

Abstract
High molecular weight heparins promote the protein Z-dependent protease inhibitor (ZPI) inhibition of factors Xa (FXa) and XIa (FXIa) by a template mechanism. To map the heparin-binding site of ZPI, the role of basic residues of the D-helix (residues Lys-113, Lys-116, and Lys-125) in the interaction with heparin was evaluated by either substituting these residues with Ala (ZPI-3A) or replacing the D-helix with the corresponding loop of the non-heparin binding serpin α1-proteinase inhibitor (ZPI-D-helixα1-PI). Furthermore, both the C-helix (contains two basic residues Lys-104 and Arg-105) and D-helix of ZPI were substituted with the corresponding loops of α1-PI (ZPI-CD-helixα1-PI). All mutants exhibited near normal reactivity with FXa and FXIa in the absence of cofactors and in the presence of protein Z and membrane cofactors. By contrast, the mutants interacted with heparin with a lower affinity and the ~48-fold heparin-mediated enhancement in the rate of FXa inhibition by ZPI was reduced to ~30-fold for ZPI-3A, ~15-fold for ZPI-D-helixα1-PI, and ~8-fold for ZPI-CD-helixα1-PI. Consistent with a template mechanism for heparin cofactor action, ZPI-CD-helixα1-PI inhibition of a FXa mutant containing a mutation in the heparin-binding site (FXa-R240A) was minimally affected by heparin. A significant decrease (~2–5-fold) in the heparin template effect was also observed for the inhibition of FXIa by ZPI mutants. Interestingly, ZPI derivatives exhibited a markedly elevated stoichiometry of inhibition with FXIa in the absence of heparin. These results suggest that basic residues of both the C and D helices of ZPI interact with heparin to modulate the inhibitory function of the serpin.
Protein Z-dependent protease inhibitor (ZPI)1 is a member of the serpin superfamily of protease inhibitors in plasma that regulates the proteolytic activity of two key proteases of the clotting cascade, factors Xa (FXa) and XIa (FXIa) (1,2). It circulates in plasma as a tight complex with its vitamin K-dependent cofactor protein Z (1,2). The complex formation with protein Z is essential for ZPI to effectively regulate the activity of membrane-bound FXa (1,2). Thus, in the presence of negatively charged phospholipid vesicles and calcium, protein Z promotes the reactivity of ZPI with FXa by greater than three orders of magnitude (1–3). The rate accelerating effect of protein Z appears to be largely mediated through a template-bridging mechanism in which the ZPI-bound cofactor binds via its Gla-domain to the negatively charged membrane surface that is also bound by FXa, thereby facilitating recognition and high-affinity interaction of the serpin with the protease (1–3). A direct Ca2+-dependent interaction between the Gla-domains of FXa and protein Z has been demonstrated to contribute to the specificity of complex formation on negatively charged phospholipid vesicles (4). In contrast to its reaction with FXa, the reactivity of ZPI with FXIa does not require the cofactor function of protein Z, but rather the serpin itself effectively inhibits the activity of FXIa in the absence of a cofactor (5). Nevertheless, recent results have indicated that high molecular weight heparin also functions as a cofactor to accelerate ZPI inhibition of both FXa and FXIa by 10–100-fold independent of protein Z and membrane (6). A bell-shaped dependence of the accelerating effect on heparin concentration further indicated that the polysaccharide cofactor promotes the inhibition of both FXa and FXIa by a template-bridging mechanism reminiscent of heparin accelerating the inhibition of thrombin by the serpin antithrombin (6,7). In this mechanism of cofactor function, heparin simultaneously binds to both the protease and the serpin, thereby enhancing the reaction by lowering the KD for bimolecular interaction (7). The binding sites for heparin on both the proteases, FXa and FXIa, have been fully characterized (8–10). However, the site on ZPI that interacts with heparin has not been identified.
In a recent study, it was demonstrated that ZPI binds heparin with a similar or higher affinity than do most other heparin-binding serpins which are known to regulate the activity of the serine proteases of the clotting cascade (6). These serpins, with the exception of protein C inhibitor which interacts with heparin via basic residues of the H-helix (11), bind heparin primarily via basic residues of the D-helix (12–14). Structural data suggests that the D-helix of ZPI contains 3 basic residues (Lys-113, Lys-116, and Lys-125) which can potentially interact with heparin (15,16). To test this possibility, in this study, we prepared two ZPI mutants in which either all three basic residues of the serpin were replaced with Ala (ZPI-3A) or the entire D-helix of the serpin was replaced with the corresponding helix of the non-heparin-binding serpin, α1-protease inhibitor (ZPI-D-helixα1-PI). Noting that the preceding C-helix of ZPI also has two basic residues, a chimeric construct was prepared in which both C and D helices of ZPI were replaced with the corresponding loops of α1-PI (ZPI-CD-helixα1-PI). The ZPI mutants were expressed in an E. coli expression system and characterized with respect to their ability to inhibit the target proteases, FXa and FXIa, in the absence and presence of cofactors. All mutants exhibited normal reactivity with FXa in the absence of cofactors or in the presence of protein Z and membrane cofactors. However, the accelerating effect of heparin on FXa inhibition by the ZPI mutants was decreased to varying degrees. The accelerating effect of heparin was essentially abolished for the ZPI-CD-helixα1-PI inhibition of the heparin-exosite mutant FXa-R240A which is known to have lost its high affinity for heparin. Direct binding studies indicated that the affinity of the mutants (in particular the ZPI-CD-helixα1-PI) for heparin has been markedly decreased. Similar results were obtained with FXIa, though, the stoichiometries of inhibition were markedly elevated (4-fold) for the mutants in the absence of heparin. These results suggest that heparin binds to both the C and D-helices of ZPI to regulate its reactivity with FXa and FXIa in the clotting cascade.
Materials and Methods
Construction and expression of recombinant proteins
Wild-type ZPI was prepared from E. coli using the SUMO fusion expression system and characterized as described (15). The ZPI mutants in which either the three basic residues Lys-113, Lys-116 and Lys-125 were substituted with Ala (ZPI-3A) or its D-helix from residues Thr-Lys-Pro-Gly-Leu-Leu-Pro-Ser-Leu-Phe-Lys-Gly-Leu-Arg-Glu-Thr-Leu-Ser were replaced with the corresponding residues of α1-PI (Glu-Ala-Gln-Ile-His-Glu-Gly-Phe-Gln-Glu-Leu-Leu-Arg-Thr-Leu-Asn) (17), were prepared by the PCR mutagenesis approach and expressed using the same expression/purification system. Another ZPI chimera was prepared in which in addition to the D-helix, the C-helix residues and those of the loop connecting the D-helix to the C-helix (Gly-Pro-Thr-Glu-Thr-Gln-Ile-Lys-Arg-Gly-Leu-His-Leu-Gln-Ala-Leu-Lys-Pro) were also replaced with the corresponding residues of α1-PI (Ala-Asp-Thr-His-Asp-Glu-Ile-Leu-Glu-Gly-Leu-Asn-Phe-Asn-Leu-Thr-Glu-Ile-Pro) (ZPI-CD-helixα1-PI). ZPI has a highly acidic N-terminal tail on the A-helix that is not conserved in other serpins, thus we deleted the first 52-residues of this loop of ZPI in another construct (ZPI-des-NT) and expressed it using the same expression system. The concentrations of ZPI derivatives were calculated from their absorbance at 280 nm using a molar absorption coefficient of 31,525 M−1 cm−1 as described (3). The expression, purification and characterization of the heparin-binding exosite mutant of FXa-R240A (8) and recombinant protein Z (4) have been described.
Human plasma proteins, FXa, FXIa, thrombin and antithrombin (AT) were purchased from Haematologic Technologies Inc. (Essex Junction, VT). Unfractionated therapeutic heparin (average MW ~15 kDa) was from Quintiles Clinical Supplies (Mt. Laurel, NJ). The chromogenic substrates, Spectrozyme FXa (MeO-CO-D-Chg-Gly-Arg-pNA, SpFXa) and Spectrozyme TH (H-D-hexahydrotyrosol-Ala-Arg-pNA, SpTH), were purchased from American Diagnostica (Greenwich, CT) and S2366 (L-pyroglutamyl-L-prolyl-L-arginine-p-nitroanilide) was purchased from Diapharma (West Chester, OH).
Determination of the second-order association rate constants
A discontinuous assay method was used to measure the second order associate rate constants (k2) for ZPI inhibition of FXa and FXIa under pseudo-first-order conditions both in the absence and presence of heparin and protein cofactors as described (6,10). Briefly, each enzyme (1 nM active site) was incubated with ZPI derivatives (100–500 nM in the absence of a cofactor and 10–100 nM in the presence of heparin) in 0.1 M NaCl, 0.02 M Tris-HCl, pH 7.5 and 2.5 mM Ca2+ (TBS/Ca2+) containing 0.1 mg/mL BSA and 0.1% PEG 8000. In the presence of protein Z (50 nM), the inhibition of FXa (0.5 nM) by ZPI (5–20 nM) was monitored in the presence of 25 μM phospholipid vesicles composed of 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) in the same TBS buffer system. All reactions were carried out in 50 μL volumes in 96-well plates and at different time points, 50 μL specific chromogenic substrate in TBS (SpFXa for FXa and S2366 for FXIa) was added to each reaction and the remaining enzyme activities were measured at 405 nm using a Vmax Kinetic Plate Reader (Molecular Devices, Menlo Park, CA) as described (10). The observed pseudo-first-order rate constants (kobs) and the second-order association rate constants for uncatalyzed and catalyzed reactions were calculated as described previously. All values are presented as the average of at least 3 independent measurements ±S.D.
Binding to Heparin-Sepharose
ZPI derivatives (~200 μg) in TBS/Ca2+ were applied on a 1-mL Heparin-Sepharose column pre-equilibrated with the same TBS buffer. The column was washed with 2 mL TBS followed by elution with a step gradient of 0.1–0.5 M NaCl using increments of 10 mM salt. To construct a heparin-affinity profile for each mutant, the OD280 of 1 mL elution fractions were measured and plotted vs. concentrations of NaCl.
Competitive binding assay
The affinity of ZPI derivatives for interaction with heparin was evaluated by the ability of ZPI derivatives to compete with antithrombin for heparin in the heparin-catalyzed inhibition of thrombin by antithrombin (AT) by a kinetic assay. In this assay, the inhibition of thrombin (0.5 nM) by AT (2.5 nM) was monitored in the presence of increasing concentrations of ZPI derivatives (0–250 nM) in the presence of a fixed concentration of heparin (5 nM) in TBS/Ca2+ containing 0.1 mg/mL BSA and 0.1% PEG 8000. Following 2–3 min incubation at room temperature, 50 μL thrombin-specific chromogenic substrate, SpTH (100 μM), containing 1 μg/mL polybrene was added to each well and the remaining activity of thrombin was measured as described above. Under these experimental conditions ~80–90% of the thrombin activity in the absence of the competitor (ZPI) was inhibited. KD values for the interaction of heparin with the ZPI derivatives were determined by non-linear regression analysis of the saturable ZPI concentration dependence of recovery of the thrombin activity according to the equation (18):
where kobs,o and kobs represent the pseudo-first-order rate constant for the heparin-accelerated antithrombin-thrombin reaction in the absence and presence of ZPI competitor, respectively (equal to the natural logarithm of the fraction of residual thrombin activity), [ZPI]o is the total ZPI concentration and KZPI,app is the apparent dissociation constant for ZPI binding to heparin. The term on the right represents the fraction of ZPI bound to heparin with an apparent dissociation constant that reflects competition with antithrombin and/or thrombin binding. This equation assumes that ZPI binds to heparin independent of whether antithrombin or thrombin alone is bound, but that this binding is sufficient to block the formation of antithrombin-thrombin-heparin ternary complexes. This is based on the fact that antithrombin binds to sequence-specific sites on a fraction of the heparin chains whereas ZPI binds nonspecifically to multiple overlapping sites on all heparin chains (6). It should be noted that the concentration of antithrombin employed in the competition assay of 2.5 nM is well below the KD of 30 nM previously measured for antithrombin binding to unfractionated heparin (19) and therefore any competitive effect of antithrombin binding to its specific sites on KZPI,app will be minimal under the experimental conditions of the assay.
Stoichiometry of inhibition (SI)
The SI values for the reaction of ZPI derivatives with FXa and FXIa were determined both in the absence and presence of heparin (1000 nM) by titrating 10–100 nM active-site titrated proteases with increasing concentrations (5–20-fold molar excess) of each serpin mutant as described previously (6). Following incubation at room temperature for sufficient time to reach maximal inhibition based on measures k2 values, the residual activity of each protease was monitored from the hydrolysis of the specific chromogenic substrate for each protease at 405 nm as described above. The SI values were determined from the x-intercept of the linear regression fit of the residual activities plotted versus the serpin/protease ratios as described (6).
Results
Expression and analysis of the reactivity of ZPI derivatives with FXa and FXIa in the absence and presence of heparin
The ZPI derivatives were expressed in E. coli using the SUMO fusion expression system and purified to homogeneity on a nickel column as described (15). SDS-PAGE analysis indicated that all ZPI derivatives had been purified to homogeneity and that they were fully reactive with their target proteases, FXa and FXIa (data not shown). The heparin concentration-dependence of second-order rate constants for the inhibition of FXa and FXIa by ZPI derivatives are presented in Figures 1 and 2. In agreement with our previous results, unfractionated heparin accelerated the ZPI inhibition of both FXa (Figure 1A) and FXIa (Figure 1B) by a template mechanism. The k2 values for ZPI inhibition of both proteases in the absence and presence of an optimal concentration of heparin are presented in Tables 1 and 2. The data presented in Table 1 for FXa suggests that heparin accelerates wild-type ZPI inhibition of the protease ~48-fold. The fold accelerating effect of heparin for ZPI inhibition of FXa was reduced to 30-fold for ZPI-3A, 15-fold for ZPI-D-helixα1-PI and 7.7-fold for ZPI-CD-helixα1-PI (Table 1). These results suggest that heparin interacts with basic residues of both C and D helices to promote ZPI inhibition of FXa by a template mechanism. In support of this hypothesis the inhibition of a FXa mutant which has a mutation in its heparin-binding exosite (FXa-R240A) and thus exhibits lower affinity for heparin, by ZPI-CD-helixα1-PI was minimally affected by heparin (Figure 1C). Thus, in contrast to the ~11-fold heparin mediated rate accelerating effect for wild-type ZPI inhibition of FXa-R240A (2.8 × 103 M−1s−1 and 3.0 × 104 M−1s−1 in the absence and presence of heparin, respectively), heparin accelerated ZPI-CD-helixα1-PI inhibition of FXa-R240A 1.8-fold (1.6 × 103 M−1s−1 and 2.9 × 103 M−1s−1 in the absence and presence of heparin, respectively).
Figure 1.
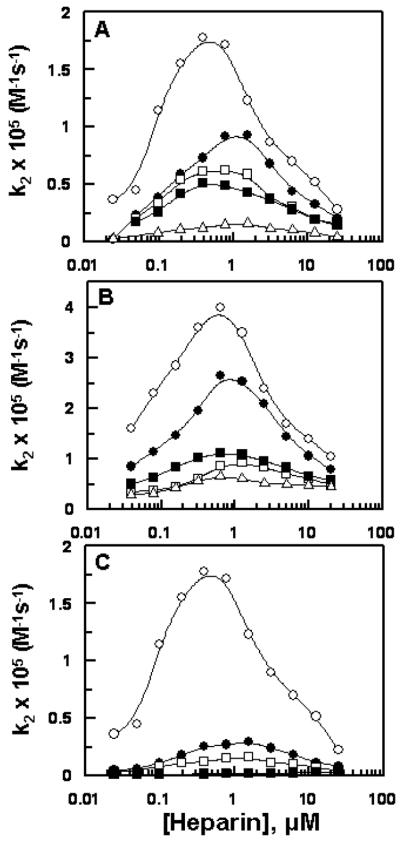
Heparin concentration dependence of the heparin-enhanced inhibition of FXa and FXIa by ZPI derivatives. (A) The heparin-accelerated k2 values for the inhibition of FXa by ZPI-WT (○), ZPI-3A (●), ZPI-des-NT (□), ZPI-D-helixα1-PI (■), and ZPI-CD-helixα1-PI (△) were determined in TBS/Ca2+ containing 0.1% PEG 8000 and 0.1 mg/mL BSA as described under “Materials and Methods”. k2 values at optimal heparin concentrations are presented in Table 1. (B) The same as panel (A) except that the heparin-accelerated k2 values for the inhibition of FXIa by the ZPI derivatives were determined. k2 values at optimal heparin concentrations are presented in Table 2. (C) The same as panel (A) except that the heparin-accelerated k2 values for the inhibition of FXa by ZPI-WT (○) and ZPI-CD-helixα1-PI (□); and FXa-R240A by ZPI-WT (●) and ZPI-CD-helixα1-PI (■) were determined. Data in all three panels are derived from at least three independent measurements ±S.D.
Figure 2.

NaCl gradient elution profiles of ZPI derivatives from Heparin-Sepharose. The ZPI derivatives (200 μg) in TBS/Ca2+ were applied on a 1-mL Heparin-Sepharose column followed by elution with a step gradient of 0.1–0.5 M NaCl using increments of 10 mM salt. The symbols are: ZPI-WT (○), ZPI-3A (●), ZPI-des-NT (□), ZPI-D-helixα1-PI (■), and ZPI-CD-helixα1-PI (△).
Table 1.
Kinetic constants and SI values for the inhibition of FXa by the ZPI derivatives in the absence and presence of heparin
| ZPI derivative | FXa | FXa (+ Hep) | Fold enhancement | ||||
|---|---|---|---|---|---|---|---|
| k2(app) × 103 M−1 S−1 | SI mol I/mol E | k2(app) × SI 104 M−1S−1 | k2(app) × 105 M−1 S−1 | SI mol I/mol E | k2(app) × SI 105 M−1S−1 | ||
| ZPI-WT | 3.4 ± 0.42 | 4.1 ± 0.23 | 1.4 ± 0.2 | 1.8 ± 0.21 | 3.8 ± 0.11 | 6.8 ± 1.0 | 48 |
| ZPI-3A | 2.6 ± 0.15 | 4.5 ± 0.34 | 1.2 ± 0.2 | 0.95 ± 0.05 | 3.8 ± 0.25 | 3.6 ± 0.4 | 30 |
| ZPI-des-NT | 2.1 ± 0.30 | 7.4 ± 0.52 | 1.5 ± 0.3 | 0.68 ± 0.10 | 7.3 ± 0.49 | 5.0 ± 1.1 | 33 |
| ZPI-D-helixα1-PI | 2.9 ± 0.25 | 5.6 ± 0.37 | 1.6 ± 0.2 | 0.51 ± 0.04 | 4.7 ± 0.42 | 2.4 ± 0.4 | 15 |
| ZPI-CD-helixα1-PI | 2.2 ± 0.14 | 6.1 ± 0.48 | 1.3 ± 0.2 | 0.16 ± 0.02 | 6.3 ± 0.55 | 1.0 ± 0.2 | 7.7 |
The apparent second-order rate constants (k2(app)) and SI values for the inhibition of FXa by the ZPI derivatives were determined in TBS/Ca2+ containing 0.1% PEG-8000 and 0.1 mg/mL BSA by a discontinuous assay method as described in Materials and Methods. The k2(app) values in the presence of heparin are derived from Fig. 1A and represent the peak values at an optimal concentration of the polysaccharide. The product k2(app) × SI represents the second-order rate constant corrected for the SI values. The fold accelerating effect of heparin is presented in the last column.
Table 2.
Kinetic constants and SI values for the inhibition of FXIa by the ZPI derivatives in the absence and presence of heparin
| ZPI derivative | FXIa | FXIa (+ Hep) | Fold enhancement | ||||
|---|---|---|---|---|---|---|---|
| k2(app) × 105 M−1 S−1 | SI mol I/mol E | k2(app) × SI 105 M−1S−1 | k2(app) × 105 M−1 S−1 | SI mol I/mol E | k2(app) × SI 106 M−1S−1 | ||
| ZPI-WT | 0.58 ± 0.15 | 10.2 ± 0.86 | 5.9 ± 2.0 | 4.2 ± 0.54 | 4.3 ± 0.27 | 1.8 ± 0.3 | 3.0 |
| ZPI-3A | 0.41 ± 0.11 | 13.3 ± 0.65 | 5.4 ± 1.7 | 2.6 ± 0.27 | 5.2 ± 0.50 | 1.4 ± 0.3 | 2.6 |
| ZPI-des-NT | 0.29 ± 0.13 | 15.6 ± 2.2 | 4.5 ± 2.7 | 1.1 ± 0.11 | 8.5 ± 0.72 | 0.94± 0.17 | 2.1 |
| ZPI-D-helixα1-PI | 0.31 ± 0.06 | 27.5 ± 3.9 | 8.5 ± 2.9 | 1.1 ± 0.12 | 6.2 ± 0.66 | 0.68 ±0.15 | 0.8 |
| ZPI-CD-helixα1-PI | 0.28 ± 0.04 | 39.4 ± 4.5 | 11.0 ± 2.8 | 0.66 ± 0.07 | 11.3 ± 1.4 | 0.74 ±0.17 | 0.67 |
The apparent second-order rate constants (k2(app)) and SI values for the inhibition of FXIa by the ZPI derivatives were determined in TBS/Ca2+ containing 0.1% PEG-8000 and 0.1 mg/mL BSA by a discontinuous assay method as described in Materials and Methods. The k2(app) values in the presence of heparin are derived from Fig. 1B and represent the peak values at an optimal concentration of the polysaccharide. The product k2(app) × SI represents the second-order rate constant corrected for the SI values. The fold accelerating effect of heparin is presented in the last column.
The non-conserved N-terminal insertion domain of ZPI has 12 negatively charged Glu and Asp residues rendering this domain highly acidic (16,20). The contribution of these residues to ZPI interaction with heparin was analyzed by deleting this acidic N-terminal tail of the serpin in the ZPI-des-NT construct. Surprisingly, the N-terminal deletion mutant of ZPI exhibited a ~1.5 to 2-fold reduced reactivity with FXa in the absence and presence of heparin, respectively, with a corresponding reduction in the accelerating effect of heparin to 33-fold (Table 1). However, the decrease in reactivity could be attributed to a ~2-fold elevation in the stoichiometry of inhibition (SI) for the ZPI mutant independent of heparin. The slightly reduced cofactor function of heparin may also be attributed to an indirect conformational effect on the heparin-binding site of the serpin caused by the deletion of N-terminal residues. These results suggest that the acidic N-terminal tail of ZPI does not have a significant role in interaction with heparin.
Similar to FXa, the rate accelerating effect of heparin on the ZPI inhibition of FXIa exhibited a bell-shaped dependence on the concentration of the polysaccharide (Figure 1B). The fold accelerating effect of heparin for the ZPI inhibition of FXIa was ~7-fold (0.58 × 105 M−1s−1 and 4.2 × 105 M−1s−1 in the absence and presence of heparin, respectively) (Table 2). However, the SI values for ZPI with FXIa were significantly greater in the absence than in the presence of heparin. Thus, the overall rate accelerating effect of heparin on ZPI inhibition of FXIa was only 3-fold (Table 2). Interestingly, the C and D helix chimeras exhibited markedly elevated SI values with FXIa in the absence of heparin (Table 2). Heparin reduced the SI values of ZPI derivatives with FXIa, such that the overall reactivity of the C and D helix chimeras with FXIa was not accelerated by heparin (Table 2). These results further support the hypothesis that both the C and D helices of ZPI interact with heparin and that the primary cofactor function of heparin in the ZPI-FXIa reaction is to lower the reactivity of FXIa with ZPI in the substrate pathway of the reaction.
Analysis of the reactivity of ZPI derivatives with FXa in the presence of protein Z
The reactivity of ZPI derivatives with FXa was also analyzed in the presence of protein Z and membrane cofactors. The results presented in Table 3 suggest that none of the basic residues of the C and D helices affect the interaction with protein Z since the mutants reacted with FXa with essentially similar k2 values and protein Z accelerated the inhibition of FXa by ZPI derivatives to a similar extent in the presence of a membrane surface. Similar results with ZPI-des-NT further suggest that the N-terminal acidic tail of ZPI also plays no role in the ability of the protein cofactor to accelerate the serpin-FXa reaction.
Table 3.
Kinetic constants and SI values for the inhibition of FXa by the ZPI derivatives in the absence and presence of protein Z and PC/PS vesicles
| ZPI derivative | FXa | FXa (+ PZ+PC/PS) | Fold enhancement | ||||
|---|---|---|---|---|---|---|---|
| k2(app) × 103 M−1 S−1 | SI mol I/mol E | k2(app) × SI 104 M−1S−1 | k2(app) × 106 M−1 S−1 | SI mol I/mol E | k2(app) × SI 106 M−1S−1 | ||
| ZPI-WT | 3.4 ± 0.42 | 4.1 ± 0.23 | 1.4±0.2 | 2.5 ± 0.51 | 2.8 ± 0.21 | 7.0±2.0 | 500 |
| ZPI-3A | 2.6 ± 0.15 | 4.5 ± 0.34 | 1.2±0.2 | 2.0 ± 0.31 | 3.3 ± 0.35 | 6.6±1.7 | 550 |
| ZPI-des-NT | 2.1 ± 0.30 | 7.4 ± 0.52 | 1.5±0.3 | 1.8 ± 0.20 | 4.5 ± 0.39 | 8.1±1.6 | 540 |
| ZPI-D-helixα1-PI | 2.9 ± 0.25 | 5.6 ± 0.37 | 1.6±0.2 | 2.1 ± 0.21 | 3.6 ± 0.28 | 7.6±1.4 | 475 |
| ZPI-CD-helixα1-PI | 2.2 ± 0.14 | 6.1 ± 0.48 | 1.3±0.2 | 0.94 ± 0.12 | 7.5 ± 0.71 | 7.1±1.6 | 546 |
The apparent second-order rate constants (k2(app)) and SI values for the inhibition of FXa by the ZPI derivatives in the absence and presence of protein Z and PC/PS vesicles were determined in TBS/Ca2+ containing 0.1% PEG-8000 and 0.1 mg/mL BSA by a discontinuous assay method as described in Materials and Methods. The product k2(app) × SI represents the second-order rate constant corrected for the SI values. The fold accelerating effect of protein Z and PC/PS vesicles is presented in the last column.
Direct binding to heparin
The affinity of ZPI derivatives for interaction with heparin was evaluated by their ability to directly bind to a heparin-Sepharose column. Analysis of the NaCl gradient elution profiles of the serpins from the column (Figure 2) suggested that the affinity of ZPI mutants for heparin has been decreased to varying degrees, with the ZPI-CD-helixα1-PI exhibiting the weakest affinity for heparin. Thus, in contrast to a NaCl concentration of 0.35 M, required to elute the wild-type ZPI, a NaCl concentration of 0.15 M was required to elute the ZPI-CD-helixα1-PI chimera from the heparin column. The ZPI derivatives interacted with the heparin column with the following order: wild-type ZPI > ZPI-des-NT > ZPI-3A ≥ ZPI-D-helixα1-PI > ZPI-CD-helixα1-PI (Figure 2). These results suggest that the basic residues of both C and D helices of ZPI interact with heparin and that the N-terminal acidic domain of ZPI somehow has a modulatory effect on the affinity of the serpin for heparin.
The affinity of ZPI derivatives for heparin was also examined by an inhibition kinetic assay monitoring the ability of the serpins to compete specifically with antithrombin and thrombin for heparin in the heparin-catalyzed inhibition of thrombin by antithrombin. The results presented in Figure 3 support the direct binding assay by demonstrating that the ZPI derivatives interact with heparin with the weaker affinity in the order of wild-type ZPI > ZPI-des-NT > ZPI-3A < ZPI-D-helixα1-PI > ZPI-CD-helixα1-PI. This is derived from the analysis of the saturable dependence of the decrease in heparin-mediated inhibition rates on the concentrations of competitors (ZPI derivatives) which yielded apparent dissociation constants for the interaction of heparin with ZPI derivatives (Figure 3). Comparison of these values (25 ± 2 nM for wild-type ZPI, 51 ± 6 nM for ZPI-des-NT, 110 ± 17 nM for ZPI-3A, 68 ± 18 nM for ZPI-D-helixα1-PI, and 393 ± 78 nM for ZPI-CD-helixα1-PI) suggest that the affinity of mutants for interaction with heparin has been reduced ~2–16-fold, with ZPI-CD-helixα1-PI exhibiting the most loss of affinity for the polysaccharide. Thus, basic residues of both the C and D helices of ZPI interact with heparin.
Figure 3.

The competitive effect of ZPI derivatives on the heparin-catalyzed inhibition of thrombin by antithrombin. The competitive effect of ZPI derivatives (0–250 nM) on the inhibition of thrombin (0.5 nM) by antithrombin (2.5 nM) was monitored in the presence of heparin (5 nM) for 2–3 min in TBS/Ca2+ containing 0.1 mg/mL BSA and 0.1% PEG 8000 as described under “Materials and Methods”. The symbols are: ZPI-WT (○), ZPI-3A (●), ZPI-des-NT (□), ZPI-D-helixα1-PI (■), and ZPI-CD-helixα1-PI (△). Solid lines are best fits of inhibition data to a hyperbolic equation. Data are derived from at least three independent measurements ±S.D.
Discussion
We recently showed that ZPI has a binding site for heparin and that the binding of long-chain heparins to the serpin promote ZPI inhibition of FXa and FXIa by a template mechanism (6). In this study we have mapped the heparin binding site of ZPI and shown that basic residues of both the C and D helices interact with the polysaccharide. The three dimensional locations of the C and D helices of ZPI along with the side chains of basic residues constituting the heparin binding sites of ZPI are shown in Figure 4. Given that most of the heparin-binding plasma serpins interact with heparin primarily through basic residues of the D-helix (12–14), we initially evaluated the contribution of each one of the basic residues of the D-helix (Lys-116, Lys-125, and Arg-128) as well as Lys-113 located on the loop connecting the D-helix to the C-helix, by substituting them with Ala in individual constructs. However, we found that while the single mutants had near normal reactivity with both FXa and FXIa in the absence of heparin, their reactivity was decreased less than 2-fold in the presence of heparin (data not presented). Thus, we decided to substitute these residues in combinations and also substitute the entire D-helix of ZPI with the corresponding D-helix of the non-heparin-activatable serpin α1-PI. Analyses of the reactivity of these mutants (ZPI-3A and ZPI-D-helixα1-PI) with FXa in the absence and presence of unfractionated heparin by kinetic assays as well as their direct interaction with heparin suggested that the D-helix of ZPI interacts with heparin. However, it is not the sole interactive-site for the polysaccharide on the serpin since the mutants exhibited only 2–3-fold decreases in their reactivity with the proteases. Following further examination of the structure of the serpin (Figure 4), we noted that the side-chains of two basic residues of the C-helix (Lys-104 and Arg-105) are in the same vicinity as D helix residues and can potentially interact with heparin. These residues are not conserved in either antithrombin or α1-PI as the corresponding sites are Glu in both serpins (12,17,20). Thus, we decided to replace both C and D helices of ZPI (including the loop connecting the helices) with the corresponding helices of α1-PI in another construct (ZPI-CD-helixα1-PI). The reactivity of this chimera with FXa in the presence of heparin was markedly impaired, the rate accelerating effect of the polysaccharide being reduced from 48-fold to 7.7-fold, suggesting that basic residues of both C and D helices constitute a heparin binding site on the serpin. Further analysis showed that the affinity of this mutant for heparin has also been dramatically impaired as evidenced by a direct binding assay and a competitive kinetic assay monitoring the binding affinity of the mutant for heparin. Thus, in contrast to an apparent KD of 25 nM for heparin interaction with ZPI, the corresponding value for the chimera exceeded 390 nM. The loss of affinity for heparin was not due to mis-folding of the chimeric serpin since the mutant exhibited normal reactivity with FXa in the absence of a cofactor and in the presence of protein Z and membrane cofactors. These results support previous kinetic and structural data demonstrating that protein Z and heparin bind to distinct non-overlapping sites on the serpin (6,15,16). The observation that heparin exhibited a minimal cofactor effect on the ZPI-CD-helixα1-PI inhibition of the FXa exosite mutant (FXa-R240A), which exhibits dramatically lower affinity for heparin (21), further supports the hypothesis that the binding of heparin to the C and D helices of ZPI and the heparin-binding exosite of FXa promotes the inhibition of the protease by a template mechanism (6). Nevertheless, the observation of a residual ~8-fold rate accelerating effect of heparin on ZPI-CD-helixa1-PI inhibition of FXa implies that the C and D helix basic residues alone do not account for all ZPI interactions with heparin. Candidate basic residues adjacent to these helices that may also contribute to binding heparin would include Lys-162 and Arg-163 on helix E and Arg-133 on the C-terminal extension of helix D.
Figure 4.
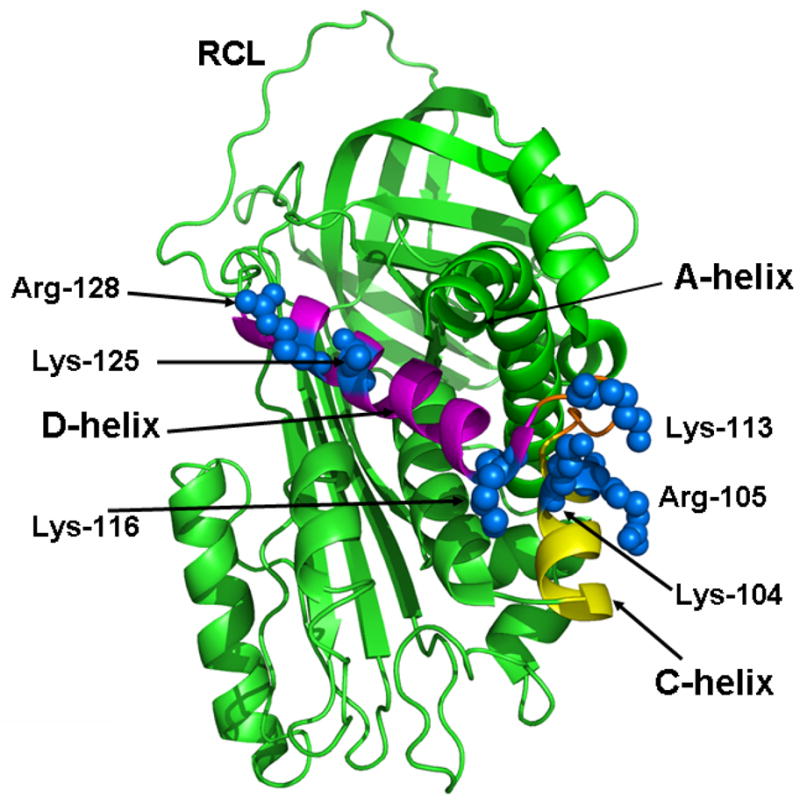
Crystal structure of ZPI. The structure of ZPI in ribbon representation is shown with the C-helix and D-helix colored in yellow and pink, respectively. The relative three dimensional locations of the side-chains of the basic residues of the C and D helices are shown in blue. The coordinates obtained from Protein Data Bank accession code 3H5C were used to prepare the figure (16).
The template cofactor effect of heparin on the ZPI-CD-helixα1-PI inhibition of FXIa was also abolished. The noteworthy observation with FXIa inhibition by ZPI derivatives was that heparin significantly reduced the SI values, suggesting that the cofactor function of heparin preferentially improves the inhibitory pathway of the serpin reaction with this protease. By contrast, heparin does not have a significant effect on SIs for the ZPI-FXa reactions and thus does not appear to influence the inhibitory pathway in this case. Similar to other serpins, ZPI inhibits its target proteases by a branched pathway, suicide substrate inhibition mechanism in which an acyl-intermediate serpin-protease complex either partitions to cleaved serpin and free enzyme or to a stable covalent complex. In the latter complex the reactive center loop of the serpin has inserted itself into β-sheet A as a new strand, and the protease has been dragged along with it to the opposite end of the molecule (22,23). The mechanism by which heparin improves the inhibitory pathway of the ZPI reaction with FXIa is not known. However, we have previously shown that heparin can induce conformational changes in the active site pocket of FXIa (10,24), thereby improving the reactivity of the protease with other serpins (i.e., antithrombin and C1-inhibitor). Noting that the SI values for both wild-type and heparin-binding site mutants of ZPI is decreased 3–4-fold in the presence of heparin, the results suggest the possibility that heparin binding to FXIa improves the inhibitory branch of the reaction pathway by an allosteric mechanism. However, the molecular basis for the overall enhancement of the SI values of the ZPI mutant reactions with FXIa in the absence of heparin is not known. Whether the mutant residues make direct interactions with FXIa or if the heparin binding C and D helices have allosteric linkages with the reactive center loop of the serpin need further investigation. Whatever the mechanism, a heparin-mediated decrease in the SI for ZPI inhibition of FXIa may have pharmacological and physiological significance. Thus, therapeutic unfractionated heparins, similar to the one used in this study, or the glycosaminoglycans lining the vascular endothelium can increase the stability of the FXIa-ZPI complex, thereby contributing to the regulation of the intrinsic clotting cascade.
The sequence alignment with other serpins indicates that the N-terminal tail A-helix of ZPI is longer and highly acidic due to the presence of 11 Glu and one Asp residues on this domain (1,17,20). This acidic tail is not ordered and has a flexible conformation explaining its lack of resolution in the crystal structures (15,16). Such an acidic N-terminal tail is only observed in the sequence of heparin cofactor II (HCII) which interacts with basic residues of the D-helix, thus reducing the affinity of the serpin for heparin (25,26). It is known that deleting this sequence in HCII improves the affinity of the serpin for heparin (26). Moreover, it has been demonstrated that heparin can disrupt the interaction of the N-terminal tail of HCII with the D-helix, thereby facilitating the interaction of the acidic tail of the serpin with anion-binding exosite-1 of thrombin (26,27). To determine whether the acidic N-terminal tail plays a similar function in ZPI, we deleted the first 52 residues of the serpin in the ZPI-des-NT mutant. Analysis of the activity of this mutant did not reveal a significant function for this tail in the reaction of the serpin with either FXa or FXIa. Paradoxically, the affinity of heparin for the deletion mutant of the serpin was slightly decreased, as evidenced by kinetic and direct binding studies. It is possible that a conformational change in the side chains of the basic residues of the C- and/or D-helix affect the affinity of the mutant for heparin, but this hypothesis needs further confirmation. Finally, the observation that the deletion mutant exhibits normal reactivity with FXa both in the absence and presence of protein Z and membrane cofactors suggests that the acidic N-terminal tail preceding the A-helix does not influence the interaction with protein Z. It should be noted that the A-helix residues, Lys-68, Met-71 and Asp-74, are known to be involved in protein Z binding (15,16), but these residues were not deleted in the ZPI-des-NT mutant which had only the first 52 residues of the serpin preceding the helix A deleted.
Acknowledgments
The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institute of Health (HL 62565 to ARR and HL 39888 to STO) and AHA Scientist Development (SDG4880022 to XH).
We thank Dr. Aiwu Zhou for providing the bacterial expression plasmid for ZPI.
Footnotes
Abbreviations – ZPI, protein Z-dependent protease inhibitor; α1-PI, α1-protease inhibitor; FXa, activated factor X; FXIa, activated factor XI; AT, antithrombin; ZPI-D-helixα1-PI, ZPI mutant in which the residues of the D-helix have been replaced with corresponding residues of α1-PI; ZPI-CD-helixα1-PI, ZPI mutant in which the residues of the C and D helices have been replaced with corresponding residues of α1-PI; ZPI-3A, ZPI mutant in which residues Lys-113, Lys-116 and Lys-125 in the α1-PI numbering (17) have been replaced with Ala; ZPI-des-NT, ZPI mutant in which the first 52 residues of the N-terminus A-helix have been deleted; FXa-R240A, FXa mutant in which Arg-240 in the chymotrypsin numbering system (28) has been replaced with Ala; PEG, polyethylene glycol; BSA, bovine serum albumin.
References
- 1.Han X, Fiehler R, Broze GJ., Jr Isolation a protein Z-dependent plasma protease inhibitor. Proc Natl Acad Sci USA. 1998;95:9250–9255. doi: 10.1073/pnas.95.16.9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han X, Fiehler R, Broze GJ., Jr Characterization of the protein Z-dependent protease inhibitor. Blood. 2000;96:3049–3055. [PubMed] [Google Scholar]
- 3.Huang X, Swanson R, Broze GJ, Jr, Olson ST. Kinetic characterization of the protein Z-dependent protease inhibitor reaction with blood coagulation factor Xa. J Biol Chem. 2008;283:29770–29783. doi: 10.1074/jbc.M805214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rezaie AR, Bae JS, Manithody C, Qureshi SH, Yang L. Protein Z-dependent protease inhibitor binds to the C-terminal domain of protein Z. J Biol Chem. 2008;283:19922–19926. doi: 10.1074/jbc.M802639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabatabai A, Fiehler R, Broze GJ., Jr Protein Z circulates in plasma in a complex with protein Z-dependent protease inhibitor. Thromb Haemostas. 2001;85:655–660. [PubMed] [Google Scholar]
- 6.Huang X, Rezaie AR, Broze GJ, Jr, Olson ST. Heparin is a major activator of the anticoagulant serpin, protein Z-dependent protease inhibitor (ZPI) J Biol Chem. 2011;286:8740–8751. doi: 10.1074/jbc.M110.188375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson ST, Shore JD. Demonstration of a two-step reaction mechanism for inhibition of alpha-thrombin by antithrombin III and identification of the step affected by heparin. J Biol Chem. 1982;257:14891–14895. [PubMed] [Google Scholar]
- 8.Rezaie AR. Identification of basic residues in the heparin-binding exosite of factor Xa critical for heparin and factor Va binding. J Biol Chem. 2000;275:3320–3327. doi: 10.1074/jbc.275.5.3320. [DOI] [PubMed] [Google Scholar]
- 9.Ho DH, Badellino K, Baglia FA, Walsh PN. A binding site for heparin in the apple 3 domain of factor XI. J Biol Chem. 1998;273:16382–16390. doi: 10.1074/jbc.273.26.16382. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Sun MF, Gailani D, Rezaie AR. Characterization of a heparin-binding site on the catalytic domain of factor XIa: mechanism of heparin acceleration of factor XIa inhibition by the serpins antithrombin and C1-inhibitor. Biochemistry. 2009;48:1517–1524. doi: 10.1021/bi802298r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shirk RA, Elisen MG, Meijers JC, Church FC. Role of the H helix in heparin binding to protein C inhibitor. J Biol Chem. 1994;269:28690–28695. [PubMed] [Google Scholar]
- 12.Ersdal-Badju E, Lu A, Zuo Y, Picard V, Bock SC. Identification of the antithrombin III heparin binding site. J Biol Chem. 1997;272:19393–19400. doi: 10.1074/jbc.272.31.19393. [DOI] [PubMed] [Google Scholar]
- 13.Belzar KJ, Zhou A, Carrell RW, Gettins PG, Huntington JA. Helix D elongation and allosteric activation of antithrombin. J Biol Chem. 2002;277:8551–8558. doi: 10.1074/jbc.M110807200. [DOI] [PubMed] [Google Scholar]
- 14.Rein CM, Desai UR, Church FC. Serpin-glycosaminoglycan interactions. Methods Enzymol. 2011;501:105–137. doi: 10.1016/B978-0-12-385950-1.00007-9. [DOI] [PubMed] [Google Scholar]
- 15.Wei Z, Yan Y, Carrell RW, Zhou A. Crystal structure of protein Z-dependent inhibitor complex shows how protein Z functions as a cofactor in the membrane inhibition of factor X. Blood. 2009;114:3662–3667. doi: 10.1182/blood-2009-04-210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Dementiev A, Olson ST, Gettins PG. Basis for the specificity and activation of the serpin protein Z-dependent proteinase inhibitor (ZPI) as an inhibitor of membrane-associated factor Xa. J Biol Chem. 2010;285:20399–20409. doi: 10.1074/jbc.M110.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber R, Carrell RW. Implications of the three-dimensional structure of alpha 1-antitrypsin for structure and function of serpins. Biochemistry. 1989;28:8951–8966. doi: 10.1021/bi00449a001. [DOI] [PubMed] [Google Scholar]
- 18.Raja SM, Chhablani N, Swanson R, Thompson E, Laffan M, Lane DA, Olson ST. Deletion of P1 arginine in a novel antithrombin variant (antithrombin London) abolishes inhibitory activity but enhances heparin affinity and is associated with early onset thrombosis. J Biol Chem. 2003;278:13688–13695. doi: 10.1074/jbc.M300062200. [DOI] [PubMed] [Google Scholar]
- 19.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Hérault JP, Herbert JM, Björk I. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb Haemost. 2004;92:929–939. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 20.Chandrasekaran V, Lee CJ, Lin P, Duke RE, Pedersen LG. A computational modeling and molecular dynamics study of the Michaelis complex of human protein Z-dependent protease inhibitor (ZPI) and factor Xa (FXa) J Mol Model. 2009;15:897–911. doi: 10.1007/s00894-008-0444-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rezaie AR, Olson ST. Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by promoting the assembly of an intermediate heparin-antithrombin-factor Xa bridging complex. Demonstration by rapid kinetics studies. Biochemistry. 2000;39:12083–12090. doi: 10.1021/bi0011126. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence DA, Ginsburg D, Day DE, Berkenpas MB, Verhamme IM, Kvassman JO, Shore JD. Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J Biol Chem. 1995;270:25309–25312. doi: 10.1074/jbc.270.43.25309. [DOI] [PubMed] [Google Scholar]
- 23.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407:923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Manithody C, Qureshi SH, Rezaie AR. Contribution of exosite occupancy by heparin to the regulation of coagulation proteases by antithrombin. Thromb Haemost. 2010;103:277–823. doi: 10.1160/TH09-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ragg H, Ulshöfer T, Gerewitz J. Glycosaminoglycan-mediated leuserpin-2/thrombin interaction. Structure-function relationships. J Biol Chem. 1990;265:22386–22391. [PubMed] [Google Scholar]
- 26.Van Deerlin VM, Tollefsen DM. The N-terminal acidic domain of heparin cofactor II mediates the inhibition of alpha-thrombin in the presence of glycosaminoglycans. J Biol Chem. 1991;266:20223–20231. [PubMed] [Google Scholar]
- 27.Rogers SJ, Pratt CW, Whinna HC, Church FC. Role of thrombin exosites in inhibition by heparin cofactor II. J Biol Chem. 1992;267:3613–3617. [PubMed] [Google Scholar]
- 28.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 Å crystal structure of human α-thrombin: interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]