

Abstract
Blood platelets are anucleate cell fragments that play a critically important role in hemostasis and thrombosis. Platelets are activated with various agonists that allow them to aggregate, thus forming either hemostatic plugs or pathologic thrombi. Recent studies have revealed that at least two activated platelet subpopulations are formed upon potent stimulation of platelets with collagen and/or thrombin. One of these subpopulations consists of so-called coated platelets that express high levels of phosphatidylserine and retain α-granule proteins, including fibrinogen, on their surface. They also have reduced levels of the main aggregation receptor-activated glycoprotein IIb-IIIa, which might indicate a defect in their proaggregatory ability. In this study, the proaggregatory abilities of coated and noncoated platelets were assessed by means of light transmission aggregometry of suspensions with varying ratios of platelets from one subpopulation to those of a different subpopulation. A mathematical model of platelet aggregation in heterogeneous mixtures was developed to assist in the analysis of experimental data. Flow cytometry was employed to monitor platelet recruitment into aggregates and the ability of platelets to bind external fibrinogen. Finally, confocal microscopy was used to image coated platelets involved into aggregates formed by mechanical shaking. The obtained data revealed to our knowledge a novel mechanism regulating aggregate formation of platelet subpopulations: coated platelets cannot aggregate with each other but can be recruited into aggregates by noncoated platelets.
Introduction
Blood platelets are small (2–4 μm) anucleate cell fragments that circulate in blood at a concentration of 2–4 × 105/μl. They have a distinct property of becoming activated by a number of physiological substances (thrombin, collagen, ADP, thromboxane A2, etc.): this endows them with the ability to aggregate, i.e., to form clusters. The ability to aggregate allows them to stop bleeding by forming hemostatic plugs and thus sustaining the integrity of the vascular system. Under pathological conditions, this proaggregatory ability leads to the formation of life-threatening thrombi that are a major cause of mortality and morbidity in developed countries.
During the last decades, all experimental and theoretical studies of platelet aggregation assumed that activated platelets are homogeneous (1). However, it was discovered that potent stimulation of platelets with collagen and thrombin (2,3), or with thrombin alone (4), leads to their segregation into two subpopulations. Members of the new subpopulation, termed coated platelets, express high levels of phosphatidylserine (PS) and retain on their outer membrane surface large amounts of several α-granule proteins, including fibrinogen, von Willebrand factor, fibronectin, factor V, and thrombospondin (3,5,6). Although there exist many studies on the activated platelet subpopulations that do not actually use the term coated (as recently summarized and reviewed in Jackson and Schoenwaelder (7) and Munnix et al. (8)), formation of a distinct, highly procoagulant PS-expressing platelet subpopulation appears to be a universal and ubiquitous phenomenon, and this subpopulation was often found to share many features with coated platelets, although exceptions were reported as well (9). Strictly speaking, platelets play two critical roles in hemostasis and thrombosis: adhering/aggregating at the site of vascular injury, and providing a negatively charged surface for thrombin generation (10). Coated and noncoated platelets are assumed to have different procoagulant activities, since they provide vastly different contents of PS, on which the procoagulant complexes are assembled (11). However, it is not precisely known whether platelets of these two subpopulations differ from each other in their second ability, that of aggregate formation.
Three lines of evidence indicate that PS-exposing, coated platelets probably have impaired proaggregatory ability compared with normal activated platelets. First, they bind 10 times less PAC-1 (a monoclonal antibody that recognizes the fibrinogen-binding site of activated glycoprotein IIb-IIIa (GPIIbIIIa) (12), a platelet integrin required for aggregation (13)) than noncoated platelets do, although the surface expression of GPIIbIIIa in coated platelets is unchanged (3,8). Second, inhibition of transglutaminase that leads to reduction of the platelet population with coated characteristics also significantly increases the rate and extent of thrombus growth on a thrombogenic surface (14). Finally, during thrombus formation under flow, different clusters of platelets appear to be responsible for aggregate formation and for coagulant activity (9). This feature may play a functional role in governing thrombus growth (14,15).
However, there were no attempts to investigate the proaggregatory ability of coated platelets directly. Here, we provide evidence from in vitro experiments that platelets from these two distinct subpopulations, coated and noncoated platelets, indeed have qualitatively different proaggregatory abilities, although the difference is more subtle than has been expected. Coated platelets cannot bind each other, but they efficiently participate in aggregate formation being recruited into aggregates by noncoated platelets.
Materials and Methods
Reagents
The following materials were obtained from the sources shown in parentheses: convulxin (Pentapharm, Basel, Switzerland); human thrombin (Haematologic Technologies, Essex Junction, VT); prostaglandin E1 (MP Biochemicals, Irvine, CA); R-phycoerythrin (PE)-conjugated annexin V (Molecular Probes, Eugene, OR, or eBioscience, San Diego, CA); fluorescein-5-isothiocyanate (FITC)-conjugated PAC-1 (BD Biosciences, San Jose, CA); FITC-conjugated annexin V (BD Biosciences); PPACK (Calbiochem, San Diego, CA); FITC-conjugated anti-human fibrinogen antibody (Labvision, Fremont, CA); peridinin-chlorophyll-protein (PerCP)-conjugated anti-human CD61 (BD Biosciences); FITC (Molecular Probes); and Sephadex G-25 (Pharmacia, Uppsala, Sweden). Collagen-related peptide (CRP) was kindly provided by Prof. R.W. Farndale (University of Cambridge, Cambridge, UK). Glycoprotein IIb-IIIa antagonist Monafram, a F(ab′)2 fragment of monoclonal antibody that blocks this receptor (16,17), was a generous gift of Prof. A.V. Mazurov (Russian Cardiology Research and Production Center, Moscow, Russia). Human fibrinogen and all other reagents were from Sigma-Aldrich (St Louis, MO).
Platelet isolation
Platelets were isolated from freshly drawn human blood of healthy volunteers collected with their written informed consent under approval of the Center for Theoretical Problems of Physicochemical Pharmacology and National Research Center for Hematology Ethical Committees essentially as described (18). Briefly, blood was collected into 3.8% sodium citrate with pH 5.5 at 9:1 blood/anticoagulant volume ratio and supplemented with prostaglandin E1 (1 μM) and usually with apyrase (0.1 unit/ml) to prevent platelet activation. It was centrifuged at 100 × g for 8 min at room temperature. The obtained platelet-rich plasma was supplemented with 3.8% sodium citrate, pH 5.5, at 1:3 citrate/plasma volume ratio to decrease pH. Platelets were concentrated by centrifugation at 400 × g for 5 min, resuspended in buffer A (150 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.4 mM NaH2PO4, 20 mM HEPES, 5 mM glucose, and 0.5% bovine serum albumin), and subjected to gel filtration on a chromatography column packed with Sepharose CL-2B and equilibrated with buffer A.
Aggregation
Washed platelets at 100,000/μl with 2.5 mM CaCl2 in buffer A were preincubated for 3 min at 37°C and stimulated by the indicated agonists for 15 min to allow formation of the coated and noncoated platelet subpopulations (9). Then, 1 μM PPACK and 1 mg/ml fibrinogen were added, and aggregation of the samples was monitored for 15 min by light transmission with the Model 490 aggregometer (Chrono-log, Havertown, PA) while stirring (800 rpm) at 37°C.
Flow cytometry
Platelets at 100,000/μl were activated by incubation with agonists in buffer A with 2.5 mM CaCl2 for 15 min, additionally incubated with annexin V (2) and appropriate antibodies (anti-fibrinogen antibody or PAC-1) for 3 min, diluted 10-fold, and immediately analyzed in a FACSCalibur flow cytometer (BD Biosciences). The acquired data were processed using WinMDI 2.8 software (Joseph Trotter, Scripps Research Institute, La Jolla, CA).
Flow-cytometry-based aggregation assay
Platelets at 20,000/μl were activated by incubation with agonists in buffer A with 2.5 mM CaCl2 for 15 min and additionally incubated with labeling antibody and annexin V for 3 min. This was followed by addition of 1 μM PPACK and 1 mg/ml fibrinogen. Then platelets were shaken (600 rpm) in MS1 Minishaker (IKA, Staufen, Germany) for 3 min, diluted 10-fold, and analyzed in a flow cytometer.
Fibrinogen labeling
Fibrinogen (at 10 mg/ml) was dissolved in 0.1 M sodium bicarbonate buffer, pH 9. Then, 10 mg/ml FITC dissolved in DMSO was added (MR = 5). This was followed by incubation for 2 h at 4°C with continuous stirring. The reaction was stopped by adding 1.5 M hydroxylamine, pH 8.5, and incubation for 30 min at 4°C. Then, the reaction mixture was centrifuged for 1 min at 16,000 × g in Sephadex G-25 spin columns to separate the conjugate from unreacted labeling reagent.
Fibrinogen binding to platelet subpopulations
Platelets at 20,000/μl were activated by incubation with agonists in buffer A with 2.5 mM CaCl2 for 15 min. Then, 1 μM PPACK was added to prevent thrombin-induced fibrin formation. This was followed by additional incubation of platelets with annexin V and 0.5 mg/ml FITC-conjugated fibrinogen for 3 min. Platelets were diluted 20-fold and analyzed in a flow cytometer.
Confocal microscopy
Glass coverslips (24 × 24 mm, Heinz Herenz, Hamburg, Germany) were cleaned with potassium dichromate, rinsed with distilled water and dried. The cleaned coverslips were coated with 20 mg/ml fibrinogen in buffer A for 40 min at room temperature, rinsed with distilled water, and then assembled as part of the flow chamber. Platelets at 20,000/μl were activated by incubation with agonists in buffer A with 2.5 mM CaCl2 and annexin V for 15 min. Then, 1 μM PPACK and 1 mg/ml fibrinogen were added, and platelets were shaken (600 rpm) in a minishaker for 3 min and allowed to spread on the fibrinogen surface in the chamber for 20–30 min. Confocal images of platelet aggregates were acquired with an Axio Observer.Z1 microscope (Carl Zeiss, Jena, Germany) with a 100× microscopic objective. A 488-nm laser was utilized.
Statistics
All experiments were performed at least in triplicate with platelets from different donors. Comparisons were carried out with the one-way ANOVA test. Statistical significance was set as P < 0.05. Values are reported as mean ± SE unless specified otherwise.
Mathematical model of platelet aggregation
To analyze experimental data on platelet aggregation in samples containing different subpopulations, a mathematical model was created. The principal model assumptions were: 1), platelet suspension with continuous stirring consists of two subpopulations, one of which (the noncoated ones) aggregates normally; 2), platelet aggregates grow in size as the platelets irreversibly bind each other; and 3), the probability of a collision between two particles (either platelets or aggregates) in suspension does not significantly depend on their size. The variables were X1, i.e., concentration of the noncoated platelets and their aggregates, and X2, concentration of the coated platelets and aggregates that contain them. Model equations were
| (1) |
| (2) |
where ki are rate constants defining the stickiness of platelets. Equations 1 and 2 have the explicit solution:
| (3) |
| (4) |
where initial concentrations of platelets from distinct subpopulations are X1(0) = X10, X2(0) = X20.
Light transmission, y, was assumed to be exponentially proportional to the number of particles in suspension (20):
| (5) |
where S is an aggregometer-specific constant. Taking into account the values of initial and final light transmission (y(0) = 0, y(t→∞) = 1), we obtained:
| (6) |
A typical aggregatory curve was approximated by Hill's equation, and thus, ki and S in the case where all platelets in suspension aggregate equally were determined:
k1 = k3 = 0.0009, k2 = 0.00091, S = 0.001.
Results
Variation of agonist concentration can be used to obtain coated-platelet percentage up to ∼90% in suspension
To investigate proaggregatory abilities of platelets from two distinct subpopulations, we activated platelets with agonists of different potency. This resulted in the formation of different percentages (up to 89.2%) of coated platelets within 15 min (Fig. 1 A). This time interval is known to be sufficient for coated-platelet formation to reach completion with regard to annexin V binding and downregulation of PAC-1 binding (9) (this was also confirmed independently by control experiments; data not shown). Indeed, activation of platelets with thrombin alone (Fig. 1, B and C), or dual agonist activation with thrombin plus convulxin (not shown), resulted in segregation of activated platelets into two distinct subpopulations. The dot plots in Fig. 1, B and C, show that, in agreement with previous reports (3,5,8,15), PS-positive platelets have decreased PAC-1 binding (Fig. 1 B) and increased fibrinogen retention (Fig. 1 C). These data indicate that varying the agonist concentration can produce up to 90% of coated platelets in suspension.
Figure 1.
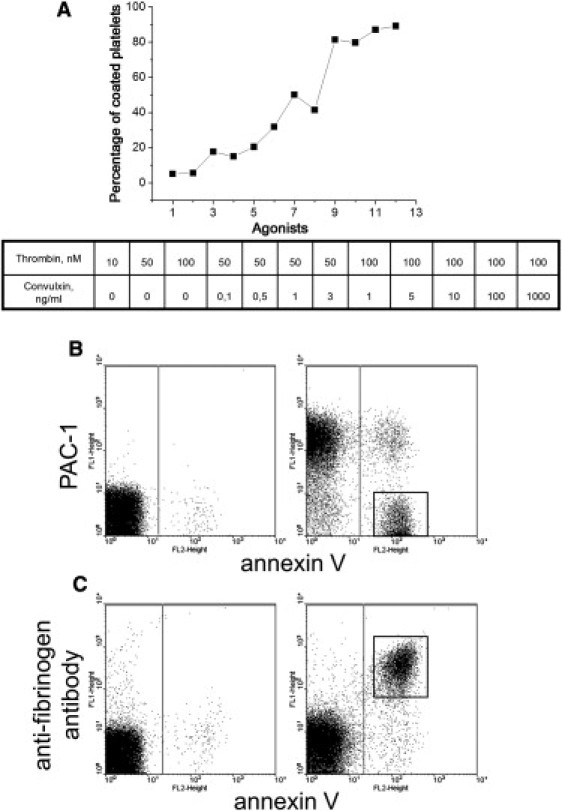
Different agonist concentrations can produce various percentages of coated platelets that have increased fibrinogen retention and decreased PAC-1 binding. (A) Correlation of coated-platelet percentage, determined by flow cytometry, with the degree of platelet stimulation. Platelets (100,000/μl) were stimulated with 10, 50, or 100 nM thrombin, 50 nM thrombin plus 0.1, 0.5, 1, or 3 ng/ml convulxin, and 100 nM thrombin plus 1, 5, 10, 100, or 1000 ng/ml convulxin (numbers 1–12 on the x axis, consistently). Typical data obtained from the experiments with platelets from three different donors are presented. (B and C) Dot plots for binding of PAC-1 (B) or anti-fibrinogen antibody (C) versus annexin V for platelets (100,000/μl) either unstimulated (left) or stimulated with 100 nM thrombin (right). The boxed area indicates the coated-platelet subpopulation. A typical experiment out of three performed with platelets from different donors is shown.
Increasing coated-platelet percentage inhibits aggregation in a nonlinear manner
To compare proaggregatory abilities of the noncoated and coated platelets, we first compared aggregation of the samples with different percentages of coated platelets in the classical light transmission aggregometry assay (Fig. 2). The speed (maximal slope of the aggregatory curve) and extent (maximal increase of light transmission) of aggregation were estimated, as shown in Fig. 2 A. The speed of aggregation indicated a uniform decline with the increasing coated-platelet percentage (Fig. 2 B), whereas the extent of aggregation remained constant in the range of ∼0–60% of coated platelets and was considerably decreased with subsequent increase of coated-platelet percentage (Fig. 2 C). These data suggest that coated platelets somehow participate in aggregate formation; otherwise, the presence of 50% of coated platelets in suspension would significantly inhibit both of the aggregation parameters. However, this intuitively obvious suggestion needs to be analyzed and proven more rigorously.
Figure 2.
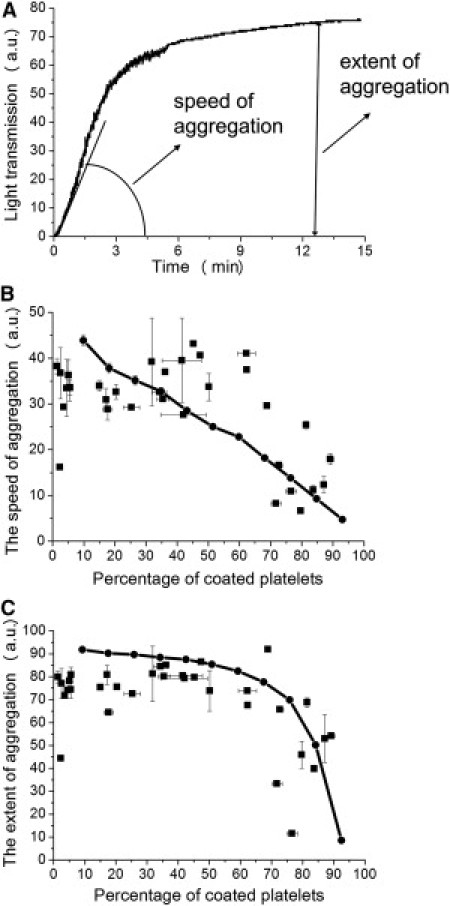
Inhibition of aggregation by the increasing number of coated platelets in the sample. (A) Typical aggregation curve and its parameters, the speed and extent of aggregation. (B and C) Dependencies of the speed (B) and extent (C) of aggregation of platelets (100,000/μl) stimulated with thrombin (10, 50, or 100 nM) or thrombin (50 or 100 nM) plus convulxin (0.1, 0.5, 1, 3, 5, 50, 60, 70, 80, 90, 100, 150, 200, 500, or 1000 ng/ml) on the percentage of coated platelets. Values are expressed as the mean ± SEM for n = 2 experiments with platelets from six different donors. Solid curves represent these dependencies based on a mathematical model for platelets from two distinct subpopulations in the case when coated platelets can bind noncoated ones, but fail to bind each other.
Mathematical model of aggregation of platelets from two distinct subpopulations
To interpret correctly the results from aggregometry experiments, we developed a model describing interactions between platelets from two subpopulations (see Methods). Aggregatory curves based on this model were plotted for three cases in which 1), coated and noncoated platelets have equal capacity to bind each other; 2), coated platelets can bind noncoated platelets but cannot form aggregates by themselves; and 3), coated platelets do not have the capacity to bind any platelets. In each case, the dependencies of the speed and extent of aggregation on the percentage of coated platelets in suspension were determined (Fig. 3). If all platelets have equal capacity to bind each other, that is, none of the constants are zero, there is no correlation between the percentage of coated platelets and the parameters of aggregation, as shown in Fig. 3, B and C. When coated platelets can bind noncoated platelets but fail to bind each other, that is, k3 = 0, the dependencies of aggregation parameters on the percentage of coated platelets (Fig. 3, B and C) strongly remind us of dependencies obtained in our aggregometry assay. An overlay of the dependencies of aggregation parameters on the percentage of coated platelets obtained in the experimental assay and based on the mathematical model is shown in Fig. 2, B and C. Finally, if coated platelets fail to bind any platelets, that is, k2 = k3 = 0, both aggregation parameters are drastically inhibited with the increasing number of coated platelets (Fig. 3, B and C). These results point to the fact that the data obtained in experiments using aggregometry are caused by the participation of coated platelets together with noncoated ones in aggregate formation. According to this model, coated platelets cannot bind each other but are involved into developing aggregates by noncoated platelets. We suggest that the binding of coated and noncoated platelets occurs most likely via coated platelet fibrinogen, known to be retained on their surface of coated platelets with great affinity (3). This may serve as a possible explanation of why coated platelets fail to bind each other, but are good at binding noncoated platelets.
Figure 3.
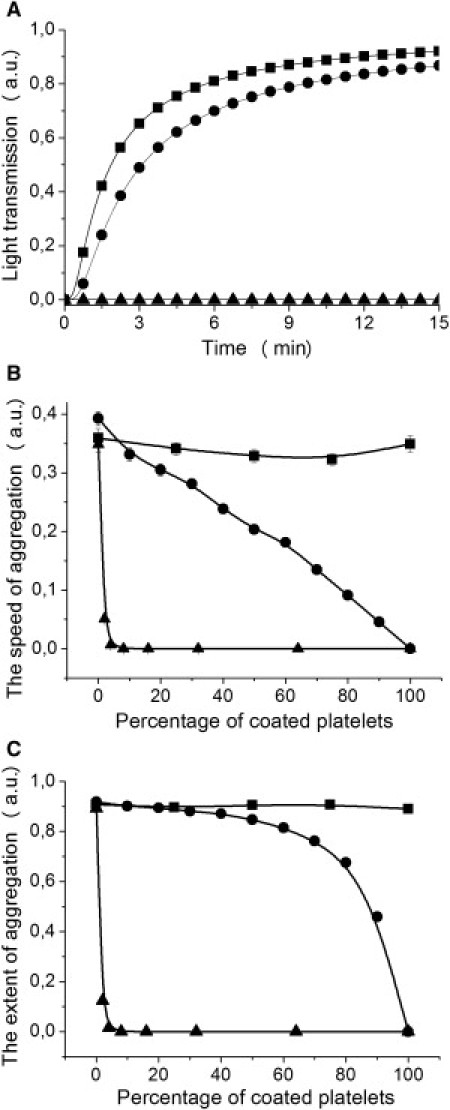
Dependencies of aggregation parameters on the percentage of coated platelets reflect the rate of coated-platelet participation in aggregate formation. (A) Dependencies of light transmission on time based on the aggregation model for platelets from two distinct subpopulations in the case where 50% of coated platelets present in suspension. (B and C) Dependencies of speed (B) and extent (C) of aggregation on the percentage of coated platelets, based on the aggregation model for platelets from two distinct subpopulations. The three curves on each panel represent the cases in which 1), all platelets have equal capacity to bind each other (squares), that is, constants are k1 = k3 = 0.0009, k2 = 0.00091, and S = 0.001; 2), coated platelets can bind noncoated ones but fail to bind each other (circles), that is, constants are k1 = 0.0009, k2 = 0.00091, k3 = 0, and S = 0.001; and 3), coated platelets cannot bind any platelets (triangles), that is, constants are k1 = 0.0009, k2 = k3 = 0, and S = 0.001.
Coated platelets fail to bind external fibrinogen
As we have just seen, the data from the classical aggregometry assay are in excellent agreement with the model wherein coated platelets cannot aggregate with each other but can be recruited into aggregates by noncoated platelets. To examine directly whether coated platelets indeed have impaired capacity for binding external fibrinogen in addition to that retained on their surface, flow cytometric studies were performed. Activated platelet binding of FITC-conjugated fibrinogen was estimated (Fig. 4). The mean fluorescence intensity of noncoated platelets was 91.4 ± 17.8 a.u., whereas those of coated and untreated platelets were 8 ± 0.4 and 3.6 ± 0.1 a.u. (n = 3), respectively. That is, only noncoated platelets readily bound external fibrinogen, whereas coated platelets almost completely failed to do so.
Figure 4.
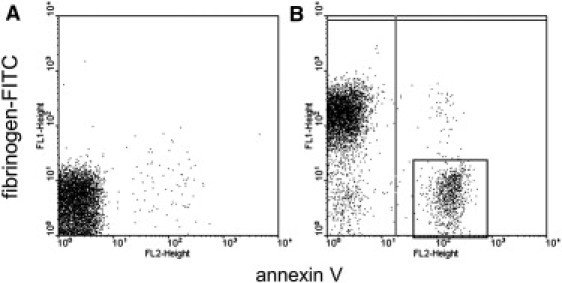
Coated platelets fail to bind external fibrinogen. (A and B) Dot plots for binding of fibrinogen-FITC versus annexin V for platelets (20,000/μl) either unstimulated (A) or stimulated with 200 nM thrombin (B). The boxed area indicates the coated-platelet subpopulation. Shown is a typical experiment out of three performed with platelets from different donors.
Coated platelets participate in aggregate formation by being recruited into aggregates by noncoated platelets
To test the second part of our hypothesis, that coated platelets participate in aggregate formation because they are recruited into developing aggregates by noncoated platelets, other flow cytometric studies were performed. Platelets were activated so, that there were both coated and noncoated platelets, allowed to form aggregates by shaking for several minutes, and analyzed by flow cytometer. Numbers of coated and noncoated platelets excluding aggregates for samples with and without the shaking were compared (Fig. 5). Percentages of noncoated and coated platelets leaving the suspension after shaking to form aggregates were 68.6 ± 7.4% (n = 3, P < 0.05) and 26.1 ± 5.3% (n = 3, P < 0.05), respectively.
Figure 5.
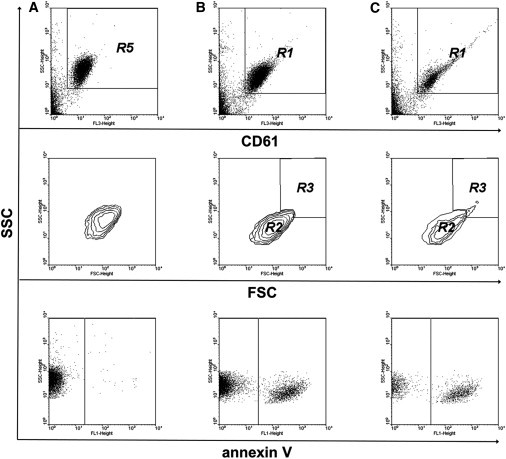
Coated platelets participate in aggregate formation being recruited into aggregates by noncoated platelets. (A–C) Dot plots for binding of CD61 (upper), contour plots (middle), and dot plots for binding of annexin V (lower) for platelets (20,000/μl) either unstimulated (A) or stimulated with 50 nM thrombin and 50 ng/ml convulxin without (B) and with (C) shaking. Events showing a high level of CD61 binding are marked as region R5 for unactivated platelets (A) and region R1 for activated platelets (B and C). R2 is the region of activated individual platelets and R3 the region of aggregates. Shown is a typical experiment out of three performed with platelets from different donors.
Participation of coated platelets in aggregate formation is most likely due to fibrinogen retention on the coated-platelet surface. The unoccupied terminus of a coated-platelet fibrinogen molecule interacts with an activated GPIIbIIIa receptor on the adjacent noncoated platelet, and thus the aggregation occurs. The assumption that aggregate formation in this experimental model is dependent on GPIIbIIIa was tested by using a GPIIbIIIa antagonist Monafram. The presence of excess Monafram during platelet activation prevented the platelets from leaving the suspension, and almost no aggregates were observed (data not shown). These results support the hypothesis that noncoated platelets act like bridges between coated ones in the developing aggregates.
The difference in the involvement of the two platelet subpopulations into the aggregates may vary quantitatively with the amount of fibrinogen added. Given our suggestion that coated platelets are recruited into developing aggregates via the fibrinogen retained on their surface, the addition of external fibrinogen may cause competition between the coated platelets and the added fibrinogen molecules for fibrinogen binding sites on noncoated platelets. A fibrinogen concentration of 1 mg/ml was chosen as comparable to its plasma concentration and was added in the experiments throughout this study to compare correctly the results obtained from different assays.
Confocal microscopy reveals coated platelets involved into the aggregates
To confirm the flow cytometric results, showing that participation of coated platelets in the aggregates does take place, the annexin-V-labeled aggregates formed upon conditions used in our cytometric studies were imaged by confocal microscopy. Fig. 6 demonstrates the typical look of aggregates thus formed. Round-shaped PS-positive platelets are held together by mushy structures of PS-negative platelets, in agreement with the concept that coated platelets can aggregate with noncoated ones but not with each other.
Figure 6.
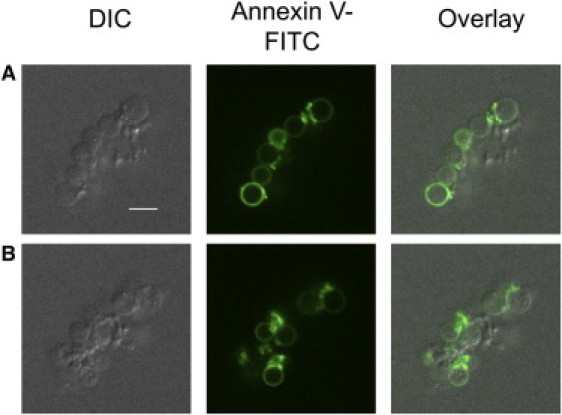
Coated platelets are incorporated into the aggregates formed by shaking. Representative confocal images of DIC, FITC (green) fluorescence of annexin V, and an overlay of the two for an aggregate of platelets stimulated with 100 nM thrombin plus 5 μg/ml CRP are shown. All panels represent one field of view. Two optical cross sections of an aggregate separated by 4.6 μm (A and B) are shown. Note that round-shaped annexin-V-positive platelets are held together by annexin-V-negative platelets with altered structure. Scale bar, 5 μm.
Discussion
This study aimed to investigate the proaggregatory abilities of two platelet subpopulations produced upon stimulation of platelets with thrombin or thrombin plus glycoprotein VI agonist. The principal finding is that highly procoagulant fibrinogen-coated platelets cannot bind each other but can be recruited into aggregates by noncoated platelets.
Four lines of evidence support this conclusion: 1), partial inhibition of platelet aggregation by coated platelets in the conventional aggregometry assay; 2), impaired ability of coated platelets to bind external fibrinogen; 3), demonstration by flow cytometry that coated platelets can be recruited into aggregates; 4), direct confocal microscopic visualization of coated platelets in the aggregates formed by shaking.
Up to 90% of coated-platelet content in suspension was achieved upon potent platelet stimulation with thrombin or thrombin plus convulxin by varying agonist concentrations. A large percentage (60% or more) of coated platelets inhibited aggregation in the sample, whereas the presence of 50% of coated platelets did not change the extent of aggregation, suggesting that coated platelets somehow participate in aggregate formation, albeit to a lesser degree. Theoretically, the differences in proaggregatory ability of suspensions with platelets from two distinct subpopulations may result not only from the different percentages of coated platelets in them, but also from some abnormalities, which platelets might have due to the stimulation of different potency. However, this possibility seems to be negligible, because flow cytometry revealed neither significant differences in platelets activated by a range of agonist concentrations nor signs of their desensitization.
The suggestion that coated platelets participate in aggregate formation was strongly confirmed by results obtained from the mathematical model of aggregation describing interactions between platelets from two subpopulations in three possible cases, in which 1), coated and noncoated platelets have equal capacity to bind each other; 2), coated platelets can bind noncoated ones but cannot form aggregates by themselves; and 3), coated platelets do not have the capacity to bind any platelets. When all platelets had equal capacity to bind each other, there was no correlation between the percentage of coated platelets and the parameters of aggregation. When coated platelets could bind noncoated ones but failed to bind each other, the dependencies of aggregation parameters on the percentage of coated platelets strongly reminded us of dependencies obtained in our aggregometry assay. Finally, when coated platelets failed to bind any platelets, the speed and extent of aggregation were drastically inhibited with the increasing number of coated platelets. These results point to the fact that the data obtained in experiments using aggregometry were caused by the participation of coated platelets in aggregate formation together with noncoated ones. According to this model, coated platelets cannot bind each other but are recruited into developing aggregates by noncoated platelets. A serious assumption was made that the probability of a collision between two particles does not depend on their size. However, the relevancy of using this simplified model arises from the fact that formation of large aggregates containing >10–20 platelets is considered to be quite slow. This means that by the time large aggregates, whose size can in no way be equated to the size of an individual platelet, were formed, measurements of light transmission had already been completed.
Second, only noncoated platelets had the capacity to bind external fibrinogen from a suspension, which molecules are known to serve as bridges between two adjacent platelets or between a platelet and the thrombogenic surface in aggregation or adhesion (21,22), while coated platelets failed to bind it.
Furthermore, flow cytometry revealed that coated platelets readily leave the suspension to form aggregates together with noncoated ones. The main drawback of this approach is similar to that of the aggregometry assay, consisting in the theoretically possible defects, which platelets might have due to the intensive shaking. These defects themselves may be the cause of coated platelets leaving the suspension, and not necessarily to form aggregates only. Finally, coated platelets in aggregates were detected by confocal microscopy, indicating their participation in aggregate formation provoked by shaking.
All these experiments were performed after 15 min of incubation with the agonists, when both platelet subpopulations, coated and noncoated platelets, had already formed. It makes our studies suitable for processes in hemostasis at low shear rates, where the aggregation mechanism primarily requires platelet activation (23–25). However, additional in vivo experiments and studies in flow chambers at different shear velocities will be needed to finalize the mechanism of coated-platelet participation in aggregate formation. Another problem, not addressed in this study, is how the increased procoagulant function of coated-platelet aggregates may affect thrombus dynamics and whether such aggregates lead to increased thrombin generation followed by fibrin formation and stabilization or propagation of thrombi.
Although PS-expressing platelets formed upon potent stimulation of platelets with thrombin or thrombin plus convulxin are known to have decreased PAC-1 binding, there has been no previous direct evidence on whether they can or cannot successfully participate in aggregate formation. It has been shown that during thrombus formation under flow, annexin-V-positive platelets assemble around the clots composed of annexin-V-negative PAC-1-binding platelets (9), which is actually very much in line with the coated-platelet aggregation mechanism revealed in this study. However, since those experiments were performed in whole blood in the presence of coagulation, it was not feasible to reliably deduce the specific mechanisms from those observations in a complex system. We suggest that coated-platelet participation in aggregate formation is provided mostly by the fibrinogen known to be retained on the surface of these platelets with an exceptional affinity (5), though von Willebrand factor may also play a role. It is noteworthy that formaldehyde-fixed platelets bearing cross-linked fibrinogen and erythrocytes with covalently bound fibrinogen were reported to participate in aggregate formation in the same manner as do coated platelets in the experiments of our study. These platelet substitutes do not aggregate by themselves but are incorporated into aggregates of activated platelets (26,27).
The mechanism of aggregation of platelets from two distinct subpopulations based on the data of this study is presented in Fig. 7. Briefly, coated platelets cannot bind each other but do bind noncoated platelets. Basic observations regarding coated platelets that are reproduced in our scheme include the following: coated platelets retain on their surface large amounts of procoagulant proteins; fibrinogen is bound to coated platelets with exceptional affinity (28); and GPIIbIIIa receptors on coated platelets are inactive or occupied with some ligand (3) (not shown).
Figure 7.
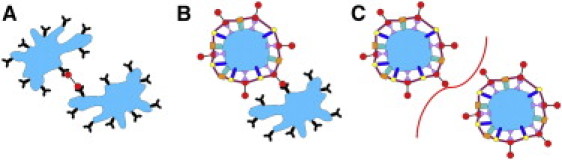
Coated platelets do not aggregate with each other but are recruited into aggregates by noncoated platelets: a possible mechanism. Capacities of platelets from two distinct subpopulations for binding each other are illustrated. Irregular shapes (A and B) depict normal activated platelets. Round shapes (B and C) depict coated platelets. Colored molecules on the coated-platelet surface represent receptors and their ligands, which form the coat of the coated platelet. (A) The interaction between two noncoated platelets occurs via fibrinogen (red) binding with GPIIbIIIa receptors (black). (B) Interaction between coated and noncoated platelets occurs when a GPIIbIIIa receptor on the noncoated platelet surface binds the free terminus of a fibrinogen molecule retained on the coated-platelet surface. (C) The interaction between two coated platelets cannot occur, since they have no active or free GPIIbIIIa receptors capable of binding the fibrinogen retained on the coated-platelet surface. The curve between platelets symbolizes the impossibility of their forming an aggregate by interaction with each other.
The most intriguing question in the platelet heterogeneity field is the physiological roles of the two platelet subpopulations. Reduced PAC-1 binding of coated platelets and the possibility of having a reduced proaggregatory ability led to suggestions that annexin-V-positive, or coated, platelets can participate in the downregulation of thrombus growth (14,15). On the other hand, coated platelets are known to sustain a procoagulant response by accelerating thrombin production by providing the PS necessary for membrane-dependent reactions catalyzed by tenase and prothrombinase complexes (29). Therefore, it was not clear how they could carry out their procoagulant function unless they have a way of incorporating into a thrombus. The data of this study suggest that they do possess such a way, via contacts with noncoated platelets, to get into a thrombus and there carry out their procoagulant function. Actually, one might speculate that this function itself could contribute to the downregulation of further platelet attachment by providing a fibrin surface with low adhesive properties (30). This seems to be a more probable mechanism than their direct anti-aggregatory properties for stopping thrombus growth, considering the significant time required to produce coated platelets, their ability to aggregate rather well with noncoated ones, and the very high percentage of coated platelets required to inhibit aggregation observed in our study. However, it is quite probable that the inability of strongly activated platelets to aggregate with each other is an additional physiological protective mechanism against thrombosis.
Acknowledgments
We thank Prof. R. Farndale and Prof. A. V. Mazurov for generously providing CRP and Monafram, respectively, and N. A. Podoplelova and V. V. Petrukhin for technical assistance with confocal microscopy.
The study was supported by the Russian Academy of Sciences Presidium Basic Research Programs Molecular and Cellular Biology, Basic Science for Medicine, Integrative Physiology, and Molecular Mechanisms of Physiologic Functions, and by the Russian Foundation for Basic Research grants 10-01-91055, 11-04-00303, 11-04-12080, 12-04-00652, and 12-04-00438.
References
- 1.Panteleev M.A., Ananyeva N.M., Saenko E.L. Mathematical models of blood coagulation and platelet adhesion: clinical applications. Curr. Pharm. Des. 2007;13:1457–1467. doi: 10.2174/138161207780765936. [DOI] [PubMed] [Google Scholar]
- 2.Dachary-Prigent J., Freyssinet J.M., Nurden A.T. Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: a flow cytometry study showing a role for free sulfhydryl groups. Blood. 1993;81:2554–2565. [PubMed] [Google Scholar]
- 3.Dale G.L., Friese P., Alberio L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002;415:175–179. doi: 10.1038/415175a. [DOI] [PubMed] [Google Scholar]
- 4.Kotova Y.N., Ataullakhanov F.I., Panteleev M.A. Formation of coated platelets is regulated by the dense granule secretion of adenosine 5′diphosphate acting via the P2Y12 receptor. J. Thromb. Haemost. 2008;6:1603–1605. doi: 10.1111/j.1538-7836.2008.03052.x. [DOI] [PubMed] [Google Scholar]
- 5.Dale G.L. Coated-platelets: an emerging component of the procoagulant response. J. Thromb. Haemost. 2005;3:2185–2192. doi: 10.1111/j.1538-7836.2005.01274.x. [DOI] [PubMed] [Google Scholar]
- 6.Alberio L.J., Clemetson K.J. All platelets are not equal: COAT platelets. Curr. Hematol. Rep. 2004;3:338–343. [PubMed] [Google Scholar]
- 7.Jackson S.P., Schoenwaelder S.M. Procoagulant platelets: are they necrotic? Blood. 2010;116:2011–2018. doi: 10.1182/blood-2010-01-261669. [DOI] [PubMed] [Google Scholar]
- 8.Munnix I.C., Cosemans J.M., Heemskerk J.W. Platelet response heterogeneity in thrombus formation. Thromb. Haemost. 2009;102:1149–1156. doi: 10.1160/TH09-05-0289. [DOI] [PubMed] [Google Scholar]
- 9.Munnix I.C., Kuijpers M.J., Heemskerk J.W. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 2007;27:2484–2490. doi: 10.1161/ATVBAHA.107.151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman M., Monroe D.M. Coagulation 2006: a modern view of hemostasis. Hematol. Oncol. Clin. North Am. 2007;21:1–11. doi: 10.1016/j.hoc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 11.Kempton C.L., Hoffman M., Monroe D.M. Platelet heterogeneity: variation in coagulation complexes on platelet subpopulations. Arterioscler. Thromb. Vasc. Biol. 2005;25:861–866. doi: 10.1161/01.ATV.0000155987.26583.9b. [DOI] [PubMed] [Google Scholar]
- 12.Shattil S.J., Cunningham M., Hoxie J.A. Detection of activated platelets in whole blood using activation-dependent monoclonal antibodies and flow cytometry. Blood. 1987;70:307–315. [PubMed] [Google Scholar]
- 13.Coller B.S., Shattil S.J. The GPIIb/IIIa (integrin αIIbβ3) odyssey: a technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112:3011–3025. doi: 10.1182/blood-2008-06-077891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulkarni S., Jackson S.P. Platelet factor XIII and calpain negatively regulate integrin αIIbβ3 adhesive function and thrombus growth. J. Biol. Chem. 2004;279:30697–30706. doi: 10.1074/jbc.M403559200. [DOI] [PubMed] [Google Scholar]
- 15.Jobe S.M., Wilson K.M., Di Paola J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood. 2008;111:1257–1265. doi: 10.1182/blood-2007-05-092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byzova T.V., Vlasik T.N., Mazurov A.V. [Inhibition of platelet aggregation with monoclonal antibodies to the glycoprotein IIb-IIIa complex] Biull. Eksp. Biol. Med. 1994;118:402–405. [PubMed] [Google Scholar]
- 17.Mazurov A.V., Pevzner D.V., Ruda M.Y. Safety, inhibition of platelet aggregation and pharmacokinetics of Fab′2 fragments of the anti-glycoprotein IIb-IIIa monoclonal antibody FRaMon in high-risk coronary angioplasty. Platelets. 2002;13:465–477. doi: 10.1080/0953710021000057839. [DOI] [PubMed] [Google Scholar]
- 18.Panteleev M.A., Ananyeva N.M., Saenko E.L. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J. Thromb. Haemost. 2005;3:2545–2553. doi: 10.1111/j.1538-7836.2005.01616.x. [DOI] [PubMed] [Google Scholar]
- 19.Reference deleted in proof.
- 20.Cronberg S. A mathematical model for optical platelet aggregation test. Acta Med. Scand. Suppl. 1971;525:49–53. doi: 10.1111/j.0954-6820.1972.tb05791.x. [DOI] [PubMed] [Google Scholar]
- 21.Kulkarni S., Dopheide S.M., Jackson S.P. A revised model of platelet aggregation. J. Clin. Invest. 2000;105:783–791. doi: 10.1172/JCI7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaidi T.N., McIntire L.V., Thiagarajan P. Adhesion of platelets to surface-bound fibrinogen under flow. Blood. 1996;88:2967–2972. [PubMed] [Google Scholar]
- 23.Jackson S.P. The growing complexity of platelet aggregation. Blood. 2007;109:5087–5095. doi: 10.1182/blood-2006-12-027698. [DOI] [PubMed] [Google Scholar]
- 24.Jackson S.P., Nesbitt W.S., Westein E. Dynamics of platelet thrombus formation. J. Thromb. Haemost. 2009;7(Suppl 1):17–20. doi: 10.1111/j.1538-7836.2009.03401.x. [DOI] [PubMed] [Google Scholar]
- 25.Maxwell M.J., Westein E., Jackson S.P. Identification of a 2-stage platelet aggregation process mediating shear-dependent thrombus formation. Blood. 2007;109:566–576. doi: 10.1182/blood-2006-07-028282. [DOI] [PubMed] [Google Scholar]
- 26.Agam G., Livne A. Passive participation of fixed platelets in aggregation facilitated by covalently bound fibrinogen. Blood. 1983;61:186–191. [PubMed] [Google Scholar]
- 27.Agam G., Livne A.A. Erythrocytes with covalently bound fibrinogen as a cellular replacement for the treatment of thrombocytopenia. Eur. J. Clin. Invest. 1992;22:105–112. doi: 10.1111/j.1365-2362.1992.tb01943.x. [DOI] [PubMed] [Google Scholar]
- 28.Szasz R., Dale G.L. Thrombospondin and fibrinogen bind serotonin-derivatized proteins on COAT-platelets. Blood. 2002;100:2827–2831. doi: 10.1182/blood-2002-02-0354. [DOI] [PubMed] [Google Scholar]
- 29.Zwaal R.F., Comfurius P., Bevers E.M. Lipid-protein interactions in blood coagulation. Biochim. Biophys. Acta. 1998;1376:433–453. doi: 10.1016/s0304-4157(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 30.Kamocka M.M., Mu J., Rosen E.D. Two-photon intravital imaging of thrombus development. J. Biomed. Opt. 2010;15:016020. doi: 10.1117/1.3322676. [DOI] [PMC free article] [PubMed] [Google Scholar]