|
10 ± 1.2 |
| 33 |
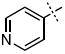
|
0.61 ± 0.12 |
| 34 |
|
2.79 ± 0.35 |
| 35 |
|
1.21 ± 0.2 |
| 36 |
|
73.2 ± 4.2 |
| 37 |
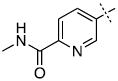
|
12.3 ± 1.6 |
| 38 |
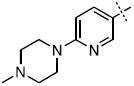
|
21.3 ± 5.4 |
| 39 |
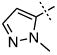
|
1.32 ± 0.27 |
| 40 |
|
24.9 ± 6.5 |
| 41 |
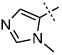
|
22.3 ± 8.1 |
| 42 |
|
1.16 ± 0.38 |
| 43 |
|
66.2 ± 5.8 |
| 44 |
|
9.11 ± 0.87 |
IC50 values were determined using the SNAPtide® assay.17
Interestingly, some of the SAR trends observed in the biphenyl series remained valid in this series (Table 2). In particular, meta substitution relative to the second aromatic ring was disfavored and resulted in poor inhibition (25, 26, and 31), as did bulky substitution at the para position (36, 37, and 38). Ortho substitution was tolerated and resulted in good inhibitors in both series (9 and 29). However, addition of a p-methoxy moiety resulted in potent inhibition within the pyridyl series, in contrast to the first generation biphenyls (27 vs. 10).
Indeed, in the pyridyl series, p-methoxy substitution had little effect on the IC50 (24 vs. 27 or 28), while this same substitution in the biphenyl series attenuated the inhibitory activity (4 vs. 10). The reason for this discrepancy could be that because of the change in the electronics of the aromatic moiety by inclusion of the heteroatom, the enzyme can now tolerate the additional steric bulk at this position. Put another way, we hypothesize that the inconsistent SAR across compound series is best explained through the known plasticity of this enzyme;16 addition of the hydrogen bond-accepting pyridyl ring leads to a structural change in the enzyme that induces a pocket capable of accommodating additional substitution that was not tolerated in the initial biphenyl series (i.e., 4 vs. 12). A further example that supports this hypothesis is given in the comparison of compounds 34 and 35; here, inclusion of two nitrogen atoms is comparable to the parent 4 and in the presence of both heteroatoms, the addition of the p-methoxy group in 35 leads to improved inhibition.
In order to explore the nature of the binding interactions between the biaryl cinnamate inhibitors and the BoNT/A LC, we performed molecular modeling experiments in which compounds were docked onto the active site of the protease (Glide). Starting from the published crystal structure of 1 with BoNT/A (PDB ID: 2IMA),16 it was possible to dock compound 1 in the proper position relative to the crystal structure; however, when using this structure for the biaryl inhibitors, significant rotational strain was introduced in the inhibitor about the cinnamate double bond and the aryl substituent extended through the back of the pocket, resulting in poor binding energies (Figure 1). We speculated that this poor docking could be corrected by allowing flexibility in the enzyme and consequently, we attempted to dock the biaryl compounds using induced fit parameters to allow for additional enzyme flexibility. Under these conditions, the compounds could be accommodated as reflected by Glide scores that indicated that these compounds would be comparable or better inhibitors to 1.
Figure 1.
Docking of 1 (red) and 24 (blue) onto BoNT/A LC (PDB ID: 2IMA). The white arrow indicates the severe steric clash with the wall of the binding pocket.
The results of this screen show that 6 of the 43 compounds tested had better potency than the parent compound (IC50 < 0.67 μM), and an additional 9 compounds had comparable potency to the parent hydroxamate (IC50 ≤ 2 μM), a “hit” rate of 35%. This is a surprisingly large number of inhibitors, particularly given that the structural data has suggested that certain modifications (e.g., removal of the 4-chloro moiety of 1) are not tolerated. In total, this study highlights the difficulty in establishing structure-activity relationships with BoNT/A LC and cautions against making generalizations across compound series, even when there is significant structural homology present.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (AI082190 to T.J.D.). S.G. was supported by an internship from the California Institute for Regenerative Medicine (TB1-01186).
Footnotes
Supplementary Material
Synthetic detail for all compounds is available free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Willis B, Eubanks LM, Dickerson TJ, Janda KD. Angew Chem Intl Ed. 2008;47:8360. doi: 10.1002/anie.200705531. [DOI] [PubMed] [Google Scholar]
- 2.Foran PG, Davletov B, Meunier FA. Trends Mol Med. 2003;9:291. doi: 10.1016/s1471-4914(03)00113-8. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharjee Y. Science. 2011;322:1491–1492. doi: 10.1126/science.332.6037.1491. [DOI] [PubMed] [Google Scholar]
- 4.Aoki KR, Smith LA, Atassi MZ. Crit Rev Immunol. 2010;30:167. doi: 10.1615/critrevimmunol.v30.i2.50. [DOI] [PubMed] [Google Scholar]
- 5.Schiavo G, Matteoli M, Montecucco C. Physiol Rev. 2000;80:717. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Zurawski TH, Meng J, Lawrence G, Olango WM, Finn DP, Wheeler L, Dolly JO. J Biol Chem. 2011;286:6375. doi: 10.1074/jbc.M110.181784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Salas E, Ho H, Garay P, Steward LE, Aoki KR. Mov Disorder. 2004;19(Suppl 8):S23. doi: 10.1002/mds.20006. [DOI] [PubMed] [Google Scholar]
- 8.Boldt GE, Kennedy JP, Janda KD. Org Lett. 2006;8:1729. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. Proc Natl Acad Sci USA. 2007;104:2602. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capek P, Zhang Y, Barlow DJ, Houseknecht KL, Smith GR, Dickerson TJ. ACS Chem Neurosci. 2011;2:288. doi: 10.1021/cn200021q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruthel G, Burnett JC, Nuss JE, Wanner LM, Tressler LE, Torres-Melendez E, Sandwick SJ, Retterer CJ, Bavari S. Toxins. 2011;3:207. doi: 10.3390/toxins3030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hermone AR, Burnett JC, Nuss JE, Tresler LE, Nguyen TL, Solaja BA, Vennerstrom JL, Schmidt JJ, Wipf P, Bavari S, Gussio R. ChemMedChem. 2008;3:1905. doi: 10.1002/cmdc.200800241. [DOI] [PubMed] [Google Scholar]
- 13.Burnett JC, Wang C, Nuss JE, Nguyen TL, Hermone AR, Schmidt JJ, Gussio R, Wipf P, Bavari S. Bioorg Med Chem Lett. 2009;19:5811. doi: 10.1016/j.bmcl.2009.01.111. [DOI] [PubMed] [Google Scholar]
- 14.Wang C, Widom J, Petronijevic F, Burnett JC, Nuss JE, Bavari S, Gussio R, Wipf P. Heterocycles. 2009;79:487. [Google Scholar]
- 15.Capkova K, Yoneda Y, Dickerson TJ, Janda KD. Bioorg Med Chem Lett. 2007;17:6463. doi: 10.1016/j.bmcl.2007.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. Chem Biol. 2007;14:533. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 17.Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, Janda KD. J Comb Chem. 2006;8:513. doi: 10.1021/cc060010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Kati W, Chen C-H, Tripathi R, Molla A, Kohlbrenner W. Anal Biochem. 1999;267:331. doi: 10.1006/abio.1998.3014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.