

Abstract
We modified a dual-cell linear ion trap mass spectrometer to perform infrared multiphoton dissociation (IRMPD) in the low pressure trap of a dual-cell quadrupole linear ion trap (dual cell QLT) and perform large-scale IRMPD analyses of complex peptide mixtures. Upon optimization of activation parameters (precursor q-value, irradiation time, and photon flux), IRMPD subtly, but significantly outperforms resonant excitation CAD for peptides identified at a 1% false-discovery rate (FDR) from a yeast tryptic digest (95% confidence, p = 0.019). We further demonstrate that IRMPD is compatible with the analysis of isobaric-tagged peptides. Using fixed QLT RF amplitude allows for the consistent retention of reporter ions, but necessitates the use of variable IRMPD irradiation times, dependent upon precursor mass-to-charge (m/z). We show that IRMPD activation parameters can be tuned to allow for effective peptide identification and quantitation simultaneously. We thus conclude that IRMPD performed in a dual-cell ion trap is an effective option for the large-scale analysis of both unmodified and isobaric-tagged peptides.
Introduction
With the advent of large-scale proteomics,1–4 a number of techniques have been developed to enable quantitation of proteins, including label-free methods (e.g., spectral counting),5 metabolic labeling,6–10 and chemical labeling.11–14 Isobaric tagging strategies (i.e., TMT and iTRAQ) have proven highly useful mainly for their ability to multiplex and compatibility with tissues and biofluids.15,16 The success of an isobaric tagging experiment, however, depends heavily on the dissociation method; the ideal method routinely produces MS/MS spectra which are informative of both peptide sequence and relative abundance. While resonant-excitation collisional-activated dissociation (CAD) is a common form of peptide dissociation for discovery proteomics,17–19 the CAD process is inefficient at low RF amplitudes. Unfortunately, low RF amplitudes are required for the retention of any low mass fragment ions, including isobaric reporter tags, rendering CAD unacceptable for isobaric tagging experiments. This shortcoming has inspired the development of alternatives. Pulsed-q dissociation and high amplitude short time excitation (HASTE) are modifications of the resonant-excitation process in which the ion trap RF amplitude is pulsed high for efficient energy deposition and then quickly lowered to retain low m/z ions.20,21 Though amenable to the study of isobaric-tagged peptides, PQD and HASTE suffer from low precursor-to-product ion conversion efficiencies. Beam-type collisional activation is efficiently performed at low RF amplitudes, enabling the successful interrogation of isobaric-tagged peptides.17,22–25 The pre-requisite for a dedicated, external RF device to serve as the collision cell, however, renders many ion trap systems unable to perform beam-type collisional activation. Work from our group has recently demonstrated that the ESI ion injection optics of stand-alone ion traps can serve this function effectively (iHCD).26 While this work cogently demonstrated that stand-alone ion traps in most cases possess the hardware to perform beam-type collisional activation, the efficiency of peptide dissociation using any beam-type collisional activation scheme is limited by ion loss due to scattering, a problem exacerbated under the activation conditions (high collision energy) optimal for the generation of isobaric reporter tags.
Photon-based fragmentation techniques, such as UVPD, IRMPD, are efficiently performed at low RF amplitudes in QLT ion traps, do not induce ion scattering, and require only a single RF device.27–29 The utility of IRMPD has been somewhat limited because the activation efficiency of peptide cations, at typical ion trap operating pressures (~1 mTorr), is relatively low.30 Efforts made to improve the efficiency include dynamic adjustment of ion trap pressure,31,32 increased photon flux,33 pre-activation prior to or during IRMPD via either resonant excitation34 or elevated bath gas temperature.35 Other approaches involve chemical modification of peptides through the attachment of chromogenic moieties or increasing the charge state peptides ionize in via ESI (supercharging).36–39 Though each of these approaches enables efficient IRMPD, an attractive and straightforward solution is to perform IRMPD on a dual-cell quadrupole linear ion trap (dual-cell QLT).40,41 This approach requires no chemical modification of peptides. Further, the hardware modifications are simple, just the addition of a viewport to the rear flange of the instrument.
Here we build on the initial description of IRMPD performed in a dual-cell ion trap by demonstrating marginally superior effectiveness compared to CAD for shotgun sequencing of complex peptide mixtures. Further, we evaluate IRMPD as a means to interrogate TMT-tagged peptides. A recent study by Glish and co-workers demonstrated the viability of IRMPD for iTRAQ-tagged peptides using a standard peptide.42 This analysis, however, did not evaluate the potential of IRMPD for large-scale isobaric tagging studies, and provided no comparison of IRMPD to beam-type collisional activation. Here we report conditions which simultaneously provide good quantitative accuracy and effective peptide sequencing. IRMPD shows strong potential to improve upon existing methods for the generation of TMT reporter ions via sequential dissociation of primary b- and y- fragments to accumulate ions in the TMT reporter channel. Moreover, a preliminary comparison of IRMPD to beam-type collisional activation suggests that IRMPD may be superior for the generation of isobaric reporter tag.
Experimental Section
Sample Preparation, Mass Spectrometry, LC separation
Wild-type yeast was grown and lysed, digested using trypsin, and labeled using six channel TMT tags as previously described (Supplemental).43–45 All experiments were performed on a modified LTQ-Velos (Thermo Fisher Scientific, San Jose, CA) dual pressure linear ion trap mass spectrometer comprising both high (~5 mTorr) and low (~0.3 mTorr) pressure linear ion traps.40,41,46 Briefly, the back flange of the mass spectrometer was modified to hold a ZnSe window and a stainless steel blocking disk with an aperture of ~2mm concentric with the trapping volume of the QLT (Supplemental Fig. 1). All IRMPD experiments were performed using a Firestar T-100 Synrad 100-W CO2 continuous wave laser (Mukilteo, WA). For all IRMPD MS/MS events, precursor peptides were dissociated within the low pressure linear ion trap; for all CAD MS/MS events, dissociation was carried out within the high pressure linear ion trap. LC separations were carried out using a NanoAcquity UPLC system and auto-sampler to load samples onto a self-packed 75 μm i.d. 5 μm particle 8 cm pre-column, and separated on a 50 μm i.d. 5 μm 25 cm analytical column (Waters, Milford, MA) as previously described.43
During the nLC-MS/MS analysis of the unmodified complex peptide mixture, the mass spectrometry method consisted of an MS analysis followed by consecutive CAD and IRMPD data dependent MS/MS scans of the 3 most intense precursors. For nLC-MS/MS analysis of TMT-tagged complex peptide mixtures, the mass spectrometry scan sequence consisted of an MS survey scan followed by ten IRMPD MS/MS events interrogating the top ten most intense precursors. Activation using CAD was performed in the high pressure cell at normalized collision energy (NCE) of 35 for 10 ms; IRMPD was carried out in the low pressure cell. IRMPD conditions for each LC analysis were varied (vida supra). Precursors were dynamically excluded for 90 s using an isolation window of ± 1.5 Th. AGC target values were 40,000 for MS and 10,000 for MS/MS analysis.
Database Searching and Data Analysis
For unmodified and TMT-labeled peptide LC- MS/MS analyses, data reduction was performed with COMPASS,47 a program which converts output files to searchable text files, as described previously.47 OMSSA (version 2.1.8,NCBI) was used to search spectra against the concatenated target-decoy SGD yeast database (www.yeastgenome.org, downloaded 01-05-2010). Average mass tolerances of ± 5 Th and ± 0.5 Th were used for precursor and product m/z respectively, with carbaminomethylation of cysteine set as a fixed modification and oxidation of methionine set as a variable modification. For TMT tagged samples, TMT 6-plex on the N-terminus and TMT 6-plex on lysine residues were set as fixed modifications, with TMT 6-plex on tyrosine residues set as a variable modification. All peptide spectrum matches (PSMs) were filtered to a false discovery rate (FDR) of 1% using expectation value (e-value) and the concatenated forward-reverse database method.48,49 Protein identifications were subsequently reduced for parsimony and filtered to 1% FDR.49 Protein quantitation was evaluated with COMPASS, which corrects for isotopic impurities, normalizes reporter ion intensities, and coalesces peptide quantitation into protein quantitation.47 B- and y- type fragment ions were searched for both CAD and IRMPD spectra.
Safety Considerations
IR safety glasses were worn at all times in the presence of the IR laser.
Results Section
Evaluation of IRMPD for shotgun proteomics
CAD is a common peptide activation method used for large-scale proteomics due to high efficiency of activation and ease of use. Both of these traits are in large part because primary b- and y- type fragment ions formed from precursor dissociation are not susceptible to further activation (secondary dissociation); this obviates the need to optimize activation conditions to avoid excessive secondary dissociation events that exist for other dissociation techniques (e.g., beam-type collisional activation, ETD). Primary product ions formed via IRMPD dissociation, however, are susceptible to secondary dissociation. Secondary dissociation events can produce product ions having analytical value (e.g., diagnostic side chain losses, immonium ions), though excessive secondary dissociation can confound spectral interpretation. To potentiate the utility of IRMPD for shotgun peptide sequencing, it is critical to control and optimize the degree of secondary dissociation. The primary variables influencing the magnitude of secondary dissociation for IRMPD MS/MS events are the photon flux (laser power), the reduced Mathieu parameter of the precursor (q-value), and the duration of time ions are subjected to IR activation. The Mathieu parameter is essentially a measure of the restorative force excerted upon ions within the ion trap; the higher the Mathieu parameter, the greater the restorative force. Ions having higher Mathieu parameters will therefore spend a proportionally greater amount of time in the center of the ion trap, where the overlap with the photon beam is the highest. This in turn leads to greater overall rates of photo-dissociation as noted previously.41,50 We performed a series of nLC-MS/MS analyses, investigating several combinations of irradiation times (3, 5, 10, 15, and 25 ms), q-values (0.10, 0.15, 0.20, and 0.25), and laser powers (36 W, 48 W, and 60 W). For each experiment, an MS survey scan was followed by MS/MS activation of the top three most intense precursors using IRMPD (conditions varied for each analysis) and resonant-excitation CAD (NCE = 35, 10 ms activation time).
In total, 60 nLC-MS/MS analyses were conducted (Table 1). For very short irradiation times, IRMPD yielded somewhat low PSM totals because the IRMPD spectra comprise mostly unreacted precursor, with few b- and y- type product ions. Conversely, long irradiation times or high q-values produce spectra dominated by low m/z ions, presumably because secondary IRMPD dissociation events are far more prevalent. For 6 of the 60 IRMPD conditions considered, IRMPD produced a slightly greater number of PSMs (1% False-discovery rate, FDR) than CAD. The conditions under which IRMPD performed the best relative to CAD was at a precursor q-value of 0.10, laser power of 48W, and irradiation time of 10 ms. To determine the reproducibility of this result we performed two additional technical replicates (Supplemental Fig. 2a). In each of the three trials, IRMPD produced slightly more PSMs than CAD (7,223 vs. 6,969; 6,617 vs. 6,432; 6,909 vs. 6,747); these data lead us to conclude that IRMPD is slightly more effective than CAD with statistical significance (> 95% confidence, paired students t-test, p = 0.019). In each trial, CAD and IRMPD largely identified similar subsets of the peptide complex mixture (Supplemental Fig. 2b), likely due to the similar ‘slow heating’ dissociation mechanisms of both CAD and IRMPD.51 While resonant-excitation CAD is a common dissociation technique for large-scale proteomics, there are well-known shortcomings associated with CAD, including inefficient energy deposition at low RF amplitudes, necessitating higher RF amplitudes and a higher low-mass cutoff (LMCO). Here we have demonstrated that IRMPD is at least as effective as CAD for shotgun sequencing of peptides derived from protein digestion using trypsin. Unlike CAD, however, IRMPD is efficiently performed at low RF amplitudes,28 alleviating LMCO limitations and allowing for the observation of potentially important low m/z product ions, namely isobaric reporter tags.
Table 1.
Results of parametric optimization of IRMPD for large-scale LC-MS/MS analysis. We varied photon flux, irradiation time, and q-value for IRMPD activation. For each experiment, CAD activation conditions were held constant to provide an internal benchmark of success. Shown in bold and red are IRMPD conditions which resulted in more peptide spectral matches (PSMs) than activation with CAD. Each number represents the number of peptide PSMs (1% FDR) resulting from the use of CAD or IRMPD for a single LC-MS/MS experiment.
| IRMPD Peptide Identifications under various conditions | |||||||
|---|---|---|---|---|---|---|---|
| q-value | IR Time (ms) | 36 W laser power
|
48 W laser power
|
60 W laser power
|
|||
| IRMPD IDs | CAD IDs | IRMPD IDs | CAD IDs | IRMPD IDs | CAD IDs | ||
| 0.10 | 3 | 2926 | 6821 | 4977 | 6782 | 5862 | 6364 |
| 5 | 5143 | 6946 | 6717 | 7003 | 6242 | 6224 | |
| 10 | 6788 | 6834 | 7223 | 6969 | 6017 | 6263 | |
| 15 | 6960 | 6742 | 6634 | 6763 | 4721 | 6315 | |
| 25 | 4224 | 5965 | 3627 | 6489 | 915 | 6480 | |
| 0.15 | 3 | 4000 | 7015 | 5968 | 6907 | 6237 | 6416 |
| 5 | 5950 | 6723 | 6667 | 6648 | 6306 | 6222 | |
| 10 | 6937 | 6862 | 6350 | 6868 | 4959 | 6824 | |
| 15 | 6001 | 6721 | 4201 | 6731 | 603 | 6243 | |
| 25 | 866 | 5962 | 163 | 6576 | 30 | 6363 | |
| 0.20 | 3 | 4881 | 7005 | 6172 | 6953 | 6204 | 6486 |
| 5 | 6582 | 6945 | 6676 | 6886 | 5937 | 6479 | |
| 10 | 6533 | 6756 | 5043 | 6670 | 2224 | 6377 | |
| 15 | 4470 | 6868 | 1191 | 6799 | 106 | 6382 | |
| 25 | 106 | 6134 | 5 | 6620 | 1 | 6356 | |
| 0.25 | 3 | 5505 | 6840 | 6481 | 6786 | 6272 | 6649 |
| 5 | 6628 | 6941 | 6320 | 6557 | 5778 | 6444 | |
| 10 | 5318 | 6875 | 3288 | 6756 | 663 | 6364 | |
| 15 | 1517 | 6317 | 147 | 6454 | 16 | 6522 | |
| 25 | 29 | 6288 | 0 | 6543 | 0 | 6207 | |
Compatibility with isobaric tagging techniques
Isobaric tagging technique (i.e., iTRAQ and TMT) usage has become increasingly widespread and important for quantitative proteomics owing to compatibility with tissues and biofluids and the ability to multiplex several samples in a single experiment. To investigate the use of IRMPD for MS/MS analysis of isobaric-tagged peptides, a synthetic peptide, WAAAKAAAK, was divided into six aliquots and labeled with TMT six-plex in 1:5:2:1.5:1:3 ratios, and subjected to IRMPD (Fig. 1). In addition to producing a near-complete series of both b- and y-type product ions, IRMPD also generates substantial reporter ion signal. The observed channels are within ~ 10% of the purity-corrected theoretical values (indicated by black dots), comparable to the accuracy that can be expected using beam-type collisional activation (HCD).25 We conclude that IRMPD conducted within a dual pressure QLT mass spectrometer is compatible with peptide quantitation using isobaric tags.
Figure 1.

Single scan IRMPD MS/MS spectrum of the doubly protonated peptide cation, WAAAKAAAK, performed at a q=0.13 relative to the precursor for 7 ms at 60 W laser power. The MS/MS AGC target was set to 1 x 104 charges. The labeled peptides were mixed in ratios of 1:5:2:1.5:1:3. The black dots indicate isotopic-purity corrected theoretical ratios.
An interesting observation was that the partitioning between reporter and b-/y- type product ions was dependent upon the irradiation time. To investigate the partitioning between reporter and b-/y- type product ions in greater detail, we generated an irradiation time-resolved plot of product ions derived from doubly protonated WAAAKAAAK. These data indicate that the optimal IRMPD activation time for the generation of b- and y- type product ions is shorter than that required to optimize production of TMT reporter ions (Fig. 2, Supplemental Figure 3). At very short IRMPD irradiation times (< 5 ms), insufficient time has elapsed for the precursor to dissociate into fragment ions. To determine whether reporter tag can be harvested from a primary product ion, we dissociated WAAAKAAAK precursor using CAD, isolated of the y6 product ion, and activated y6 by IRMPD (Fig. 3). Intriguingly, photoactivation of the y6 product ion primarily produces TMT reporter products. IRMPD of the peptide precursor generates b- and y- type product ions along with the TMT reporter ion; continued exposure of the b- and y-type products to photons induces secondary dissociation and concentrates signal in TMT reporter region (126–131 Th). This is of importance because maximizing TMT reporter ion intensity likewise improves quantitative accuracy. CAD activation of the y6 ion would likely produce, among other things, TMT reporter ions. Unfortunately, CAD of the y6 product ion (m/z ~ 1018) at a q-value of 0.25 would place the low-mass cutoff at m/z ~ 280, precluding the retention of such reporter ions. While the q-value of CAD activation could potentially be lowered, doing so would lower the efficiency of CAD activation by favoring resonance ejection over resonant activation.52
Figure 2.

IRMPD time-resolved plot of precursor, b-/y- type, and isobaric reporter tag intensities. The maximum yield of b- and y- type product ions occurs and a shorter time than the maximum yield of TMT reporter tag.
Figure 3.

Single-scan IRMPD MS3 spectra of the y6 ion generated via CAD from the doubly protonated peptide WAAAKAAAK. The isolated y6 ion was subjected to IRMPD for 1, 5, 10, and 15 ms at 60W laser power. Increasing IR activation times produces increasingly intense reporter ions.
LC-MS/MS isobaric tagging experiments
Encouraged by the results of our shotgun analyses of unmodified peptides and the apparent compatibility of IRMPD for MS/MS of TMT-tagged peptides, we investigated IRMPD for shotgun analysis of complex, TMT-tagged peptide mixtures. To ensure that the reporter ions necessary for peptide quantitation are retained for all peptide precursors, we set the QLT RF amplitude to a fixed value during IRMPD (low-mass cutoff was ~ m/z 100). We interrogated un-fractionated yeast whole-cell lysate digested with trypsin and labeled with TMT in the known ratios (1:5:2:1.5:1:3). In each experiment, IRMPD was performed on the ten most abundant precursors identified from the MS survey scan; we performed six individual analyses using irradiation times from 3 to 25 ms.
By fixing the RF amplitude during IRMPD the optimal irradiation time roughly scales with precursor m/z (Supplemental Fig. 4). To evaluate quantitative accuracy, the intensity of each individual TMT quantitation channel was compared to the individual intensity of all the other channels (e.g., TMT126 was compared to TMT127 through TMT131; TMT127 was compared to TMT126 and TMT128 through TMT131, etc.). We averaged the absolute value of the percentage deviation between theoretical (e.g., TMT126/TMT127 should be 0.2) and observed ratios for each of the 15 comparisons, producing a metric of approximate quantitative accuracy for each MS/MS spectrum. We conclude that quantitative accuracy is likewise dependent on precursor m/z, with accuracy generally superior for peptide precursors having low m/z values (Supplemental Fig. 5), although this can be remedied (vide infra). High quantitative accuracy generally correlated with high TMT reporter tag intensity (Supplemental Fig. 6). We reason that the uneven PSM production and quantitative accuracy across the precursor m/z range is largely a result of performing IRMPD at fixed QLT RF amplitude; by using this strategy, precursor q-value is inversely related to precursor m/z. The precursor q-value influences the proportion of time that precursor peptides spend in the center of QLT, exerting a strong influence over the magnitude of secondary dissociation and optimal irradiation time. To make IRMPD performance for both PSM production and quantitative accuracy uniform for precursors having a wide range of m/z values, we executed a set of nLC-MS/MS experiments where IRMPD time was set in a data-dependent manner, depending on precursor m/z. The general strategy we employed was to normalize the degree of secondary dissociation for all peptide precursors by manipulating the IRMPD irradiation time. Low m/z precursors (having a high q-value) require short irradiation times; in contrast, high m/z precursors (having a low q-value) require longer times (Supplemental Fig. 4). For precursor m/z ~ 550, 5 ms represents the optimal irradiation time for production of PSMs. To produce similarly favorable results for all precursor m/z values, we varied the irradiation time as shown below (Eqn. 1).
| Eq. 1 |
where t is the IRMPD irradiation time, and t0 is 5 ms. Reducing this equation further results in a straightforward, data-dependent IRMPD irradiation time set by a single normalized IRMPD irradiation time coefficient multiplied by the precursor m/z value (Eqn. 2).
| Eq. 2 |
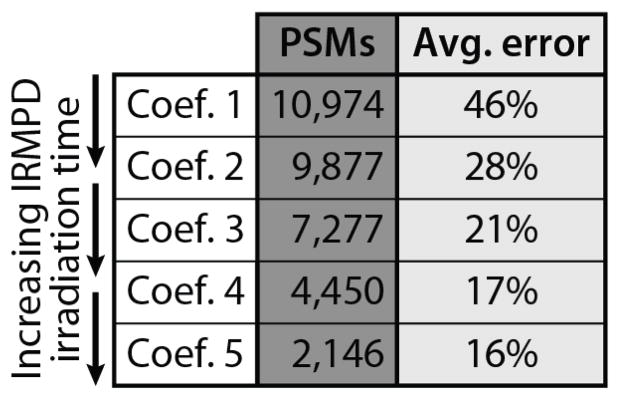
where t is the IRMPD irradiation time (ms) and ci is the coefficient. In our initial experiment (designed to optimize PSM production), this coefficient is 0.0091. To investigate conditions more favorable for TMT reporter tag generation and higher quantitative accuracy, we also conducted experiments in which this coefficient was set to 0.0136, 0.0182, 0.0227, and 0.0273; these coefficients correspond to irradiation times of between 5 and 15 ms for precursor m/z = 550. We term these coefficients 1, 2, 3, 4, and 5 respectively. By setting the IRMPD irradiation time dynamically, depending on precursor m/z, we successfully normalized the magnitude of secondary dissociation, ensuring homogenous IRMPD quantitative accuracy (Fig. 4) and PSM production across a wide range of precursor m/z values. Peptide precursors possessing higher m/z values are subjected to longer irradiation times, compensating for lower q-values (and slower IRMPD reaction kinetics); conversely, low m/z peptides having higher q-values are subjected to shorter IRMPD activation times. To examine the difference in identification rate for each coefficient, we binned each individual PSM by precursor m/z and divided by the total number of spectral features sampled over the course of the analysis. The resulting data provide a metric for how often a PSM is produced for each coefficient as a function of precursor m/z (Fig. 4). There is a slight falloff in PSM probability for peptides having either low or high m/z values; we attribute this to the sampling of non-peptidic features (low m/z) and decreased QLT trapping efficiency at very low q-values (high precursor m/z). For peptides having intermediate m/z values (comprising the vast majority of PSMs), the identification rate is consistent over a wide m/z range, with coefficient 1 representing the optimal setting for PSM production. TMT reporter tag intensity (Supplemental Fig 7) is likewise more uniform for all precursor peptides. Using coefficient 1 (shortest activation times) to set the irradiation time results in the most PSMs (10,974), but the worst quantitative accuracy (average error ~ 46%). Conversely, using coefficient 5 (longest irradiation times), we obtain the best quantitative accuracy (average error ~ 16%), but the lowest number of PSMs (2,146). Coefficients 2–4 represent intermediate irradiation times, offering a compromise between PSM production and quantitative accuracy (Table 2).
Figure 4.

Average probability of error (A) and average quantitative error (B) binned by precursor m/z value for IRMPD experiments. For each experiment, the RF amplitude was held constant, resulting in a fixed low-mass cutoff (LMCO) and dynamic precursor q- value. IRMPD activation times were set in a data-dependent manner by multiplying the precursor m/z by a coefficient. Using this strategy, we are able to normalize IRMPD PSM production and quantitative accuracy across a wide range of precursor m/z values. Coefficients 1 – 5 correspond to increased IRMPD irradiation time. Increasing irradiation time increases secondary dissociation, resulting in better quantitative accuracy, but confounding spectral interpretation.
Table 2.
PSMs and approximate quantitative error for IRMPD using different irradiation times. Using a straightforward data-dependent IRMPD irradiation time to normalize performance vs. precursor m/z, we see that the highest numbers of PSMs occur using shorter irradiation times, with best quantitative accuracy at longer irradiation times. We attribute this to more secondary dissociation at longer times producing greater reporter tag intensity at the expense of b- and y- type fragment ions.
 |
The most common means of interrogating isobaric-tagged peptides, beam-type collisional dissociation, HCD, has similar tradeoffs.25 A series of shotgun experiments using HCD to interrogate isobaric-tagged peptides reveals HCD activation conditions can be tailored to either optimize PSMs or reporter tag intensity, but not both (Supplementary Fig. 8).44 If we plot the IRMPD data displayed in figure 4 and table 2 similarly, treating the coefficient as an analogue to HCD collision energy, the two plots are remarkably similar (Supplementary Fig. 9). This is presumably because HCD dissociation of b- and y-type product ions results in the formation of reporter tag, increasing the overall reporter tag intensity and quantitative accuracy.53 The optimal activation conditions of both IRMPD and HCD require careful consideration of the overall goals of the experiment.
Ultimately, we conclude that the use of static QLT RF amplitude and dynamic IRMPD irradiation time represents an effective strategy to both identify and quantify complex mixtures of isobaric-tagged peptides. Ideal IRMPD activation conditions for the generation of reporter tag result in a high degree of secondary dissociation, depleting b- and y- type product ions and complicating spectral interpretation. Fragmentation, whether in an ion trap or collision cell, can produce reporter signal; however, neither is optimized for exclusive tag production. IRMPD for as little as 30 ms results in near-exclusive conversion of precursor peptides into TMT reporter ions. To determine how this compares to iHCD and pulsed-q dissociation (PQD), we interrogated MAAAKAAAK under conditions which maximize precursor-to-reporter tag conversion efficiency for each method. We find that IRMPD converts precursor signal to TMT reporter ions with over two-fold greater efficiency than iHCD, and with nearly ten-fold greater efficiency than PQD (Supplemental Fig. 10). An enticing future application of the present work is the divorcing of the dissociation event used for peptide sequencing from that used to generate reporter tag. IRMPD performed under conditions optimized for exclusive TMT reporter tag generation followed by either IRMPD under different activation conditions or other dissociation techniques to provide peptide sequence information would result in both peptide sequence information and highly accurate quantitative information.
Conclusion
We modified a standalone dual-cell quadrupole linear ion trap (QLT) mass spectrometer to perform IRMPD in the low-pressure cell. We demonstrated that IRMPD performs at least as well as resonant-excitation CAD for shotgun peptide sequencing. The proper combinations of IRMPD activation parameters result in marginally more PSMs generated than CAD for a complex peptide mixture generated using trypsin (paired student’s t-test, p=0.019) while maintaining roughly the same overall activation time (~10 ms for both CAD and IRMPD). We also evaluated IRMPD performed on a dual-cell QLT in the context of isobaric-tagged peptides. We demonstrate that IRMPD produces reporter tags with fidelity to the theoretical ratios and that the partitioning between b- and y- type ions and reporter tags is heavily dependent upon irradiation time as secondary dissociation of b- and y- type ions produces isobaric report tag.
We performed a series of nLC-MS/MS analyses of complex mixtures of isobaric-tagged peptides. Using fixed QLT RF amplitude results in consistent retention of TMT reporter ions, but also strong dependence between precursor m/z and the ability to both identify peptides and provide accurate quantitation. We reason that such uneven performance is a result of precursor q-value (dependent upon precursor m/z at fixed RF amplitudes) exerting influence in the degree of IRMPD secondary dissociation for a given irradiation time. To counter this, we developed an algorithm to set the IRMPD irradiation time in a data-dependent manner, based on precursor m/z value, the ability to sequence precursor peptides and gain quantitative information are normalized across a wide precursor m/z range. While optimal settings for peptide identification are somewhat different than optimal activation settings for quantitation that an effective compromise can be reached and that IRMPD represents a viable option for the interrogation of isobaric-tagged peptides. This tradeoff is highly similar to what is encountered when using beam-type collisional activation for interrogation of isobaric-tagged peptides. Future work will focus on the use of IRMPD for isobaric tagging on high-resolution mass spectrometers, as well as a more comprehensive comparison of IRMPD to beam-type collisional activation for the interrogation of isobaric-tagged peptides.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support from Thermo Fisher Scientific, NSF career grant 0747990, and NIH grant R01 GM080148.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Hunt DF, Yates JR, Shabanowitz J, Winston S, Hauer CR. PROTEIN SEQUENCING BY TANDEM MASS-SPECTROMETRY. Proc Natl Acad Sci U S A. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 4.de Godoy LMF, et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–U1260. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- 5.Liu HB, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 6.Ong SE, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Jiang H, English AM. Quantitative analysis of the yeast proteome by incorporation of isotopically labeled leucine. J Proteome Res. 2002;1:345–350. doi: 10.1021/pr025523f. [DOI] [PubMed] [Google Scholar]
- 8.Du Y, Parks BA, Sohn S, Kwast KE, Kelleher NL. Top-down approaches for measuring expression ratios of intact yeast proteins using Fourier transform mass spectrometry. Anal Chem. 2006;78:686–694. doi: 10.1021/ac050993p. [DOI] [PubMed] [Google Scholar]
- 9.Washburn MP, Ulaszek R, Deciu C, Schieltz DM, Yates JR. Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal Chem. 2002;74:1650–1657. doi: 10.1021/ac015704l. [DOI] [PubMed] [Google Scholar]
- 10.MacCoss MJ, Wu CC, Liu HB, Sadygov R, Yates JR. A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal Chem. 2003;75:6912–6921. doi: 10.1021/ac034790h. [DOI] [PubMed] [Google Scholar]
- 11.Ross PL, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Thompson A, et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 13.Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 14.Hsu JL, Huang SY, Chow NH, Chen SH. Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem. 2003;75:6843–6852. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- 15.Choe L, et al. 8-Plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer’s disease. Proteomics. 2007;7:3651–3660. doi: 10.1002/pmic.200700316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dayon L, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 17.Hunt DF, Buko AM, Ballard JM, Shabanowitz J, Giordani AB. SEQUENCE-ANALYSIS OF POLYPEPTIDES BY COLLISION ACTIVATED DISSOCIATION ON A TRIPLE QUADRUPOLE MASS-SPECTROMETER. Biomedical Mass Spectrometry. 1981;8:397–408. doi: 10.1002/bms.1200080909. [DOI] [PubMed] [Google Scholar]
- 18.Louris JN, et al. INSTRUMENTATION, APPLICATIONS, AND ENERGY DEPOSITION IN QUADRUPOLE ION-TRAP TANDEM MASS-SPECTROMETRY. Anal Chem. 1987;59:1677–1685. doi: 10.1021/ac00140a021. [DOI] [Google Scholar]
- 19.McLuckey SA. PRINCIPLES OF COLLISIONAL ACTIVATION IN ANALYTICAL MASS-SPECTROMETRY. J Am Soc Mass Spectrom. 1992;3:599–614. doi: 10.1016/1044-0305(92)85001-z. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz JCS, JEP, Quarmby ST. 53rd ASMS Conference on Mass Spectrometry and Allied Topics. [Google Scholar]
- 21.Cunningham C, Glish GL, Burinsky DJ. High amplitude short time excitation: A method to form and detect low mass product ions in a quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2006;17:81–84. doi: 10.1016/j.jasms.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Morris HR, et al. High sensitivity collisionally-activated decomposition tandem mass spectrometry on a novel quadrupole/orthogonal-acceleration time-of-flight mass spectrometer. Rapid Commun Mass Spectrom. 1996;10:889–896. doi: 10.1002/(sici)1097- 0231(19960610)10:8<889::aid-rcm615>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- 23.Tang XJ, Ens W, Standing KG, Westmore JB. DAUGHTER ION MASS-SPECTRA FROM CATIONIZED MOLECULES OF SMALL OLIGOPEPTIDES IN A REFLECTING TIME-OF-FLIGHT MASS-SPECTROMETER. Anal Chem. 1988;60:1791–1799. doi: 10.1021/ac00168a029. [DOI] [PubMed] [Google Scholar]
- 24.Verentchikov AN, Ens W, Standing KG. REFLECTING TIME-OF-FLIGHT MASSSPECTROMETER WITH AN ELECTROSPRAY ION-SOURCE AND ORTHOGONAL EXTRACTION. Anal Chem. 1994;66:126–133. doi: 10.1021/ac00073a022. [DOI] [PubMed] [Google Scholar]
- 25.Olsen JV, et al. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 26.McAlister GC, Phanstiel DH, Brumbaugh J, Westphall MS, Coon JJ. Higher-energy Collision-activated Dissociation Without a Dedicated Collision Cell. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.O111.009456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madsen JA, Boutz DR, Brodbelt JS. Ultrafast Ultraviolet Photodissociation at 193 nm and its Applicability to Proteomic Workflows. J Proteome Res. 2010;9:4205–4214. doi: 10.1021/pr100515x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brodbelt JS, Wilson JJ. INFRARED MULTIPHOTON DISSOCIATION IN QUADRUPOLE ION TRAPS. Mass Spectrom Rev. 2009;28:390–424. doi: 10.1002/mas.20216. [DOI] [PubMed] [Google Scholar]
- 29.Reilly JP. ULTRAVIOLET PHOTOFRAGMENTATION OF BIOMOLECULAR IONS. Mass Spectrom Rev. 2009;28:425–447. doi: 10.1002/mas.20214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Black DM, Payne AH, Glish GL. Determination of cooling rates in a quadrupole ion trap. J Am Soc Mass Spectrom. 2006;17:932–938. doi: 10.1016/j.jasms.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Boue SM, Stephenson JL, Yost RA. Pulsed helium introduction into a quadrupole ion trap for reduced collisional quenching during infrared multiphoton dissociation of electrosprayed ions. Rapid Commun Mass Spectrom. 2000;14:1391–1397. doi: 10.1002/1097- 0231(20000815)14:15<1391::aid-rcm36>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 32.Hashimoto Y, Hasegawa H, Waki L. High sensitivity and broad dynamic range infrared multiphoton dissociation for a quadrupole ion trap. Rapid Commun Mass Spectrom. 2004;18:2255–2259. doi: 10.1002/rcm.1619. [DOI] [PubMed] [Google Scholar]
- 33.Newsome GA, Glish GL. Improving IRMPD in a Quadrupole Ion Trap. J Am Soc Mass Spectrom. 2009;20:1127–1131. doi: 10.1016/j.jasms.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Hashimoto Y, Hasegawa H, Yoshinari K, Waki I. Collision-activated infrared multiphoton dissociation in a quadrupole ion trap mass spectrometer. Anal Chem. 2003;75:420–425. doi: 10.1021/ac025866x. [DOI] [PubMed] [Google Scholar]
- 35.Payne AH, Glish GL. Thermally assisted infrared multiphoton photodissociation in a quadrupole ion trap. Anal Chem. 2001;73:3542–3548. doi: 10.1021/ac010245+. [DOI] [PubMed] [Google Scholar]
- 36.Kjeldsen F, Giessing AMB, Ingrell CR, Jensen ON. Peptide sequencing and characterization of post-translational modifications by enhanced ion-charging and liquid chromatography electron-transfer dissociation tandem mass spectrometry. Anal Chem. 2007;79:9243–9252. doi: 10.1021/ac701700g. [DOI] [PubMed] [Google Scholar]
- 37.Pikulski M, Wilson JJ, Aguilar A, Brodbelt JS. Amplification of infrared multiphoton dissociation efficiency in a quadruple ion trap using IR-active ligands. Anal Chem. 2006;78:8512–8517. doi: 10.1021/ac061472k. [DOI] [PubMed] [Google Scholar]
- 38.Madsen JA, Brodbelt JS. Comparison of Infrared Multiphoton Dissociation and Collision- Induced Dissociation of Supercharged Peptides in Ion Traps. J Am Soc Mass Spectrom. 2009;20:349–358. doi: 10.1016/j.jasms.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Gardner MW, Vasicek LA, Shabbir S, Anslyn EV, Brodbelt JS. Chromogenic cross-linker for the characterization of protein structure by infrared multiphoton dissociation mass spectrometry. Anal Chem. 2008;80:4807–4819. doi: 10.1021/ac800625x. [DOI] [PubMed] [Google Scholar]
- 40.Second TP, et al. Dual-Pressure Linear Ion Trap Mass Spectrometer Improving the Analysis of Complex Protein Mixtures. Anal Chem. 2009;81:7757–7765. doi: 10.1021/ac901278y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardner MW, et al. Infrared Multiphoton Dissociation of Peptide Cations in a Dual Pressure Linear Ion Trap Mass Spectrometer. Anal Chem. 2009;81:8109–8118. doi: 10.1021/ac901313m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Enyenihi AA, Griffiths JR, Glish GL. Tandem mass spectrometric methods for the analysis of iTRAQ labeled peptides in a quadrupole ion trap. Int J Mass Spectrom. 2011;308:260–264. doi: 10.1016/j.ijms.2011.08.014. [DOI] [Google Scholar]
- 43.McAlister GC, Phanstiel D, Wenger CD, Lee MV, Coon JJ. Analysis of Tandem Mass Spectra by FTMS for Improved Large-Scale Proteomics with Superior Protein Quantification. Anal Chem. 2010;82:316–322. doi: 10.1021/ac902005s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wenger CD, et al. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods. 2011;8:933–935. doi: 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee MV, et al. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol Syst Biol. 2011;7:514. doi: 10.1038/msb.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madsen JA, et al. Top-Down Protein Fragmentation by Infrared Multiphoton Dissociation in a Dual Pressure Linear Ion Trap. Anal Chem. 2009;81:8677–8686. doi: 10.1021/ac901554z. [DOI] [PubMed] [Google Scholar]
- 47.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ. COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11:1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore RE, Young MK, Lee TD. Qscore: An algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom. 2002;13:378–386. doi: 10.1016/s1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- 49.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 50.Remes PM, Glish GL. Mapping the Distribution of Ion Positions as a Function of Quadrupole Ion Trap Mass Spectrometer Operating Parameters to Optimize Infrared Multiphoton Dissociation. J Phys Chem A. 2009;113:3447–3454. doi: 10.1021/jp808955w. [DOI] [PubMed] [Google Scholar]
- 51.Sleno L, Volmer DA. Ion activation methods for tandem mass spectrometry. J Mass Spectrom. 2004;39:1091–1112. doi: 10.1002/jms.703. [DOI] [PubMed] [Google Scholar]
- 52.Charles MJ, McLuckey SA, Glish GL. COMPETITION BETWEEN RESONANCE EJECTION AND ION DISSOCIATION DURING RESONANT EXCITATION IN A QUADRUPOLE ION-TRAP. J Am Soc Mass Spectrom. 1994;5:1031–1041. doi: 10.1016/1044-0305(94)85065-8. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, et al. A Robust Error Model for iTRAQ Quantification Reveals Divergent Signaling between Oncogenic FLT3 Mutants in Acute Myeloid Leukemia. Mol Cell Proteomics. 2010;9:780–790. doi: 10.1074/mcp.M900452-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.