

Abstract
Purpose
To investigate the effects of genetic factors on idiopathic scoliosis (IS) and genetic modes through genetic epidemiological survey on IS in Chongqing City, China, and to determine whether SH3GL1, GADD45B, and FGF22 in the chromosome 19p13.3 are the pathogenic genes of IS through genetic sequence analysis.
Methods
214 nuclear families were investigated to analyse the age incidence, familial aggregation, and heritability. SH3GL1, GADD45B, and FGF22 were chosen as candidate genes for mutation screening in 56 IS patients of 214 families. The sequence alignment analysis was performed to determine mutations and predict the protein structure.
Results
The average age of onset of 10.8 years suggests that IS is a early onset disease. Incidences of IS in first-, second-, third-degree relatives and the overall incidence in families (5.68%) were also significantly higher than that of the general population (1.04%). The U test indicated a significant difference, suggesting that IS has a familial aggregation. The heritability of first-degree relatives (77.68 ±10.39%), second-degree relatives (69.89 ±3.14%), and third-degree relatives (62.14 ±11.92%) illustrated that genetic factors play an important role in IS pathogenesis. The incidence of first-degree relatives (10.01%), second-degree relatives (2.55%) and third-degree relatives (1.76%) illustrated that IS is not in simple accord with monogenic Mendel’s law but manifests as traits of multifactorial hereditary diseases. Sequence alignment of exons of SH3GL1, GADD45B, and FGF22 showed 17 base mutations, of which 16 mutations do not induce open reading frame (ORF) shift or amino acid changes whereas one mutation (C→T)occurred in SH3GL1 results in formation of the termination codon, which induces variation of protein reading frame. Prediction analysis of protein sequence showed that the SH3GL1 mutant encoded a truncated protein, thus affecting the protein structure.
Conclusion
IS is a multifactorial genetic disease and SH3GL1 may be one of the pathogenic genes for IS.
Introduction
Idiopathic scoliosis (IS) is a three-dimensional spinal deformity that manifests as coronal scoliosis accompanied by vertebral rotation [1]. The aetiology remains unclear. Previous studies focused on histological factors such as cartilage, skeleton, joint, nerve and muscle but remain inconclusive [2–5]. In recent years, several studies have indicated that genetic factors play an important role in IS pathogenesis. These studies also show a high genetic heterogeneity of either autosomal dominant inheritance with a major gene or multifactorial modes of inheritance. Autosomal dominant [3–5], X-linked dominant [6] and multifactorial inheritance [1, 7, 8] are commonly reported. In this study, we investigated 214 IS families recuited in Southwest Hospital (Chongqing, China) by using the genetic epidemiological method and analysed effects of genetic factors and the mode of inheritance through patients’ age, IS incidence, familial aggregation and heritability, as well as traits of hereditary diseases.
IS-related susceptible genes currently being researched include melatonin receptor 1A [14], melatonin receptor 1B [15], Matrilin-1 (MATN1) [16], γ-1-syntrophin (SNTG1) [17], COL1A1 [18], COL1A2, COL2A1, elastic fibre [19] and aggrecan [20] genes. Studies showed that these genes are related with IS but cannot explain all IS patients. Chromosomal regions involved in IS susceptibility include 6p, 10q, 18q [21], 6p, 6q, 17p11.2 [5], 8q [22], 9q, 16q [23], 19p13 [3], 19p13.3 [9], 15q25-26 [20], 9q31.2, 9q34.2, 17q25.3-qtel [24], 12p [25] and Xq23-26 [26]. There are considerable numbers of genes in these foci. However, specific IS-susceptible genes still need to be identified by deeper investigation. Two scholars reported pathogenic foci on chromosome 19p13 [3, 9]. Furthermore, Chan et al. [9] defined the critical region of IS in the vicinity of D19S216, flanked by D19S894 and D19S1034. By April 2009, 213 genes had been identified in 19p13.3 in the Human Genome Organisation (HUGO) data base [32]. We screened SH3GL1 [28], GADD45B [29] and FGF22 [30] in the vicinity of D19S216 on chromosome 19p13.3 [9] as IS candidate genes and conducted a study using the case–sibling and case–relative control designs on the basis of families. Primer pairs were designed according to SH3GL1, GADD45B, and FGF22 gene exons for polymerase chain reaction (PCR) amplification, cloning, and sequencing. Sequence alignment among siblings and relatives was conducted, and the protein structure was predicted.
Materials and methods
Patient information
For this study, we selected 214 IS (68 male, 146 female) patients, with a male:female ratio of 1:2.14 treated in the Southwest Hospital from January 2003 to December 2009. All probands had typical clinical manifestations and were confirmed by radiographs and surgery. Patients’ perioperative imaging information was reviewed in case records according chart number. Fifty-six IS patients of 214 families (15 male and 41 female patients) with a mean age of 15 (range 8–22) years and a mean scoliotic Cobb angle of 67.5° (40–110°) were selected as the trial group, including 44 probands treated in the hospital, seven followed up in clinic, two of whom were proband fathers and three were proband mothers. They were categorised to Lenke type 1 (n = 26), 2 (n = 9), 3 (n = 11), 4 (n = 2), 5 (n = 6),and 6 (n = 2). All diagnoses were confirmed by physical and imaging examinations. Other spinal diseases, such as congenital scoliosis, hemivertebrae deformity, Marfan’s syndrome and central nervous system (CNS) diseases, were excluded. Ninety-three relatives (43 male and 50 female) with a mean age of 30 (range 8–52) years and normal phenotype served as the control group, including six proband siblings, 40 fathers and 47 mothers.
Epidemiological investigation
Proband families were examined by trained investigators using an independently designed IS genetic epidemiological questionnaire, and the family tree was mapped. Investigation methods included face-to-face enquiry in clinic and wards and/or telephone inquiry. Before entering this study, participants were informed about the objective and were provided with informed consent. Investigated individuals were IS probands and parents. Participants included first-degree relatives (probands, their parents, children and siblings), second-degree relatives (grandparents, uncles and aunts) and third-degree relatives (cousins). Patients’ general situation (including health) and scoliosis history were reviewed. Suspected diseased individuals and susceptible individuals were confirmed by inspection, Adam’s Forward Bend Test and radiographs.
IS diagnostic criteria
Diseased individuals were identified based on a Cobb angle over 10° in the standing anteroposterior radiographs, and susceptible individuals were identified by Cobb angle under 10°. Children less than 14 years old with a normal phenotype were regarded as nondiseased relatives [10].
Statistical analysis
Familial aggregation was analysed using the sample- and population-rate-comparing U test:
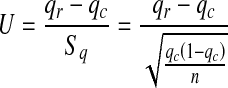 |
Standard error of population rate 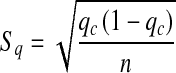
Incidence of IS in the first-, second-, and third-degree relatives and all the families: 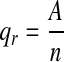 A number of IS patients in various-degree relatives and all families; n total number of persons investigated in various-degree relatives and all families; qc incidence of IS in the control population and 1.04% reported in literature [11].
A number of IS patients in various-degree relatives and all families; n total number of persons investigated in various-degree relatives and all families; qc incidence of IS in the control population and 1.04% reported in literature [11].
IS heritability was estimated by the Falconer regression method [12]. Heritability  Regression coefficient
Regression coefficient 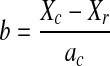 Variance
Variance 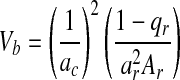 Standard error
Standard error 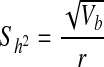 , 95% confidence interval
, 95% confidence interval  Weighted mean of heritability h2 of first-, second-, and third-degree relatives:
Weighted mean of heritability h2 of first-, second-, and third-degree relatives: 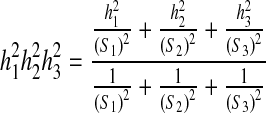 Standard error of
Standard error of 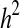 weighted mean
weighted mean 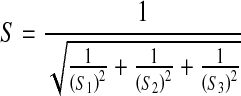
r relationship coefficient (1/2 for first-degree relatives, 1/4 for second-degree relatives, and 1/8 for third-degree relatives), A absolute number of relatives having IS, Xr average threshold of susceptibility of probands’ relatives, Xc average threshold of susceptibility of control population, a ratio of threshold probability density to incidence of general population, X and a obtained from the X and Falconer table according to prevalence, qc and r control population and proband relatives, respectively, qr and c incidence of probands’ relatives and control population, respectively.
Analytical method of exon sequence alignment of candidate genes SH3GL1, GADD45B and FGF22
Genome DNA extraction
We collected 10 ml of peripheral venous blood from 56 probands and 93 relative controls. Ethylenediaminetetraacetic acid (EDTA) was added to the blood sample for anticoagulation. Genomic DNA was extracted using the Wizard whole-blood genome DNA extraction kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Resulting products were assayed with agarose gel electrophoresis and imaged with Bio-RAD Gel Doc 2000 gel imaging system (Bio-Rad Laboratories, Hercules, CA, USA). Purity was determined with the WD-94038B ultraviolet (UV) analyser.
PCR amplification and sequencing
Six pairs of primers were designed using sequences of ten exons of SH3GL1 as the template. Two pairs of primers were designed and synthesised using sequences of four exons of GADD45B as the template. One pair of primers was designed and synthesised using sequences of three exons of FGF22 as the template. Each primer pair was independently amplified with PTC-100 PCR [America MJ Research Company (Bio-Rad Laboratories) using genomic DNA as the template]. The reaction system consisted of 25 μl solution composed of 10.0 μl ultrapure water, 13.0 μl Master Mix, 1.5 μl DNA template, and 0.5 μl forward plus reverse primers. Conditions and primer pairs for PCR are available upon request. PCR products were assayed by electrophoresis [1% agarose gel, 1× Tris-acetate EDTA (TAE) electrophoretic liquid, 100 V, 30 minutes]. PCR products were extracted and purified using column-centrifugation DNA gel extraction kit and purification kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instruction. Purified PCR products were sequenced with a 3130 genetic analyser (Applied Biosystems, Carlsbad, CA, USA).
Statistical analysis
Sequence alignment of siblings and relatives was analysed using Chromas software (Technelysium Pty Ltd, Tewantin, QLD, Australia) and DNA sequence analysis software Vector NTI Advance 10.3 (Invitrogen, Carlsbad, CA, USA).
Results
Epidemiological investigation results
Onset age
We analysed 214 families with a total of 2,732 members, consisting of 214 probands and 2,518 relatives (589 first-degree, 1,532 second-degree, 397 third-degree); 109 relatives had IS. Investigated IS patients (including probands) had an onset age of three to 20 years and 78.91% between ten and 14 years, with a mean age of 11.8 years. The ratio of males to females was 1:2.59, suggesting that females are more susceptible to this disease than are males. IS usually occurs in the early years, suggesting that it is very likely to be a hereditary disease (Table 1).
Table 1.
Number of idiopathic scoliosis (IS) patients in different age segments
| Onset age (years) | Male | Female | Total number | Proportion in IS patients |
|---|---|---|---|---|
| 0–3 | 3 | 0 | 3 | 0.96% |
| 4–9 | 11 | 13 | 24 | 7.67% |
| 10–14 | 47 | 200 | 247 | 78.91% |
| >15 | 19 | 20 | 39 | 12.46% |
| Together | 80 | 233 | 313 |
Comparison of IS incidence among various-degree relatives and analysis of familial aggregation
In this study, 214 probands had a total of 2,518 first-, second-, and third-degree relatives, and 109 relatives developed IS. Overall IS incidence in the investigated families was (214 + 109)/(2518 + 214) × 100% = 11.82% and 1.04% in the control population [11]. IS in families was assayed with the U test, and results showed U = 2.58 and corresponding P < 0.01. A significant difference in incidence was noted between the total investigated families and the control population, promoting the familial aggregation of IS. Incidence was 10.01% for first-degree, 2.55% for second-degree and 1.76% for third-degree relatives, and overall incidence in relatives was 109/2,518 × 100% = 4.33%. The incidence in first- and second-degree relatives and all relatives was significantly higher than that of the control population (1.04%). U test results showed that incidence in first- and second-degree relatives and overall were significantly higher than that of the general population, illustrating the familial aggregation of IS. Despite a higher incidence in third-degree relatives than in the general population, the difference was not statistically significant, suggesting that the incidence in third-degree relatives was consistent with the control population (Table 2).
Table 2.
Idiopathic scoliosis (IS) incidence in various-degree relatives
| No. | Onset number A | Incidence (q%) | U value | P value | |
|---|---|---|---|---|---|
| First-degree relatives | 589 | 59 | 10.01% | 21.46 | <.01 |
| Second-degree relatives | 1,532 | 39 | 2.55% | 5.81 | <.01 |
| Third-degree relatives | 397 | 7 | 1.76% | 1.38 | >.05 |
| Total number of relatives | 2,518 | 109 | 4.25% | 15.89 | <.01 |
| Total investigated families | 2,732 | 321 (including probands) | 11.74% | 56.32 | <.01 |
| Control population | 1.04% |
A absolute number of relatives with IS
Estimation of heritability (h2 ± sh2)
IS heritability was estimated by the Falconer threshold method [10]. Heritability was 77.68 ± 10.39% for first-degree relatives (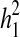 ), 69.89 ± 3.14% for second-degree relatives (
), 69.89 ± 3.14% for second-degree relatives (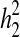 ), and 62.14 ±11.92% for third-degree relatives (
), and 62.14 ±11.92% for third-degree relatives ( ). The weighted mean
). The weighted mean 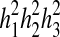 of first-, second- and third-degree relatives (h2) was 49.17 ± 2.92% (Table 3). IS incidence in relatives and heritability estimation (h2 ± sh2) showed that IS is a multifactorial hereditary disease.
of first-, second- and third-degree relatives (h2) was 49.17 ± 2.92% (Table 3). IS incidence in relatives and heritability estimation (h2 ± sh2) showed that IS is a multifactorial hereditary disease.
Table 3.
Idiopathic scoliosis heritability in various-degree relatives
| Relative | No. | Patient number A | Incidence (q%) | 1-q | Average threshold of susceptibility X | Ratio of threshold a | b | h2 ± 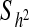 (%) (%) |
|---|---|---|---|---|---|---|---|---|
| First-degree relatives | 589 | 59 | 10.01% | 0.8999 | 1.282 | 1.755 | 0.3884 | 77.68 ±10.39 |
| Second-degree relatives | 1532 | 43 | 2.81% | 0.9732 | 1.911 | 2.295 | 0.1747 | 69.89 ±3.14 |
| Third-degree relatives | 397 | 7 | 1.76% | 0.9824 | 2.106 | 2.467 | 0.0777 | 62.14 ±11.92 |
| Control population | 1.04% | 0.9896 | 2.312 | 2.652 |
A absolute number of IS relatives,X average threshold of susceptibility of proband relatives and control population
aRatio of threshold probability density to population incidence
X and a, obtained through normal-distribution X and a table (Falconer table) according to q,
Algorithm (refer to “Method”)
Sequence alignment
The amplified product bands corresponded to the objective DNA fragment band in gel electrophoresis (Fig. 1). Cloning sequence alignment analysis of ten exons of SH3GL1 of 56 IS patients showed gene mutation sites in all patients and a total of 12 mutant alleles in the second, fourth, fifth, sixth, eighth and tenth exons, of which ten mutations were located in the coding region and two (both in the tenth exon) in the noncoding region (Table 4). Cloning sequence alignment analysis of the four exons of GADD45B of 56 IS patients indicated gene mutation sites in all 56 patients and a total of three mutant alleles in the first, third and fourth exons (Table 4). Cloning sequence alignment analysis of the three exons of FGF22 of 56 IS patients showed gene mutation sites in all 56 patients and a total of two mutant alleles in the first and third exons (Table 4).
Fig. 1.

Electrophoretogram of polymerase chain reaction (PCR) products of primer E3/E4, E5/E6, E7/E8, E9/E10, E1/E2, and E11-1/E11-2 of blood samples
Table 4.
Results of cloning sequence alignment of ten exons of SH3GL1, GADD45B and FGF22 exon
| Exon no. | Primer pair | Starting and stopping points of exon mRNA (1–2,460) | Position of mutant sites in mRNA | IS patient genotype | Sibling and relative genotypes with normal genotype | NCBI allele | Significant mutant site (ORF or amino acid change) |
|---|---|---|---|---|---|---|---|
| 1 | E1/E2 | 1–151 | None | None | None | None | None |
| 2 | E3/E4 | 152–220 | 164 | AT | AA | A | None |
| 184 | CG | CG | G | None | |||
| 186 | AT | AA | C | None | |||
| 3 | E3/E4 | 221–293 | None | None | None | None | None |
| 4 | E5/E6 | 294–437 | 422 | GG | AA | G | None |
| 5 | E7/E8 | 438–571 | 439 | GC | TT | T | None |
| 492 | TT | TA | A | None | |||
| 515 | TT | CC | C | C→T, ORF | |||
| 524 | AT | AA | A | None | |||
| 6 | E7/E8 | 572–730 | 579 | TA | TA | T | None |
| 7 | E7/E8 | 731–834 | None | None | None | None | None |
| 8 | E9/E10 | 835–959 | 918 | CG | CC | C | None |
| 9 | E9/E10 | 960–1,016 | None | None | None | None | None |
| 10 | E11-1/E11-2 | 1,017–2,460 | 2,313 | GG | GA | G | None |
| 2,384 | AG | AA | A | None | |||
| GADD45B exon | |||||||
| 1st/2nd | GADD45B-12 | 1–380 | 138 | CG | GG | G | None |
| 3rd | FGF22–23 | 319–513 | 444 | CG | GG | G | None |
mRNA messenger RNA, NCBI National Center for Biotechnology Information, ORF open reading frame
Prediction analysis of protein sequence
The 515th alleles of SH3GL1 was CC in 93 relatives, TT in five of 56 probands (8.93%) (proband and the mother with IS, proband and the father with IS and another male proband) (Fig. 2) and CC in the remaining 51 probands (91.07%). If the proband’s gene base is T, the stop codon (TAG) is formed and the protein reading frame also changes and encodes truncated proteins
Fig. 2.

Mutant site of the 515th allele of SH3GL1 439th base
439th base  492nd base Significant mutant site in the
492nd base Significant mutant site in the  515th base. 78-10f was amplification results of E7/E8 primer of SH3GL1 including 5th/6th/7th exons; 78- was the No. of primer E7/E8 and 10f was the forward sequencing results of No. 10 sample
515th base. 78-10f was amplification results of E7/E8 primer of SH3GL1 including 5th/6th/7th exons; 78- was the No. of primer E7/E8 and 10f was the forward sequencing results of No. 10 sample
Prediction analysis of open reading frame of SH3GL1
The coding regions of SH3GL1 of normal controls produced an open reading frame (ORF) of 1,107 bp and encoded 369 amino acids (Fig. 3). Coding regions of SH3GL1 of IS patients produced two ORF regions (Fig. 4). The first was 411 bp and coded 137 amino acids. The second was 507 bp and coded 169 amino acids. The intermediate sequence was 189 bp and coded 62 amino acids. Consequently, coding-region mRNA sequence of SH3GL1 in IS patients generated two proteins, which were encoded by ORF, and the two proteins encoded by either the first or second ORF were truncated proteins, which induced loss of some functions. SH3GL1 base sequence analysis showed that the 515th allele of SH3GL1 was CC in normal families but CT in IS patients. If the 515th base was T, the stop codon (TAG) was formed and the protein reading frame also changed (possibly 107–518 bases or 707–1,213 bases). Prediction analysis of protein sequence showed the truncated protein, which affected the primary structure of the protein.
Fig. 3.

Prediction chart of open reading frame (ORF) of SH3GL1 of normal relative and sibling controls ■ Overall length of messenger RNA (mRNA)  ORF that could translate protein
ORF that could translate protein
Fig. 4.

A prediction chart of open reading frame (ORF) of SH3GL1 of idiopathic scoliosis (IS) patients ■ Overall length of messenger RNA (mRNA)  ORF that could translate protein
ORF that could translate protein
For the coding sequence that could translate proteins, the starting–stopping points in mRNA were 107–1213. For the two coding sequences that could translate proteins, the starting–stopping points in mRNA were 107–517 and 707–1213, respectively.
Discussion
Hereditary disease is induced by abnormal genetic material (genes and chromosomes) and is characterised by congenital, familial, rare and lifetime traits. Contribution of genetic factors to diseases is assessed by incidence, familial aggregation and heritability in genetic studies [10]. In this study, there were families in which only probands, probands and first-degree relatives, probands and second-degree relatives or probands and third-degree relatives with IS, but there were no big families in which probands and first-, second-, third-degree relatives had IS. Analysis of familial incidence showed the frequency of various-degree relatives was significantly higher than in the control population and that the genetic relationship was correlated with the incidence: first-degree relatives greater than second-degree relatives, and second-degree relatives greater than third-degree relatives (i.e. a closer relationship represented a higher incidence). These showed that IS had a significant familial aggregation. IS incidence in second-degree relatives (2.25%) was significantly lower than that in the first-degree relatives (10.01%) and did not reduce by 50%, although the incidence in third-degree relatives (1.76%) was significantly lower than that in first-degree relatives (10.01%) and did not reduce by 25%, suggesting that despite a significant familial aggregation, IS did not abide by Mendel’s law. Therefore, it is speculated that IS is not a monogenic hereditary disease. The incidence of IS families in this study was consistent with results reported by Harrington [10], who investigated 207 IS families and found that the incidence was 11% for first-degree, 2.4% for second-degree and 1.4% for third-degree relatives.
We analysed IS onset age and found that 78.91% of patients developed the disease between ten and 14 years, with mean age of 10.84 years, indicating that IS was a disease occurring at an early age and had the features of hereditary diseases [33]. The overall incidence of IS in investigated families was 11.74%, although the incidence in the control population was 1.04%. A significant difference between overall and population incidence was detected by the U test, suggesting that IS had a typical familial aggregation. The incidence of IS in various-degree relatives was also significantly higher than that of the control population. U test analysis showed a significantly higher incidence in first- and second-degree relatives than in the control population, suggesting a familial aggregation. Despite a higher incidence in third-degree relatives than in the control population, the difference was not statistically significant, suggesting that the incidence in third-degree relatives was consistent with the control population. Effects of genetic factors on IS pathogenesis can be assessed with IS heritability. Heritability IS incidences over 60% indicates an important effect of genetic factors; heritability incidences under 60% means that environmental factors are of great influence. In our study, heritability IS occurrences in first-, second- and third-degree relatives was 77.68 ± 10.39%, 69.89 ± 3.14% and 62.14 ± 11.92%, respectively. Furthermore, all incidences over 60% suggest that genetic factors play an important role in IS pathogenesis.
Multifactorial hereditary diseases are caused synergistically by multiple pairs of minor genes and environmental factors and also have a low incidence. Their inheritance principle remains unknown but has common features [13], such as a higher incidence in various-degree relatives than in the general population; familial aggregation; the incidence in proband siblings is much less than 50% or 100%; the incidence in proband siblings younger than probands is higher than other relatives; relatives with a closer relationship with probands has a higher incidence; the proportion of consanguineous marriage of proband’s parents is slightly higher than the general population; and the pairwise concordance rate of monozygotic twins is higher than that of dizygotic twins. A hereditary disease meeting the above features is defined as the multifactorial hereditary disease. This study, performed by assaying IS incidence of various-degree relatives, familial aggregation, and heritability, shows that IS is a multifactorial hereditary disease.
In genetics, susceptible IS genes are commonly investigated using two strategies: candidate-gene and positional-gene strategy. a considerable number of studies have reported on candidate genes, such as melatonin receptor 1A gene [14], melatonin receptor 1B gene [15], MATN1 gene [16], SNTG1 gene [17], COL1A1 [18], COL1A2, COL2A1, elastic fibre [19] and aggrecanase [20]. It was shown that these genes are related to IS but cannot explain all IS patients. A positional cloning strategy study showed that chromosomal foci encoding genes involved in susceptibility to IS included 6p, 10q, 18q [21], 6p, 6q, 17p11.2 [5], 8q [22], 9q, 16q [23], 19p13 [3], 19p13.3 [9], 15q25-26 [20], 9q31.2,-9q34.2, 17q25.3-qtel [24], 12p [25] and Xq23-26 [26]. These experiments identified these IS-susceptible genes in the specific region of chromosome (19p13 [3]), but these regions contain billions of base pairs and thousands of known genes. Therefore, determining the specific IS-susceptible gene requires considerable work. So far, CHD7 is the exclusive IS-susceptible gene positioned by the positional cloning method. Pathogenic foci related to IS have been reported on chromosome 19p13 [3, 9]. Furthermore, Chan et al. [9] defined the IS critical region in the vicinity of D19S216, flanked by D19S894 and D19S1034. On the basis of Chan et al.’s study, we conducted further research on positional cloning, and a total of 213 known genes were identified in 19P13.3 by searching the HUGO [32]. We screened structure- and function-related SH3GL1 [28], GADD45B [29] and FGF22 [30] in the vicinity of D19S216, conducted a study with the case–siblings and case–relatives control designs on the basis of families; designed primer pairs according to exons of SH3GL1, GADD45B and FGF22 for PCR amplification, cloning and sequencing; analysed sequence alignment among siblings and relatives; and predicted the protein structure. According to the genetic theory, the homology of nucleotide sequence among individuals in families (similarity of base sequences) is positively correlated with relationship (i.e. monozygotic twins more than dizygotic twins, more than sibling pair, more than proband and parents, more than proband and first-degree relatives, more than proband and second-degree relatives, more than proband and third-degree relatives). The similarity of base sequences in monozygotic and dizygotic twins is the highest and is the best sample of base sequence alignment. Such samples are difficult to collect within a short timeframe, however. Therefore, case–sibling and relative–control design on the basis of families was chosen to reduce the heterogenicity of aligned sequences and to find mutations.
In this study, ten exons of SH3GL1 in IS patients were found to have 12 mutations, which were positioned in the second, fourth, fifth, sixth, eighth and tenth exons. Four exons of GADD45B were observed for three mutations in the first, third and fourth exons. Three exons of FGF22 were observed for two mutations in the first and third exons. Eleven mutations in SH3GL1, three in GADD45B and two in FGF22 did not induce ORF translocation or amino acid changes and therefore are nonsense mutations. The 515th alleles of SH3GL1 was CC in 93 relatives, TT in five of the 56 probands (8.93%) (proband and the mother with IS, proband and the father with IS, and another male proband) (Fig. 3) and CC in the remaining 51 probands (91.07%). If the proband’s gene base is T, the TAG is formed and the protein reading frame also changes and encodes truncated proteins, affecting protein structure. This protein may be the diseased protein of IS, demonstrating that SH3GL1 is possibly one of the main gene candidates. Because SH3GL1 [3] is strongly related with osteoclast mechanism, it is inferred that the original structure of IS is the sclerotic structure forming the vertebral column and related with osteoclast activity, which defines the direction of further studies. However, whether patients’ genes encode truncated proteins and, if so, which category of truncated proteins, await confirmation by further gene cloning studies. Our experiment also confirmed that although SH3GL1 may be one of the main candidate genes, it was not abnormal in all IS patients, as reported by Gao et al. [22]. CHD7 also exists in a small number of families [31]. Therefore, our study also indirectly proves that IS was a multifactorial hereditary disease, has genetic heterogenicity [27] and may include multiple main genes.
Contributor Information
Tao Yang, Phone: +86-23-65340297, FAX: +86-23-65340297, Email: y07yxy1998@126.com.
Jianzhong Xu, Email: xjzslw@126.com.
Yun Bai, Email: baiyungene@gmail.com.
References
- 1.Ocaka L, Zhao C, Reed JÁ, et al. Assignment of two loci for autosomal dominant adolescent idiopathic scoliosis to chromosomes 9q31.2-q34.2 and 17q25.3-qtel. J Med Genet. 2008;45(2):87–92. doi: 10.1136/jmg.2007.051896. [DOI] [PubMed] [Google Scholar]
- 2.Lonstein JE. Adolescent idiopathic scoliosis. Lancet. 1994;344(8934):1407–1412. doi: 10.1016/S0140-6736(94)90572-X. [DOI] [PubMed] [Google Scholar]
- 3.Alden KJ, Marosy B, Nzegwu N, et al. Idiopathic scoliosis: identification of candidate regions on chromosome 19p13. Spine. 2006;31(16):1815–1819. doi: 10.1097/01.brs.0000227264.23603.dc. [DOI] [PubMed] [Google Scholar]
- 4.Lonstein J. Idiopathic scoliosis. In: Bradford D, Winter R, Ogilvie J, editors. LonsteinJ. Philadelphia WB Saunders: Moe’s Textbook of scoliosis and other spinal deformities; 1995. pp. 219–256. [Google Scholar]
- 5.Salehi LB, Mangino M, Serio S, et al. Assignment of a locus for autosomal dominant idiopathic scoliosis (IS) to human chromosome 17p11. Hum Genet. 2002;111(4–5):401–404. doi: 10.1007/s00439-002-0785-4. [DOI] [PubMed] [Google Scholar]
- 6.Wajchenberg M, Puertas EB, Zatz M. Estudo da prevalência da escoliose idiopática do adolescente em pacientes brasileiros. Coluna. 2005;4(3):127–131. [Google Scholar]
- 7.Axenovich TI, Zaidman AM, Zorkoltseva IV, et al. Segregation analysis of idiopathic scoliosis: demonstration of a major gene effect. Am J Med Genetic. 1999;86(4):389–394. doi: 10.1002/(SICI)1096-8628(19991008)86:4<389::AID-AJMG15>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 8.Wise CA, Barnes R, Gillum J, et al. Localization of susceptibility to familial idiopathic scoliosis. Spine. 2000;25(18):2372–2380. doi: 10.1097/00007632-200009150-00017. [DOI] [PubMed] [Google Scholar]
- 9.Chan V, Fong GC, Luk KD, et al. A genetic locus for adolescent idiopathic scoliosis linked to chromosome 19p13.3. Am J Hum Genet. 2002;71(2):401–406. doi: 10.1086/341607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrington PR.(1977) The etiology of idiopathic scoliosis. Clin Orthop Relat Res. (l26):17–25. [PubMed]
- 11.Liu SL, Huang DS. (1996) Scoliosis in China. A general review. Clin Orthop Relat Res. (323):113–118. [PubMed]
- 12.Falconer DS. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann Hum Genetics. 1965;29(1):51–76. doi: 10.1111/j.1469-1809.1965.tb00500.x. [DOI] [Google Scholar]
- 13.Dipple KM, McCabe ER. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66(6):1729–1735. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morcuende JA, Minhas R, Dolan L, et al. Allelic variants of human melatonin 1A receptor in patients with familial adolescent idiopathic scoliosis. Spine. 2003;28(17):2025–2029. doi: 10.1097/01.BRS.0000083235.74593.49. [DOI] [PubMed] [Google Scholar]
- 15.Qiu XS, Tang NL, Yeung HY, et al. Melatonin receptor 1B (MTNR1B) gene polymorphism is associated with the occurrence of adolescent idiopathic scoliosis. Spine. 2007;32(16):1748–1753. doi: 10.1097/BRS.0b013e3180b9f0ff. [DOI] [PubMed] [Google Scholar]
- 16.Montanaro L, Parisini P, Greggi T, et al. Evidence of a linkage between matrilin-1 gene (MATN1) and idiopathic scoliosis. Scoliosis. 2006;1:21. doi: 10.1186/1748-7161-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bashiardes S, Veile R, Allen M, et al. SNTG1, the gene encoding gamma1-syntrophin: a candidate gene for idiopathic scoliosis. Hum Genet. 2004;115(1):81–89. doi: 10.1007/s00439-004-1121-y. [DOI] [PubMed] [Google Scholar]
- 18.Carr AJ, Ogilvie DJ, Wordsworth BP, et al. Segregation of structural collagen genes in adolescent idiopathic scoliosis. Clin Orthop Relat Res. 1992;274:305–310. [PubMed] [Google Scholar]
- 19.Miller NH, Mims B, Child A, et al. Genetic analysis of structural elastic fiber and collagen genes in familial adolescent idiopathic scoliosis. J Orthop Res. 1996;14(6):994–999. doi: 10.1002/jor.1100140621. [DOI] [PubMed] [Google Scholar]
- 20.Marosy B, Justice C, Nzegwu N, et al. Lack of association between the aggrecan gene and familial idiopathic scoliosis. Spine. 2006;31(13):1420–1425. doi: 10.1097/01.brs.0000219944.18223.52. [DOI] [PubMed] [Google Scholar]
- 21.Wise CA, Barnes R, Gillum J, Herring JA, et al. Localization of susceptibility to familial idiopathic scoliosis. Spine. 2000;25(18):2372–2380. doi: 10.1097/00007632-200009150-00017. [DOI] [PubMed] [Google Scholar]
- 22.Gao X, Gordon D, Zhang D, et al. CHD7 gene polymorphisms are associated with susceptibility to idiopathic scoliosis. Am J Hum Genet. 2007;80(5):957–965. doi: 10.1086/513571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller NH, Justice CM, Marosy B, et al. Identification of candidate regions for familial idiopathic scoliosis. Spine. 2005;30(10):1181–1187. doi: 10.1097/01.brs.0000162282.46160.0a. [DOI] [PubMed] [Google Scholar]
- 24.Raggio CL, Giampietro PF, Dobrin S, et al. A novel locus for adolescent idiopathic scoliosis on chromosome 12p. J Orthop Res. 2009;27(10):1366–1372. doi: 10.1002/jor.20885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Justice CM, Miller NH, Marosy B, et al. Familial idiopathic scoliosis: Evidence of an X-linked susceptibility locus. Spine. 2003;28(6):589–594. doi: 10.1097/01.BRS.0000049940.39801.E6. [DOI] [PubMed] [Google Scholar]
- 26.Miller NH. Genetics of familial idiopathic scoliosis. Clin Orthop Relat Res. 2007;462:6–10. doi: 10.1097/BLO.0b013e318126c062. [DOI] [PubMed] [Google Scholar]
- 27.Bonaïti C, Feingold J, Briard ML, et al. Genetics of idiopathic scoliosis. Helv Paediatr Acta. 1976;31(3):229–240. [PubMed] [Google Scholar]
- 28.Chen Y, Deng L, Maeno-Hikichi Y, et al. Formation of an endophilin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell. 2003;115(1):37–48. doi: 10.1016/S0092-8674(03)00726-8. [DOI] [PubMed] [Google Scholar]
- 29.Ma DK, Jang MH, Guo JU, et al. (2009) Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. (5917)323:1074–1077. [DOI] [PMC free article] [PubMed]
- 30.Umemori H, Linhoff MW, Ornitz DM, Sanes JR. FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell. 2004;118(2):257–270. doi: 10.1016/j.cell.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 31.Qiu Y. Current trends in genetic etiology research of adolescent idiopathic scoliosis. Zhonghua Wai Ke Za Zhi. 2010;48(6):401–404. [PubMed] [Google Scholar]
- 32.http://www.genenames.org/
- 33.Riseborough EJ, Wynne-Davies R. A genetic survey of idiopathic scoliosis in Boston, Massachusetts. J Bone Joint Surg Am. 1973;55(5):974–982. [PubMed] [Google Scholar]