

Abstract
PTEN gene is considered one of the most mutated tumor suppressor genes in human cancer, and it's likely to become the first one in the near future. Since 1997, its involvement in tumor suppression has smoothly increased, up to the current importance. Germline mutations of PTEN cause the PTEN hamartoma tumor syndrome (PHTS), which include the past-called Cowden, Bannayan-Riley-Ruvalcaba, Proteus, Proteus-like, and Lhermitte-Duclos syndromes. Somatic mutations of PTEN have been observed in glioblastoma, prostate cancer, and brest cancer cell lines, quoting only the first tissues where the involvement has been proven. The negative regulation of cell interactions with the extracellular matrix could be the way PTEN phosphatase acts as a tumor suppressor. PTEN gene plays an essential role in human development. A recent model sees PTEN function as a stepwise gradation, which can be impaired not only by heterozygous mutations and homozygous losses, but also by other molecular mechanisms, such as transcriptional regression, epigenetic silencing, regulation by microRNAs, posttranslational modification, and aberrant localization. The involvement of PTEN function in melanoma and multistage skin carcinogenesis, with its implication in cancer treatment, and the role of front office in diagnosing PHTS are the main reasons why the dermatologist should know about PTEN.
1. PTEN Gene: What It Is and How It Works
PTEN stands for phosphatase and tensin homolog deleted in chromosome 10, and it is considered one of the most mutated tumor suppressor genes in human cancer. In the near future, it is likely to become the first one overcoming the current leader, p53 gene [1]. The involvement of PTEN's alteration in tumorigenesis has been first suspected and subsequently proven in 1997 [2], when high frequency of loss of heterozygosity (LOH) at 10q23 chromosome band was observed in several human tumors. Furthermore, the suppression of tumorigenesis in glioblastoma murine cells by the wildtype chromosome 10 led to envision a tumor suppressor gene mapping in 10q23. Such gene was eventually isolated by the above-mentioned authors and called PTEN. They detected homozygous deletions, frame shift, or nonsense mutations in PTEN in 63% (5/8) of glioblastoma cell lines, 100% (4/4) of prostate cancer cell lines, and 10% (2/20) of breast cancer cell lines. Steck et al. [3] independently isolated the same gene and called it mutated in multiple advanced cancers-1 (MMAC-1). Indeed, a common feature of PTEN somatic mutations, already presented in 10q LOH, is the association with advanced-stage tumors (mainly glial and prostate cancers), whereas this is not true for endometrial cancer, being affected equally at all the stages. This has led to the suggestion that the activation of PTEN is at an early stage in endometrial carcinogenesis, but later on in glial and prostatic carcinogenesis. This mechanism is the cornerstone of the classical two-hit Knudson' hypothesis [4]: a single mutation in one homolog of a tumor-suppressor gene is not sufficient to initiate tumor growth; however, deletion or disabling of the allele on the homologous chromosome results in unregulated cell growth. Both sporadic and hereditary tumors can be explained by such mechanism. In sporadic tumors, both alleles are normal at conception; subsequently, a postzygotic mutation (first hit) in one cell creates the heterozygosity (one mutant and one normal allele); thereafter, a deletion or a new mutation (second hit) in the other allele of that cell provokes the LOH, starting the uncontrolled tumor growth. In hereditary tumors, the heterozygosity for mutant allele (first hit) is present at conception, and is sufficient that a postzygotic mutation (second hit) during life creates the LOH for the onset of uncontrolled tumor growth.
Liaw et al. [5] found germline mutations of PTEN gene in families with Cowden syndrome [6] (CS), showing the function of tumor suppressor gene also in the germline. Furthermore, germline PTEN mutations lead to increased breast cancer incidence, but do not frequently cause familial breast cancer [7], notwithstanding 10% of breast cancer cell lines have inactivated PTEN [2, 3]. Recently it has been shown that PTEN loss is a common event in breast cancers caused by BRCA1 deficiency [8]. Marsh et al. [9] defined PTEN hamartoma tumor syndrome (PHTS) as a syndromic condition including one or more hamartomas which has its biological basis in a germline mutation of the PTEN gene. Following such assumption, PHTS includes patients with the previous diagnosis of CS, Bannayan-Riley-Ruvalcaba syndrome [10] (BRRS), Proteus syndrome [11] (PS), Proteus-like syndrome [12] (PLS), and Lhermitte-Duclos syndrome [13] (LDS).
Li et al. [2] have shown that PTEN gene is a human cdc14 homolog, like CDC14A and CDC14B. The cdc14 gene is a key point for the progression of cell cycle in Saccharomyces cerevisiae. Its protein acts in late nuclear division preparing for subsequent DNA replication. The human PTEN gene spans 103,207 bases, is made up of 9 exons, and codes for a 1212-bp transcript and a 403-amino-acid protein.
The PTEN product has the kinetic properties of dual-specific phosphatases [14] and acts on G1 cell cycle progression through negative regulation of the PI3-kinase/Akt or PKB signalling pathway [15]. PTEN is a member of the protein-tyrosine phosphatase (PTP) gene superfamily [2, 3]. These are genes consisting of conserved catalytic domains, flanked by noncatalytic regulatory sequences [16]. The PTP catalytic domains show a “signature motif,” which is the canonical sequence HCXXGXXRS/T. Among the PTP superfamily genes, a further split is made in “classic” PTP (acting only towards phosphothyrosine residues) and dual-specificity phosphatase families (dephosphorylating phosphotyrosine, phosphoserine and/or phosphotreonine). PTEN is a dual-specificity phosphatase. The catalytic domain of PTEN has been proven to be essential for its function, which is lost following any mutation within the signature motif [17].
PTEN gene in glioblastoma-derived cell lines regulates hypoxia- and IGF-1-induced angiogenic gene expression by regulating Akt activation of HIF-1 activity [18]. Restoration of wild-type PTEN to glioblastoma cell lines lacking functional PTEN ablates hypoxia and IGF-1 induction of HIF-1-regulated genes. In addition, Akt activation leads to HIF-1α stabilization, whereas PTEN attenuates hypoxia-mediated HIF-1 α stabilization. Loss of PTEN during malignant progression contributes to tumor expansion through the deregulation of Akt activity and HIF-1-regulated gene expression. PTEN abnormalities have been found also in primary acute leukemias and non-Hodgkin's lymphomas [19].
PTEN and phosphorylated Akt levels are inversely correlated in the large majority of the examined samples, suggesting that PTEN regulates phosphatidylinositol 3,4,5-triphosphates and may play a role in apoptosis. Overexpression of PTEN inhibits cell migration, whereas antisense PTEN enhances migration [20]. The phosphatase domain of PTEN is essential because its inactivation does not allow the downregulation of integrin-mediated cell spreading and formation of focal adhesions, peculiar of wild-type PTEN. Overexpression of focal adhesion kinase (FAK) partially antagonizes the effects of PTEN. Thus, the negative regulation of cell interactions with the extracellular matrix could be the way PTEN phosphatase acts as a tumor suppressor.
PTEN gene plays an essential role in human development. Indeed, the additional effect of three homozygotic mutations together (e.g., mutations in both alleles) produces early embryonic lethality in mice, whereas heterozygosis (e.g., mutations in one allele) increases tumor incidence [21–23]. Furthermore, PTEN antagonizes growth factor-induced Shc phosphorylation and inhibits the MAP kinase (MAPK) signalling pathway [24]. The way this inhibition is accomplished is currently understood as a suppression by the PTEN protein phosphatase activity [25].
The function of PTEN gene and the way its mutation can cause a disease can barely be understood if one does not see the PTEN protein inside the PI3K/Akt/mTOR signalling pathway [26] (Figure 1). The activation of phosphoinositide 3′ kinase (PI3K) is the primary event in this pathway [27]. This can occur from several growth factor receptors (GFRs), such as PDGFR, EGFR, FGFR, IGF-1R, VEGFR, IL-R, interferon receptors (IF-Rs), integrin receptors, and the Ras pathway [28–30]. The major role of PI3K is the phosphorylation of phosphatidylinositol (4,5) P (PIP2) to phosphatidylinositol (3,4,5) P (PIP3). PIP3 binds and translocates Akt near the cell membrane where it can be phosphorylated and activated by phosphatidylinositol(3,4,5)P-dependent kinase 1 (PDK1) and phosphatidylinositol(3,4,5)P-dependent kinase 2 (PDK2). Akt has several downstream effectors which mediate its ability to promote cell survival and growth. Then, activation of PI3K/Akt pathway is observed in several human cancers. PTEN is the antagonist of PI3K because it dephosphorylates PIP3 to PIP2. The PI3K/PTEN imbalance, caused for instance by a mutation of PTEN, is then responsible for the progression to human cancer.
Figure 1.
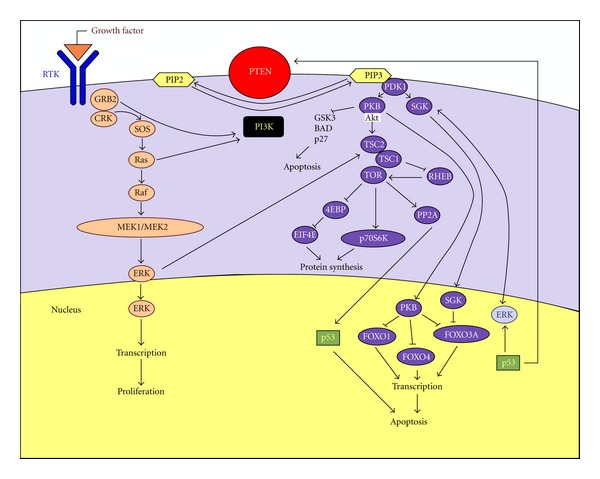
Pathway involving the PTEN protein.
Salmena et al. [1] have recently proposed a model for the causes and consequences of PTEN loss. While retinoblastoma (RB) gene has been the foundation of the Knudson's hypothesis [4], showing that only the homozygous loss of such gene can start the retinoblastoma initiation, PTEN behaves in a different way. There is compelling evidence in mice confirming PTEN as a haploinsufficient tumor suppressor gene: loss of one allele leads to the progression of a lethal polyclonal autoimmune disorder [31]; epithelial cancers, such as prostate cancer, are driven by PTEN heterozygosity [32]; cellular levels of PTEN protein inversely correlate with the occurrence of invasive prostate cancer [1]. Consequently, functional loss of one PTEN allele is critical for the onset of cancer in mice. Things in humans are little less compelling, but the evidence is growing. The association between PTEN heterozygous germline mutations and the so-called PHTS is the first proof. Further evidence for haploinsufficiency is supported by the following observations: some tumors arisen in patients with CS do not show biallelic mutations of PTEN gene [33]; primary prostate cancers are associated with loss or alteration of one PTEN allele in 70% of cases [34], whereas homozygous deletion is present in 10% of cases [35]; the occurrence of monoallelic mutation of PTEN in breast cancer is much more frequent (30–40% versus 5%) than that of biallelic loss [36–38]. Besides the above-mentioned proofs of the contribution of PTEN haploinsufficiency to tumor progression and cancer syndromes, the identification of CS and tumor-derived PTEN mutations preserving partial or full PTEN lipid phosphatase function [39] allows the notion that even minor impairments of PTEN function can lead to cancer. The model of Santena et al. [1] springs from the above results: the loss of PTEN function, caused not only by classical genetic mutations leading to heterozygous (50% of function) or homozygous (0% of function) loss, but also by other molecular mechanisms, such as transcriptional regression, epigenetic silencing, regulation by microRNAs, posttranslational modification, and aberrant localization, can lead to subtle and/or dramatic losses of PTEN function, behaving as a stepwise gradation of function. An unexpected result has been put forward by Chen et al. [40], who studied the relationship between PTEN dose and tumor progression in mouse models of prostate specific loss of PTEN: complete acute loss of PTEN promoted a strong senescence response opposing tumor progression. Campisi and d'Adda di Fagagna [41] interpreted senescence as an antitumor mechanism set off by tumor suppressor genes in response to triggers including DNA damage and oncogene activation.
While heterozygous and homozygous mutations are now widely understood, the other molecular mechanisms deserve some clarification. We will do so, showing that all these mechanisms have a significant impact in the way PTEN functions, as a model of tumor suppressor gene.
While transforming growth factor β (TGFβ) was considered the single transcriptional regulator of PTEN gene in 1997 [42], acting in a negative way, today the story is much more complicated. Several factors have been discovered to upregulate, such as early growth-regulated transcription factor-1 (EGR-1) [43] which is a downstream effector of Insulin-like growth factor 2 (IGF-2) [44], and peroxisome proliferation-activated receptor-γ (PPAR-γ) [45], p53 [46], and MYC [47], or downregulate PTEN, such as c-Jun [48], NFκB [49], and HES-1 [47]. Furthermore, a peculiar way of PTEN's transcriptional regulation is that of NOTCH1, which increases the transcription of PTEN at least in two fashions: activating MYC [47] or repressing CBF-1 [50, 51], which is a downregulator of PTEN. Conversely, NOTCH1 represses PTEN's transcription, activating the known PTEN's downregulator HES-1 [47]. Another complication of the action of NOTCH1 on PTEN is the tissue specificity, which allows upregulation or downregulation according to the involved tissues.
Epigenetic silencing means that the function of a gene is broken, not by means of DNA mutations, but for other mechanisms, which are mainly the DNA methylation of the gene promoter and the histone modification. The promoter methylation of PTEN gene has been associated to several cancers [52–54].
MicroRNAs (miRNAs) are single-stranded RNAs, made up of a few (usually 22) nucleotides, which repress the mRNA translation. One of the most studied miRNAs, miR-21, has been reported as a repressor of PTEN, exerting its oncogenic activity, at least partially, downregulating PTEN expression [55–59].
Posttranslational modification is a further way of regulation of the action of a gene, without any change in its DNA. It can be defined as the chemical modification of a protein after its translation. The four main chemical reactions are phosphorylation, acetylation, oxidation, and ubiquitination. PTEN has six phosphorylation sites [1], which have been involved in the modulation of its tumor suppressor functions, subcellular distribution, and stability. They are Threonine 366 (Thr366), Serine 370 (Ser370), Ser385, Ser380, Thr382, and Thr383. The last three are collectively referred to as the STT cluster, which has the most important effects on PTEN function. The wild-type or phosphorylated STT cluster is maintained to stabilize PTEN in a closed state, whereas the mutation at these phosphorylation sites opens the protein conformation, making it less stable [60]. A dephosphorylation of the STT cluster is currently considered a common way leading to cancer activation [61]. PTEN interacts with the nuclear histone acetyltransferase-associated PCAF protein, promoting PTEN acetylation at Lysine 125 (Lys125) and Lys128 sites, which in turn negatively regulates PTEN catalytic activity [62]. What is now called reactive oxygen species (ROS), which is made of oxygen ions, free radical, and peroxides, are very small molecules sharing a high reactivity due to presence of unpaired valence shell electrons. ROS modulates PTEN catalytic activity by oxidative-stress-induced formation of a disulfide bond between the active site cysteine 124 (Cys124) and Cys71 [63]. Ubiquitin [64] is a highly conserved regulatory protein that is widely (ubiquitously) expressed in eukaryotes. Ubiquitination refers to the posttranslational modification of a protein by the covalent attachment (via an isopeptide bond) of one or more ubiquitin monomers. The most prominent function of ubiquitin is labeling proteins for proteasomal degradation. Proteasome inhibition increases the half-life of PTEN [65], and the exposure of human bronchial cells to zinc ions promotes ubiquitin-dependent degradation of PTEN [66], but inhibitors of the proteasome may destabilize PTEN [67]. Notwithstanding such incoherent results, ubiquitination to lysine 13 (Lys13) and Lys289 is needed for the nuclear-cytoplasmic shuttling of PTEN [68]. Such last fact implies the notion that PTEN protein can move from cytoplasm to nucleus, and backward. However, this was not clear at the very beginning, when everyone thought it was exclusively localized to the cytoplasm. PTEN is abundantly localized in the nucleus of primary, differentiated, and resting cells, while there is a sharp decrease in cancer cells [69–72]. Cell cycle stage and differentiation status are, consequently, related to PTEN localization. The fact that PTEN protein is localized in the nucleus, besides the cytoplasm, has an important role in the tumor suppressor function of such protein. This is proven by the report of patients with more aggressive tumors, such as esophageal squamous cell carcinoma [73], cutaneous melanoma [74, 75], colorectal cancer [76], and pancreatic islet cell tumors [72], having their PTEN protein absent in the nucleus.
2. Why Should the Dermatologist Know about PTEN?
Besides using PTEN as a model for genetic disease, the dermatologist should deepen his knowledge of such fascinating pathway for some very practical reasons.
First of all, think of melanoma. Approximately 70% of melanomas have elevated Akt3 signaling both for increased gene copy number and PTEN loss. Consequently, the targeting of (V600E)B-Raf and Akt3 signalling can prevent or treat cutaneous melanocytic lesions. The development of agents specifically targeting these proteins would be very useful, because they have fewer side effects than those inhibiting both normal and mutant B-Raf protein or targeting all three Akt isoforms. Recently [77], a nanoliposomal-ultrasound-mediated approach reported for delivering small interfering RNA (siRNA) specifically targeting (V600E)B-Raf and Akt3 into melanocytic tumors present in skin to retard melanoma development. Novel cationic nanoliposomes stably encapsulate siRNA targeting (V600E)B-Raf or Akt3, providing protection from degradation and facilitating entry into melanoma cells to decrease expression of these proteins. Low-frequency ultrasound using a lightweight four-cymbal transducer array enables penetration of nanoliposomal-siRNA complex throughout the epidermal and dermal layers of laboratory-generated or animal skin. Nanoliposomal-mediated siRNA targeting of (V600E)B-Raf and Akt3 led to a cooperatively acting approximately 65% decrease in early or invasive cutaneous melanoma compared with inhibition of each singly with negligible associated systemic toxicity. Thus, cationic nanoliposomes loaded with siRNA targeting (V600E)B-Raf and Akt3 provide an effective approach for targeted inhibition of early or invasive cutaneous melanomas. Furthermore, it is currently thought that the progression of human cutaneous melanomas behaves in a stepwise fashion, due to accumulating genetic and epigenetic alterations. The combination of PTEN deficiency and Braf activation induces a melanoma in-situ-like phenotype without dermal invasion. Further addition of cell autonomous TGF-β activation in the context of PTEN deficiency and Braf activation promotes dermal invasion in skin cultures without significantly promoting proliferation in vitro and in vivo. This proinvasive phenotype of cell autonomous TGF-β activation is genetic context dependent, as hyperactivating the TGF-β type I receptor without PTEN deficiency and Braf activation failed to induce an invasive behavior. Evidence of genetic interactions among PTEN deficiency, Braf activation, and cell autonomous TGF-β activation shows that distinct stages of human melanoma are genetically tractable in the proper tissue architecture [78].
Secondly, multistage skin carcinogenesis is prone to gene synergism. The ablation of PTEN function in mouse epidermis expressing activated Fos leads to hyperplasia, hyperkeratosis, and tumors that move forward to highly differentiated keratoacanthomas, rather than to carcinomas [79].
Thirdly, the dermatologist can be the front office in diagnosing a PHTS. Acral papular neuromatosis [80] and mucocutaneous neuromas [81] can be the first sign of a disease caused by a PTEN mutation. Katona et al. [82] analysed a series of patients affected by mycosis fungoides, showing that LOH studies are a robust method for evaluating genetic abnormalities in mycosis fungoides, and several loci associated with the PTEN appear to be associated with progression from plaque to tumor stage. It has been shown that PTEN was significantly higher in depigmented epidermis, implying that vitiliginous keratinocytes may be more susceptible to TNF-alpha-mediated apoptosis through impaired Akt and NF-kappaB activation. Keratinocytes showing impaired Akt activation demonstrated increased apoptosis with less activation of NF-kappaB. Thus, reduced activation of NF-kappaB via impaired PI3K/Akt activation under increased TNF-alpha levels could result in increased apoptosis of vitiliginous keratinocytes [83].
Finally, the pathogenic role of mutated PTEN involves some conditions collectively called PTEN hamartoma tumor syndrome (PHTS) [84, 85], such as Cowden syndrome [86], Bannayan-Riley-Ruvalcaba syndrome (BRRS) [87], and Proteus syndrome (PS) [88].
Such rare diseases highlight tumoral degeneration in many body organs. Affected patients show usually variable degrees of intellective disability. The cutaneous surface of PHTS patients leads one to suspect the disease being present, many times in a peculiar way. Main examples are speckled penis in BRRS [87] (Figure 2), plantar cerebriform hyperplasia in 70–80% of PS patients [88, 89] (Figure 3), and three or more trichilemmomas in CS [86].
Figure 2.
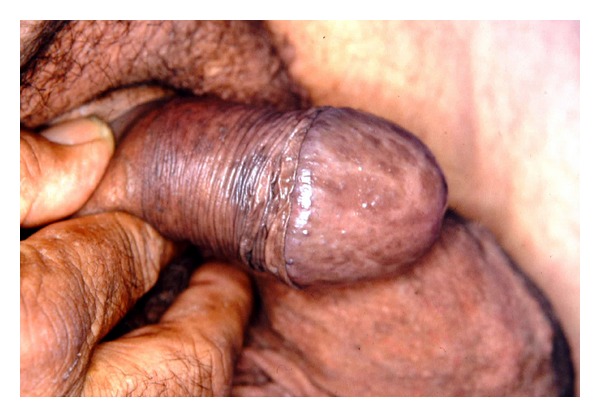
Speckled penis in a patient affected by Bannayan-Riley-Ruvalcaba syndrome.
Figure 3.
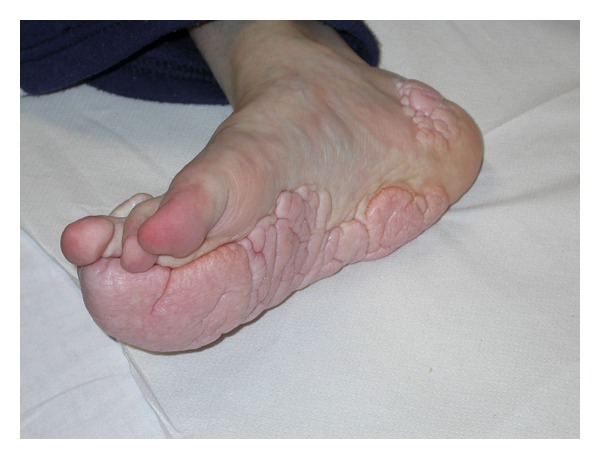
Cerebriform plantar hyperplasia in a girl affected by Proteus syndrome.
The spectrum of clinical findings associated with PTEN tumor suppressor gene germline mutations includes also mucocutaneous neuromas, as reported by Shaffer et al., who stated that this is an underrecognized manifestation of the gene [81].
These are only some reasons why the knowledge of PTEN is very much needed for the dermatologist, not only for his/her culture, but also for practical issues, such as a targeted therapy in the near future.
Conflict of Interests
The authors have no conflict of interests to declare.
References
- 1.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133(3):403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 3.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genetics. 1997;15(4):356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 4.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature Genetics. 1997;16(1):64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 6.Weary PE, Gorlin RJ, Gentry WC, Comer JE, Greer KE. Multiple hamartoma syndrome (Cowden’s disease) Archives of Dermatology. 1972;106(5):682–690. [PubMed] [Google Scholar]
- 7.Chen J, Lindblom P, Lindblom A. A study of the PTEN/MMAC1 gene in 136 breast cancer families. Human Genetics. 1998;102(1):124–125. [PubMed] [Google Scholar]
- 8.Saal LH, Gruvberger-Saal SK, Persson C, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nature Genetics. 2008;40(1):102–107. doi: 10.1038/ng.2007.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Human Molecular Genetics. 1999;8(8):1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 10.Cohen MM., Jr. Bannayan-Riley-Ruvalcaba Syndrome: renaming three formerly recognized syndromes as one etiologic entity. American Journal of Medical Genetics. 1990;35(2):291–2292. doi: 10.1002/ajmg.1320350231. [DOI] [PubMed] [Google Scholar]
- 11.Wiedemann HR, Burgio GR, Aldenhoff P, Kunze J, Kaufmann HJ, Schirg E. The proteus syndrome. Partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, microcephaly and other skull anomalies and possible accelerated growth and visceral affections. European Journal of Pediatrics. 1983;140:5–12. doi: 10.1007/BF00661895. [DOI] [PubMed] [Google Scholar]
- 12.Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Human Molecular Genetics. 2000;9(5):765–768. doi: 10.1093/hmg/9.5.765. [DOI] [PubMed] [Google Scholar]
- 13.Dastur HM, Pandya SK, Deshpande DH. Diffuse cerebellar hypertrophy. (Lhermitte Duclos disease) Neurology India. 1975;23(1):53–56. [PubMed] [Google Scholar]
- 14.Myers MP, Stolarov JP, Eng C, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15406–15411. doi: 10.1073/pnas.95.26.15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denu JM, Stuckey JA, Saper MA, Dixon JE. Form and function in protein dephosphorylation. Cell. 1996;87(3):361–364. doi: 10.1016/s0092-8674(00)81356-2. [DOI] [PubMed] [Google Scholar]
- 17.Myers MP, Tonks NK. PTEN: sometimes taking it off can be better than putting it on. American Journal of Human Genetics. 1997;61(6):1234–1238. doi: 10.1086/301659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zundel W, Schindler C, Haas-Kogan D, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes and Development. 2000;14(4):391–396. [PMC free article] [PubMed] [Google Scholar]
- 19.Dahia PLM, Aguiar RCT, Alberta J, et al. PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanisms in haematological malignancies. Human Molecular Genetics. 1999;8(2):185–193. doi: 10.1093/hmg/8.2.185. [DOI] [PubMed] [Google Scholar]
- 20.Tamura M, Gu J, Matsumoto K, Aota SI, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280(5369):1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 21.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nature Genetics. 1998;19(4):348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki A, de La Pompa JL, Stambolic V, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Current Biology. 1998;8(21):1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 23.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(4):1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weng LP, Smith WM, Brown JL, Eng C. PTEN inhibits insulin-stimulated MEK/MAPK activation and cell growth by blocking IRS-1 phosphorylation and IRS-1/Grb-2/Sos complex formation in a breast cancer model. Human Molecular Genetics. 2001;10(6):605–616. doi: 10.1093/hmg/10.6.605. [DOI] [PubMed] [Google Scholar]
- 25.Weng LP, Brown JL, Baker KM, Ostrowski MC, Eng C. PTEN blocks insulin-mediated ETS-2 phosphorylation through MAP kinase, independently of the phosphoinositide 3-kinase pathway. Human Molecular Genetics. 2002;11(15):1687–1696. doi: 10.1093/hmg/11.15.1687. [DOI] [PubMed] [Google Scholar]
- 26.Newton HB. Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 2: PI3K/Akt/PTEN, mTOR, SHH/PTCH and angiogenesis. Expert Review of Anticancer Therapy. 2004;4(1):105–128. doi: 10.1586/14737140.4.1.105. [DOI] [PubMed] [Google Scholar]
- 27.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nature Reviews Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 28.Martin KA, Blenis J. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Advances in Cancer Research. 2002;86:1–39. doi: 10.1016/s0065-230x(02)86001-8. [DOI] [PubMed] [Google Scholar]
- 29.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochimica et Biophysica Acta. 1998;1436(1-2):127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 30.Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. The Journal of Biological Chemistry. 1999;274(13):8347–8350. doi: 10.1074/jbc.274.13.8347. [DOI] [PubMed] [Google Scholar]
- 31.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten(+/−) mice. Science. 1999;285(5436):2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 32.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, P Pandolfi P. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nature Genetics. 2001;27(2):222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 33.Dahia PLM. PTEN, a unique tumor suppressor gene. Endocrine-Related Cancer. 2000;7(2):115–129. doi: 10.1677/erc.0.0070115. [DOI] [PubMed] [Google Scholar]
- 34.Gray IC, Stewart LMD, Phillips SMA, et al. Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. British Journal of Cancer. 1998;78(10):1296–1300. doi: 10.1038/bjc.1998.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whang YE, Wu X, Suzuki H, et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(9):5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. Journal of the National Cancer Institute. 1999;91(22):1922–1932. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 37.Bose S, Wang SI, Terry MB, Hibshoosh H, Parsons R. Allelic loss of chromosome 10q23 is associated with tumor progression in breast carcinomas. Oncogene. 1998;17(1):123–127. doi: 10.1038/sj.onc.1201940. [DOI] [PubMed] [Google Scholar]
- 38.Feilotter HE, Coulon V, McVeigh JL, et al. Analysis of the 10q23 chromosomal region and the PTEN gene in human sporadic breast carcinoma. British Journal of Cancer. 1999;79(5-6):718–723. doi: 10.1038/sj.bjc.6690115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waite KA, Eng C. Protean PTEN: form and function. American Journal of Human Genetics. 2002;70(4):829–844. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campisi J, d’Adda Di Fagagna F. Cellular senescence: when bad things happen to good cells. Nature Reviews Molecular Cell Biology. 2007;8(9):729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 42.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β . Cancer Research. 1997;57(11):2124–2129. [PubMed] [Google Scholar]
- 43.Virolle T, Adamson ED, Baron V, et al. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nature Cell Biology. 2001;3(12):1124–1128. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 44.Moorehead RA, Hojilla CV, De Belle I, et al. Insulin-like growth factor-II regulates PTEN expression in the mammary gland. The Journal of Biological Chemistry. 2003;278(50):50422–50427. doi: 10.1074/jbc.M306894200. [DOI] [PubMed] [Google Scholar]
- 45.Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Current Biology. 2001;11(10):764–768. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 46.Stambolic V, MacPherson D, Sas D, et al. Regulation of PTEN transcription by p53. Molecular Cell. 2001;8(2):317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 47.Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nature Medicine. 2007;13(10):1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hettinger K, Vikhanskaya F, Poh MK, et al. c-Jun promotes cellular survival by suppression of PTEN. Cell Death and Differentiation. 2007;14(2):218–229. doi: 10.1038/sj.cdd.4401946. [DOI] [PubMed] [Google Scholar]
- 49.Xia D, Srinivas H, Ahn YH, et al. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NFκB-dependent pathway. The Journal of Biological Chemistry. 2007;282(6):3507–3519. doi: 10.1074/jbc.M610141200. [DOI] [PubMed] [Google Scholar]
- 50.Chappell WH, Green TD, Spengeman JD, McCubrey JA, Akula SM, Bertrand FE. Increased protein expression of the PTEN tumor suppressor in the presence of constitutively active notch-1. Cell Cycle. 2005;4(10):1389–1395. doi: 10.4161/cc.4.10.2028. [DOI] [PubMed] [Google Scholar]
- 51.Whelan JT, Forbes SL, Bertrand FE. CBF-1 (RBP-Jκ) binds to the PTEN promoter and regulates PTEN gene expression. Cell Cycle. 2007;6(1):80–84. doi: 10.4161/cc.6.1.3648. [DOI] [PubMed] [Google Scholar]
- 52.García JM, Silva J, Peña C, et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes and Cancer. 2004;41(2):117–124. doi: 10.1002/gcc.20062. [DOI] [PubMed] [Google Scholar]
- 53.Goel A, Arnold CN, Niedzwiecki D, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Research. 2004;64(9):3014–3021. doi: 10.1158/0008-5472.can-2401-2. [DOI] [PubMed] [Google Scholar]
- 54.Kang YH, Hye SL, Woo HK. Promoter methylation and silencing of PTEN in gastric carcinoma. Laboratory Investigation. 2002;82(3):285–291. doi: 10.1038/labinvest.3780422. [DOI] [PubMed] [Google Scholar]
- 55.Meng F, Henson R, Lang M, et al. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130(7):2113–2129. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 56.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Research. 2005;65(14):6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 58.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26(19):2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 59.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochemical Journal. 2004;382(1):1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. The Journal of Biological Chemistry. 2001;276(52):48627–48630. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- 62.Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. The Journal of Biological Chemistry. 2006;281(36):26562–26568. doi: 10.1074/jbc.M605391200. [DOI] [PubMed] [Google Scholar]
- 63.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2 . The Journal of Biological Chemistry. 2002;277(23):20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 64.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. Journal of Biosciences. 2006;31(1):137–155. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 65.Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. The Journal of Biological Chemistry. 2001;276(2):993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 66.Wu W, Wang X, Zhang W, et al. Zinc-induced PTEN protein degradation through the proteasome pathway in human airway epithelial cells. The Journal of Biological Chemistry. 2003;278(30):28258–28263. doi: 10.1074/jbc.M303318200. [DOI] [PubMed] [Google Scholar]
- 67.Tang Y, Eng C. p53 down-regulates phosphatase and tensin homologue deleted on chromosome 10 protein stability partially through caspase-mediated degradation in cells with proteasome dysfunction. Cancer Research. 2006;66(12):6139–6148. doi: 10.1158/0008-5472.CAN-06-0772. [DOI] [PubMed] [Google Scholar]
- 68.Trotman LC, Wang X, Alimonti A, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128(1):141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gimm O, Perren A, Weng LP, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. American Journal of Pathology. 2000;156(5):1693–1700. doi: 10.1016/s0002-9440(10)65040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Research. 2003;63(2):282–286. [PubMed] [Google Scholar]
- 71.Lachyankar MB, Sultana N, Schonhoff CM, et al. A role for nuclear PTEN in neuronal differentiation. Journal of Neuroscience. 2000;20(4):1404–1413. doi: 10.1523/JNEUROSCI.20-04-01404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perren A, Komminoth P, Saremaslani P, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. American Journal of Pathology. 2000;157(4):1097–1103. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tachibana M, Shibakita M, Ohno S, et al. Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer. 2002;94(7):1955–1960. doi: 10.1002/cncr.0678. [DOI] [PubMed] [Google Scholar]
- 74.Whiteman DC, Zhou XP, Cummings MC, Pavey S, Hayward NK, Eng C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. International Journal of Cancer. 2002;99(1):63–67. doi: 10.1002/ijc.10294. [DOI] [PubMed] [Google Scholar]
- 75.Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ, Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. American Journal of Pathology. 2000;157(4):1123–1128. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou XP, Loukola A, Salovaara R, et al. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. American Journal of Pathology. 2002;161(2):439–447. doi: 10.1016/S0002-9440(10)64200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tran MA, Gowda R, Sharma A, et al. Targeting V600EB-Raf and Akt3 using nanoliposomal-small interfering RNA inhibits cutaneous melanocytic lesion development. Cancer Research. 2008;68(18):7638–7649. doi: 10.1158/0008-5472.CAN-07-6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lo RS, Witte ON. Transforming growth factor-β activation promotes genetic context-dependent invasion of immortalized melanocytes. Cancer Research. 2008;68(11):4248–4257. doi: 10.1158/0008-5472.CAN-07-5671. [DOI] [PubMed] [Google Scholar]
- 79.Yao D, Alexander CL, Quinn JA, Chan WC, Wu H, Greenhalgh DA. Fos cooperation with PTEN loss elicits keratoacanthoma not carcinoma, owing to p53/p21WAF-induced differentiation triggered by GSK3β inactivation and reduced AKT activity. Journal of Cell Science. 2008;121(10):1758–1769. doi: 10.1242/jcs.021147. [DOI] [PubMed] [Google Scholar]
- 80.Ferran M, Bussaglia E, Lazaro C, Matias-Guiu X, Pujol RM. Acral papular neuromatosis: an early manifestation of Cowden syndrome. British Journal of Dermatology. 2008;158(1):174–176. doi: 10.1111/j.1365-2133.2007.08237.x. [DOI] [PubMed] [Google Scholar]
- 81.Schaffer JV, Kamino H, Witkiewicz A, McNiff JM, Orlow SJ. Mucocutaneous neuromas: an underrecognized manifestation of PTEN hamartoma-tumor syndrome. Archives of Dermatology. 2006;142(5):625–632. doi: 10.1001/archderm.142.5.625. [DOI] [PubMed] [Google Scholar]
- 82.Katona TM, O’Malley DP, Cheng L, et al. Loss of heterozygosity analysis identifies genetic abnormalities in mycosis fungoides and specific loci associated with disease progression. American Journal of Surgical Pathology. 2007;31(10):1552–1556. doi: 10.1097/PAS.0b013e3180408d76. [DOI] [PubMed] [Google Scholar]
- 83.Kim NH, Jeon S, Lee HJ, Lee AY. Impaired PI3K/Akt activation-mediated NF-κB inactivation under elevated TNF-α is more vulnerable to apoptosis in vitiliginous keratinocytes. Journal of Investigative Dermatology. 2007;127(11):2612–2617. doi: 10.1038/sj.jid.5700900. [DOI] [PubMed] [Google Scholar]
- 84.Eng C. PTEN: one gene, Many syndromes. Human Mutation. 2003;22(3):183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 85.Romano C. Genetics of PTEN hamatoma tumor syndrome (PHTS) In: Ruggieri M, Pascual Castroviejo I, Di Rocco C, editors. Neurocutaneous Disorders. Phakomatoses and Hamartoneoplastis Syndromes. Springer; 2008. [Google Scholar]
- 86.Hildenbrand C, Burgdorf WHC, Lautenschlager S. Cowden syndrome—diagnostic skin signs. Dermatology. 2001;202(4):362–366. doi: 10.1159/000051684. [DOI] [PubMed] [Google Scholar]
- 87.Hendriks YMC, Verhallen JTCM, Van der Smagt JJ, et al. Bannayan-Riley-Ruvalcaba syndrome: further delineation of the phenotype and management of PTEN mutation-positive cases. Familial Cancer. 2003;2(2):79–85. doi: 10.1023/a:1025713815924. [DOI] [PubMed] [Google Scholar]
- 88.Satter E. Proteus syndrome: 2 Case reports and a review or the literature. Cutis. 2007;80(4):297–302. [PubMed] [Google Scholar]
- 89.Schepis C, Greco D, Siragusa M, Romano C. Cerebriform plantar hyperplasia: The major cutaneous feature of Proteus syndrome. International Journal of Dermatology. 2008;47(4):374–376. doi: 10.1111/j.1365-4632.2008.03440.x. [DOI] [PubMed] [Google Scholar]