

Abstract
An emerging infectious pathogen Hymenoscyphus pseudoalbidus has spread across much of Europe within recent years causing devastating damage on European common ash trees (Fraxinus excelsior) and associated plant communities. The present study demonstrates the presence of additive genetic variation in susceptibility of natural F. excelsior populations to the new invasive disease. We observe high levels of additive variation in the degree of susceptibility with relatively low influence of environmental factors (narrow-sense heritability = 0.37–0.52). Most native trees are found to be highly susceptible, and we estimate that only around 1% has the potential of producing offspring with expected crown damage of <10% under the present disease pressure. The results suggest that the presence of additive genetic diversity in natural F. excelsior populations can confer the species with important ability to recover, but that low resistance within natural European populations is to be expected because of a low frequency of the hypo-sensitive trees. Large effective population sizes will be required to avoid genetic bottlenecks. The role of artificial selection and breeding for protection of the species is discussed based on the findings.
Keywords: adaptation, conservation genetics, ecological, emerging infectious disease genetics, invasive species
Introduction
Evolution in natural populations is driven by a dynamic interaction between multiple biotic and abiotic factors including continuous competition within and between species (Valen 1973). Host-specific pathogens are among the powerful selective forces driving evolution by influencing individuals and species at genetic, ecological, spatial and temporal scales (Burdon et al. 2006). Dramatic effects are often seen when emerging infectious diseases (EIDs) suddenly cause severe damage to species of ecological or anthropogenic importance (Stukenbrock and Mcdonald 2008). In general, the risk of severe effects is assumed to be highest in host populations with narrow genetic background, as adaptation to a new pathogen will depend on the presence of genetic variation in resistance to the particular pathogen within the host species (Burdon 2001). However, the genetic architecture of host resistance in natural plant populations can take various forms (Barrett et al. 2008; Burdon and Thrall 2009), and the severity of damage often depends on demographic and landscape management features (Pautasso et al. 2010). The relation between genetic diversity and susceptibility is therefore not easily predicted, but evidence exists which supports that the likelihood of species survival is higher when a population is genetically diverse (Burdon et al. 2006; Jump et al. 2009). In species with longer lifespans, such as forest trees, the importance of conserving genetic diversity becomes even more pronounced. The number of new genotypes established during a given time period is relatively low because of the long generation time. In contrast, pathogens have a much shorter generation time with a strong potential for evolutionary shifts. Pathogens as well as environmental conditions are likely to change over a few tree generations, making adaptation challenging (Petit and Hampe 2006; Kuparinen et al. 2010). The risk of pathogens overcoming resistance controlled by single R-genes is an obvious concern, but difficult to predict. Ersoz et al. (2010) studied patterns of polymorphism in candidate genes for the host–pathogen interaction between Pinus taeda and Fusarium circinatum and Cronartium quercuum sp. Fusiforme and found indications for both active coevolution (arms-race model) and presence of more stable resistance gene polymorphisms (trench warfare model). Also, Dowkiw and Bastien (2007) found evidence that alleles in R-genes in a Populus sp. –Melampsora larici-populina leaf rust pathosystem that have been overcome by the pathogen (defeated alleles) still influenced the level of quantitative resistance.
The importance of genetic diversity is in general recognized in both operational forestry and conservation (Eriksson et al. 1993; Roberds and Bishir 1997; Hansen et al. 2005; Aitken et al. 2008; Funda et al. 2009). Recently, the potential functioning of genetic diversity as a buffer against new diseases has become highly relevant as one of the major European forest species, the European common ash, Fraxinus excelsior L., is threatened by an EID. The disease is caused by the pathogenic fungus, Hymenoscyphus pseudoalbidus (Queloz et al. 2010), the asexual form Chalara fraxinea (Kowalski 2006), which causes extensive dieback throughout East, North and Central Europe rapidly spreading towards the southern and western limits of F. excelsior's natural distribution (Bakys et al. 2009a,b; Husson et al. 2011; Rytkönen et al. 2011). F. excelsior is a species of major economical importance in European forestry (FRAXIGEN 2005). In Danish forests, it is the 5th most important broadleaved species in terms of area coverage (Nord-Larsen et al. 2009) and dominant in forests on moist, fertile soils with good drainage. F. excelsior is an important keystone species in natural plant communities protected under EU regulations (European Commission 2007). The short- and long-term effects of H. pseudoalbidus on the F. excelsior trees therefore present a major economical and ecological concern. In Denmark, the problem is now so severe that the species is no longer planted in the forestry sector. Furthermore, mature trees are being logged intensively to harvest timber before the quality deteriorates. This previously common tree species is disappearing from forest ecosystems.
In a recent study, we concluded that the majority of clones (genets) in natural Danish populations of F. excelsior must be expected to be highly susceptible to H. pseudoalbidus. However, a small fraction of clones exhibited relatively high levels of partial resistance to the pathogen (McKinney et al. 2011). The implication of this finding beyond the present generation of F. excelsior trees depends on the degree to which resistance is inherited from parents to offspring. Of particular importance is the level of additive variation and narrow-sense heritability, which are essential for estimating the expected response from selection (Falconer and Mackay 1996). Through its influence on the lifetime fitness of the single trees, additive genetic variation in susceptibility will determine the potential impact of natural selection on contemporary and long-term evolution. The heritability and level of additive genetic variation will further determine the gains from artificial selection (Walsh 2008), and the objective of the present study is therefore to estimate these parameters based on progeny from trees of putative native Danish origin. We estimate the distribution of trees in relation to the expected susceptibility of their offspring to H. pseudoalbidus, we separate the variation between progeny into between- and within-populations and families and we compare the phenotypic expression of closely related individuals at two different sites. Finally, we discuss the likely importance of the observed genetic variation and heritability for the recovery of F. excelsior in Europe, and the potential for restoring health by artificial selection and breeding.
Materials and methods
Progeny tests at two sites
We studied the degree of damage on progeny from 101 open-pollinated (OP) trees (mother trees) over three subsequent years. The 101 mother trees were selected from 14 F. excelsior populations, all of putative Danish origin located in the western part of Denmark. Five to nine trees were selected from each population. The largest distance between any two populations is around 150 km.
All 101 tested mother trees were mature and characterized by good health at the time of their selection. The selection was made just prior to the seed collection in 2001, which was 2 years before the first disease symptoms of H. pseudoalbidus were observed in Denmark, and 4 years before widespread and severe damage was reported in 2005 (Skovsgaard et al. 2009). We therefore expect that the collection sampled the gene pool prior to any selective effects induced by the new disease.
Seeds were collected from the trees; seedlings were raised in nursery and planted in autumn 2004 at two sites, Nordre Faelled (56.50 N, 10.04 E) situated near Randers and Hvinningedal (56.2 N, 9.5 E) situated near Silkeborg. At the time of establishment, 64 seedlings were planted from each OP-family, 32 at each of the two sites. The field trials were established as randomized complete block design, with four trees from each mother tree planted next to each other in row-plots, replicated eight times (blocks) at each of the two sites. Two of the 101 OP-families were not included at the Hvinningedal test site owing to lack of seedlings. In total, more than 6400 trees were planted at the two sites.
Damage was recorded for all living trees in June 2008, 2009 and 2010. Following McKinney et al. (2011), each of the approximately 6000 living trees were classified into one of five classes of crown damage. Class 0: trees with no symptoms; Class 1: trees with few insignificant damages and <10% loss of crown foliage; Class 2: trees affected with higher than 10% but <50% crow damage; and Class 3: trees that had lost more than 50% of the crown. Trees that had died from infection were scored as Class 4 (100% damage). In the data analysis, the classes were converted to percent damage score (PDS) corresponding to the median percentage of the class’ range; class 0: 0%, class 1: 5%, class 2: 35%, class 3: 75% and class 4: 100%. The observed H. pseudoalbidus symptoms included the occurrence of stem necroses and/or dead branches and twigs with a purple-brown discolouration. The symptoms were easily recognizable in the field, and we have previously verified the association between symptoms and infection of H. pseudoalbidus based on controlled inoculations followed by PCR assays of the developed symptoms (McKinney et al. in press). It cannot be excluded that a minor part of the assessed crown damage in some of the 6000 assessed trees were attributed to other factors than H. pseudoalbidus infection, but we expect the level of such ‘noise’ in the assessment to be low because of the easily recognized symptoms. Trees that were dead prior to the first specific assessment of the disease (2008) were excluded from the analysis. Trees that were killed by the pathogen during 2008 or 2009 (recorded as Class 4, 100% damage) retained this value throughout the study. During the 2008 and 2009 assessments, we undertook a careful assessment of stem necroses (a typical H. pseudoalbidus symptom) of all trees to estimate genetic parameters for the necrosis development and severity and to determine whether necrosis development correlated closely with crown damage class. Here, the presence of necroses were classified as follows: absent (Score = 0), signs of early stage of necrosis development observed as abnormal coloured patches on the stem typically in association with a small dry dead twig (Score = 1), few and/or scattered clearly recognizable necroses observed as necrotic bark associated with dead branches or twigs (Score = 2) and substantial areas on the stem with necrotic bark, or fewer but severe necrotic areas on the stem (Score = 3).
Test of relatedness of progeny
The quantitative analysis that we apply (cf. below) assumes that the progeny within families are half-sibs. As the estimation of genetic parameters is highly sensitive to deviation from this assumption (Walsh 2008), we tested a subset of the families with genetic markers. We genotyped 10 progeny at three SSR loci from each of 7 OP-families to test whether the offspring indeed were half-sibs. Leaf material was collected from 10 trees for each of the seven putative half-sib families. The material was stored at −20°C until DNA extraction. 35–40 mg leaf tissue per individual was frozen in liquid nitrogen and ground on a bead mill (MM301; Retsch, Haan, Germany) without any prior preparation. DNA was extracted using the DNeasy 96 Plant kit, Qiagen, Hilden, Germany, following the manufacturer's protocol for frozen material. The DNA extractions were maintained undiluted for the polymerase chain reaction. Three polymorphic, easy interpretable microsatellite loci were genotyped: FEMSATL11, FEMSATL19 [cf Lefort et al. (1999) and FEMSATL12 (cf. Gerard et al. 2006)]. PCRs were carried out using the Qiagen Multiplex PCR kit according to the manufacturer's instructions, but with the reaction volumes scaled down to 15 μL. PCR amplifications were completed on Applied Biosystems (Carlsbad, CA, USA) Thermo cyclers (models 9700 and 2700) under the following conditions: initial denaturation of 15 min at 95°C, 30 cycles of denaturation at 94°C for 30 s, annealing at 57°C for 90 s and extension at 72°C for 60 s, and a final extension step at 60°C for 30 min. Each amplified product was diluted with 30 μL H2O and visualized with an ABI3130xl sequencer from Applied Biosystems.
Outcrossing rate and effective number of pollen parents were estimated according to Ritland (2002) as implemented in the MLTR program (version 3.2, 2008, http://genetics.forestry.ubc.ca/ritland/programs.html). Under the hypothesis of half-sibs, the expected outcrossing rate is 100% (no selfing) and the correlation between male gametes within families should be low corresponding to high effective number of pollen parents. Genotypic distances between progeny were separated into within and between families based on an amova approach as implemented in GeneAlEx vers. 6 (Peakall and Smouse 2006) Here, we would expect 25% of variation to be between families and 75% within, according to the hypothesis that each mother tree offspring group consists of half-sibs.
Expected susceptibility of progeny from the mother trees
The 101 examined progeny groups (families) originate from trees from 14 populations. We first tested whether the populations were different and whether the relative health level of the families was similar at both sites (i.e. no family × sites interaction). Secondly, we estimated the genetic parameters for variation in susceptibility. The linear model (1) was applied to test interaction between the two sites, while genetic parameters were estimated within each site using the linear model (2).
| (1) |
| (2) |
where Y is value of the trait in question (PDS or Necrosis), μ is the grand mean, s is the effect of site, b is the effect of replication, P is the effect of population k, λ is the random interaction between site and population, ν is interaction between block (within site) and population, f is the random effect of family l within population k, ϕ is the random interaction between site and family, ρ is the random effect of the plots and e is the residual error (within plot error). The analyses of variance were performed by application of the Satterthwaite's approximation as implemented in the procedure GLM in the SAS® software (SAS Institute Inc. 2008).
The above-mentioned analysis of variance is based on a number of standard assumptions such as normal distribution and independence of residuals. We checked the assumption by visual inspection of errors used for tests of provenance, family and site by family within provenance effects and found an acceptable fit. Additionally, family means of PDS in 2008–2010 showed distributions very close to a normal distribution, making it reasonable to apply quantitative genetic models. Of special interest was spatial structure, because our data consists of observed symptoms following natural infection. However, all parts of both field trials were highly infected, and we found no indications in the residuals to support uneven spatial infection pressure.
In the present context, genotype by environment interaction is of specific interest, if it is expected to lead to divergent selection. We therefore studied the ranking stability of genotypes across sites by estimating the genetic correlation between the two sites using a multivariate approach where the same trait measured at two sites is considered as two different traits (Burdon 1977). The covariance between the traits will then be the genetic covariance across sites (Falconer and Mackay 1996), and high positive genetic correlation between sites will correspond to similar health ranking at the two sites, while low (or negative) correlation will correspond to a situation where the ranking of progeny-tested trees is site specific.
To estimate the genetic variation within natural populations in resistance, we estimated breeding values of the 101 sampled mother trees. The breeding value is a measure of the degree of genetic susceptibility that a parent tree is expected to transfer to its offspring following random mating (Falconer and Mackay 1996). The distribution of breeding values for susceptibility thus provides a presentation of the expected frequency of trees with high and low ability to produce healthy offspring. Breeding values of the tested 101 mothers were estimated from the performance of their progenies as implemented in the software program ASReml (Gilmour et al. 2009). Breeding values of the OP mother trees were estimated as twice the deviation between the family average and the population average, assuming mother trees were pollinated by a large number of pollen donors that on average represented the population mean (White and Hodge 1989).
To elucidate specifically the implications for potential adaptive response, we estimated the additive genetic variance (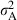 ) and the narrow-sense heritability
) and the narrow-sense heritability 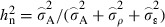 , where
, where 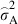 is the estimated additive genetic variance,
is the estimated additive genetic variance, 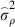 is the estimated plot variance and
is the estimated plot variance and 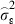 is the estimated within plot variance. We further calculated the additive genetic coefficient of variation (CVA) as the square root of
is the estimated within plot variance. We further calculated the additive genetic coefficient of variation (CVA) as the square root of 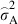 divided by the average PDS to assess the implication of genetic variation for adaptive potential (Houle 1992). Phenotypic, genetic and environmental correlations between traits were estimated according to Falconer and Mackay (1996), based on variance and covariance components obtained from the software program ASReml (Gilmour et al. 2009).
divided by the average PDS to assess the implication of genetic variation for adaptive potential (Houle 1992). Phenotypic, genetic and environmental correlations between traits were estimated according to Falconer and Mackay (1996), based on variance and covariance components obtained from the software program ASReml (Gilmour et al. 2009).
To quantify the relative degree of population differentiation in the analysed traits, we calculated 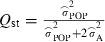 following Spitze (1993) based on the estimated variance components, where
following Spitze (1993) based on the estimated variance components, where 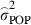 is the estimated variance between populations, that is considering populations as random in model 2. Standard errors for parameters were estimated from Taylor series approximations, and significance of the genetic variances of first order was tested using a log-likelihood ratio test (Gilmour et al. 2009).
is the estimated variance between populations, that is considering populations as random in model 2. Standard errors for parameters were estimated from Taylor series approximations, and significance of the genetic variances of first order was tested using a log-likelihood ratio test (Gilmour et al. 2009).
Results
Relationships within the OP-families
The multilocus outcrossing rate was estimated to be t = 0.96 (0.03), not significantly different from 1. The estimated multilocus correlation between pollen gametes (rp) within families was low, rp=0.005 (0.008), corresponding to an estimated high number of effective pollen donors, Nep =200. The amova estimated that 26% of the variation between genotypes was distributed between families and 74% within families. These results support that tested trees were outcrossed, pollinated by multiple pollen donors, and that the majority of 64 sibs within each OP-family are therefore likely to be half-sibs.
Adaptive potential and distribution of breeding values
Differences between populations were small and nonsignificant (Table 1). The estimated variation between populations was small compared with the large variation between the OP-families within the populations. This corresponds to low and nonsignificant Qst values 0.01-0.02 for PDS and 0.02–0.06 for necrosis score (Table 2). The genetic variation for susceptibility was thus mainly within populations.
Table 1.
F-tests for significance of genetic differences within and between populations in damage score (PDS) and necroses score
| Populations | Site × populations | Familes within populations | Site × population × family | |||||
|---|---|---|---|---|---|---|---|---|
| Trait | F | P > F | F | P > F | F | P > F | F | P > F |
| PDS 2008 | 1.2 | 0.302 | 1.2 | 0.299 | 6.6 | <0.001 | 1.0 | 0.430 |
| PDS 2009 | 1.2 | 0.326 | 2.23 | 0.017 | 6.4 | <0.001 | 1.0 | 0.420 |
| PDS 2010 | 1.2 | 0.307 | 2.05 | 0.032 | 7.3 | <0.001 | 0.8 | 0.840 |
| Necrosis 2008 | 1.6 | 0.122 | 1.82 | 0.065 | 2.8 | <0.001 | 1.2 | 0.170 |
| Necrosis 2009 | 2.8 | 0.010 | 0.91 | 0.544 | 1.9 | 0.002 | 1.3 | 0.066 |
| Site1: Randers | Site 2: Silkeborg | |||||||
|---|---|---|---|---|---|---|---|---|
| Populations | Family within population | Populations | Family within population | |||||
| F | P > F | F | P > F | F | P > F | F | P > F | |
| PDS 2008 | 1.4 | 0.174 | 4.6 | <0.001 | 1.2 | 0.275 | 3.8 | <0.001 |
| PDS 2009 | 2.0 | 0.028 | 4.2 | <0.001 | 1.2 | 0.266 | 3.8 | <0.001 |
| PDS 2010 | 1.7 | 0.076 | 4.1 | <0.001 | 1.4 | 0.197 | 3.3 | <0.001 |
Table 2.
Variance components and genetic parameters
| Source of variation | PDS 2008 (%) | PDS 2009 (%) | PDS 2010 (%) | Necrosis 08 (score) | Necrosis 09 (score) |
|---|---|---|---|---|---|
| Site: Silkeborg | |||||
| Population | 13 | 12 | 10 | 0.0069 | 0.0196 |
| Family within population | 180 | 157 | 115 | 0.0419 | 0.0423 |
| Plot | 122 | 102 | 114 | 0.0458 | 0.0595 |
| Within plot | 1285 | 1034 | 1015 | 0.6579 | 0.9433 |
| VA | 722 | 630 | 461 | 0.1678 | 0.17 |
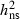
|
0.45 | 0.49 | 0.37 | 0.23 | 0.16 |
SE 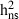
|
0.09 | 0.09 | 0.08 | 0.06 | 0.06 |
| Qst | 0.01 | 0.01 | 0.01 | 0.02 | 0.06 |
| SE Qst | 0.01 | 0.01 | 0.02 | 0.02 | 0.05 |
| Mean | 44 | 57 | 58 | 0.70 | 1.45 |
| CVA (%) | 61 | 44 | 37 | 59 | 28 |
| Site: Randers | |||||
| Population | 16 | 24 | 18 | 0.0177 | 0.0145 |
| Family within population | 152 | 123 | 149 | 0.0533 | 0.0629 |
| Plot | 2 | 0 | 0 | 0 | 0.0243 |
| Within plot | 1009 | 901 | 1112 | 1.1243 | 0.9768 |
| VA | 607 | 492 | 596 | 0.2132 | 0.2515 |
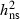 . . |
0.52 | 0.48 | 0.47 | 0.18 | 0.24 |
SE 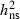
|
0.09 | 0.09 | 0.09 | 0.05 | 0.06 |
| Qst | 0.01 | 0.02 | 0.01 | 0.04 | 0.03 |
| SE Qst | 0.02 | 0.02 | 0.02 | 0.04 | 0.04 |
| Mean | 51 | 55 | 62 | 1.43 | 2.05 |
| CVA (%) | 48 | 40 | 40 | 32 | 24 |
VA, Additive genetic variation; 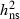 , Narrow sense heritability, Qst, Genetic differentiation between populations; CVA, Additive genetic coefficient of variation.
, Narrow sense heritability, Qst, Genetic differentiation between populations; CVA, Additive genetic coefficient of variation.
Highly significant levels of additive genetic variation were observed within populations for all traits. Narrow-sense heritability (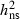 ) estimates for PDS ranged from 0.37 to 0.52 (Table 2). Heritability estimates for the necrosis scores were lower (Table 2), but necrosis scores were highly genetically correlated with PDS at both sites (Table 3), verifying that PDS in the crown is a reliable approach to assess damages caused by H. pseudoalbidus. No significant genotype by environment interaction variation was present in any of the years 2008–2010 for PDS or for necrosis score 2008–2009 (not shown). This corresponds to very high additive genetic correlations between PDS assessed at the two different sites, rA = 1.00 (2008), 0.97 (2009) and 1.00 (2010), estimated with a standard error of 0.06 in all 3 years. The ranking of the mother trees based on susceptibility (PDS) of their OP progeny was thus independent of test site. The corresponding values for necrosis score were also relatively high, rA = 0.91 (0.20) and 0.73 (0.20) for 2008 and 2009, respectively. Additionally, the relative rank in susceptibility was consistent among the progeny-tested trees over years in correspondence with high additive genetic correlations between years (Table 3).
) estimates for PDS ranged from 0.37 to 0.52 (Table 2). Heritability estimates for the necrosis scores were lower (Table 2), but necrosis scores were highly genetically correlated with PDS at both sites (Table 3), verifying that PDS in the crown is a reliable approach to assess damages caused by H. pseudoalbidus. No significant genotype by environment interaction variation was present in any of the years 2008–2010 for PDS or for necrosis score 2008–2009 (not shown). This corresponds to very high additive genetic correlations between PDS assessed at the two different sites, rA = 1.00 (2008), 0.97 (2009) and 1.00 (2010), estimated with a standard error of 0.06 in all 3 years. The ranking of the mother trees based on susceptibility (PDS) of their OP progeny was thus independent of test site. The corresponding values for necrosis score were also relatively high, rA = 0.91 (0.20) and 0.73 (0.20) for 2008 and 2009, respectively. Additionally, the relative rank in susceptibility was consistent among the progeny-tested trees over years in correspondence with high additive genetic correlations between years (Table 3).
Table 3.
Phenotypic (rp), additive genetic (rA) and environmental (re) correlation estimates between percent damage score (PDS) and necroses scores in the 3 years
| Trait | Year | Trait | Year | rp | rA | re | |||
|---|---|---|---|---|---|---|---|---|---|
| Site: Silkeborg | |||||||||
| PDS | 2008 | PDS | 2009 | 0.75 | 0.01 | 0.98 | 0.02 | 0.55 | 0.06 |
| PDS | 2010 | 0.68 | 0.01 | 1.00 | 0.02 | 0.47 | 0.06 | ||
| Necrosis | 2008 | 0.37 | 0.02 | 0.79 | 0.09 | 0.19 | 0.07 | ||
| 2009 | 0.44 | 0.02 | 0.96 | 0.08 | 0.20 | 0.07 | |||
| PDS | 2009 | PDS | 2010 | 0.74 | 0.01 | 0.97 | 0.02 | 0.58 | 0.05 |
| Necrosis | 2008 | 0.35 | 0.02 | 0.82 | 0.09 | 0.14 | 0.08 | ||
| 2009 | 0.50 | 0.02 | 1.00 | 0.07 | 0.28 | 0.07 | |||
| PDS | 2010 | Necrosis | 2008 | 0.28 | 0.02 | 0.73 | 0.11 | 0.10 | 0.06 |
| 2009 | 0.38 | 0.02 | 0.99 | 0.08 | 0.14 | 0.07 | |||
| Site: Randers | |||||||||
| PDS | 2008 | PDS | 2009 | 0.71 | 0.01 | 1.00 | 0.01 | 0.41 | 0.09 |
| PDS | 2010 | 0.65 | 0.01 | 0.91 | 0.04 | 0.39 | 0.09 | ||
| Necrosis | 2008 | 0.40 | 0.02 | 0.79 | 0.10 | 0.25 | 0.07 | ||
| 2009 | 0.32 | 0.02 | 0.80 | 0.09 | 0.04 | 0.10 | |||
| PDS | 2009 | PDS | 2010 | 0.74 | 0.01 | 0.96 | 0.02 | 0.54 | 0.07 |
| Necrosis | 2008 | 0.31 | 0.02 | 0.71 | 0.11 | 0.16 | 0.07 | ||
| 2009 | 0.28 | 0.02 | 0.73 | 0.10 | 0.04 | 0.09 | |||
| PDS | 2010 | Necrosis | 2008 | 0.32 | 0.02 | 0.65 | 0.13 | 0.20 | 0.06 |
| 2009 | 0.27 | 0.02 | 0.69 | 0.11 | 0.05 | 0.08 | |||
Standard deviation in italics.
The estimated breeding values of PDS of the mother trees revealed large tree-to-tree variation. Values based at both test sites ranged from −8% to 110% in (2008 data), 1 to 112% (2009 data) and 7% to 112% (2010-data), as shown in Fig. 1. Low values of PDS-breeding values correspond to high levels of resistance. Values below 0% (or above 100%) are attributed to estimation error and should be truncated to 0 or 100%, respectively. The substantial variation in breeding values corresponds to high coefficients of genetic variation (CVA) ranging from 37% to 61% in different years and sites (Table 2). Mean PDS increased at both sites from 2008 to 2010 (Table 2), indicating that trees at the two sites on average experienced declining health with a high level of damage in 2010. The distribution of breeding values therefore shifted towards increased damage score during 2008–2010, reflecting progressed damage of the trees in the field trials (Fig. 1). Only a single of the 101 tested mother trees had breeding value below 10% based on the 2010 assessment.
Figure 1.
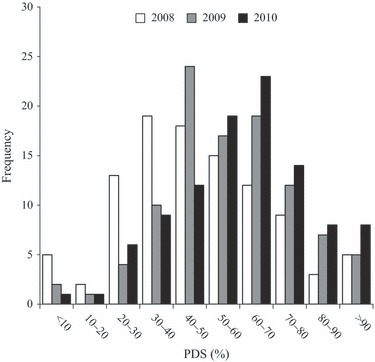
Frequency of individual breeding values of the 101 tested mother trees (PDS, Percent Damage Score, combined from both test sites).
Discussion
Levels of genetic variation in susceptibility
The present study revealed high levels of damage in offspring from Danish populations of F. excelsior corresponding to symptoms caused by H. pseudoalbidus. Previously, we have found substantial variation in the degree of susceptibility among clones from Danish forests with broad-sense heritability estimates and coefficients of genetic variation, 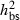 = 0.25–0.54 and CVG = 0.38–0.87, depending on year and site (McKinney et al. 2011). Stener (2007) analysed 100 clones in a Swedish trial and found similar broad-sense heritability estimates (
= 0.25–0.54 and CVG = 0.38–0.87, depending on year and site (McKinney et al. 2011). Stener (2007) analysed 100 clones in a Swedish trial and found similar broad-sense heritability estimates (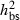 = 0.28–0.52). In the present study, we confirm the presence of genetic variation and show that this variation is to a large degree of additive nature with narrow-sense heritability estimates of similar size
= 0.28–0.52). In the present study, we confirm the presence of genetic variation and show that this variation is to a large degree of additive nature with narrow-sense heritability estimates of similar size 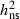 = 0.37–0.52 and CVA = 37–61%. The results are in line with the estimates of narrow-sense heritability recently found in two Lithuanian progeny trials
= 0.37–0.52 and CVA = 37–61%. The results are in line with the estimates of narrow-sense heritability recently found in two Lithuanian progeny trials 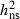 = 0.40–0.49 (Pluiura et al. 2011). The adaptive potential of the mother trees, measured as the expected susceptibility of their progeny if grown under conditions similar to the field sites, varied in our study almost from 0% to 100%. No trees are expected to produce completely healthy offspring, because all trees had breeding values >0% 6 year after planting (2010). This corresponds to results from a previous study on clones, where all clones showed signs of infection, but symptoms and damage levels developed very differently among clones (McKinney et al. 2011, in press). The estimated heritability for resistance of F. excelsior against H. pseudoalbidus is relatively high when compared with the published heritabilities of resistance in North American pines against rust fungi (Dieters et al. 1996), but similar high heritability was reported by Milgate et al. (2005) on Eucalyptus globulus susceptibility to Mycosphaerella nubilosa.
= 0.40–0.49 (Pluiura et al. 2011). The adaptive potential of the mother trees, measured as the expected susceptibility of their progeny if grown under conditions similar to the field sites, varied in our study almost from 0% to 100%. No trees are expected to produce completely healthy offspring, because all trees had breeding values >0% 6 year after planting (2010). This corresponds to results from a previous study on clones, where all clones showed signs of infection, but symptoms and damage levels developed very differently among clones (McKinney et al. 2011, in press). The estimated heritability for resistance of F. excelsior against H. pseudoalbidus is relatively high when compared with the published heritabilities of resistance in North American pines against rust fungi (Dieters et al. 1996), but similar high heritability was reported by Milgate et al. (2005) on Eucalyptus globulus susceptibility to Mycosphaerella nubilosa.
Presence of genetic variation in susceptibility to emerging pathogens has been observed in natural populations of other woody species
Clones of European Ulmus species have been reported to exhibit difference in resistance to Ophiostoma ulmi and Ophiostoma novo-ulmi (Solla et al. 2005). Ulmus glabra has been severely decimated because of the disease, but old U. glabra trees are occasionally found and small trees are still commonly found in Denmark (Nielsen and Kjaer 2010). In Castanea dentata (American chestnut), genetic variation in susceptibility towards the blight fungus Cryphonectria parasitica was also observed (Griffin 2000), but C. dentata has still been severely decimated since the early 1900s. The frequency distribution of breeding values for susceptibility to the above-mentioned diseases on broadleaved species have, to our knowledge, not been estimated in natural populations of these species in a way comparable to that used in the present study. Several studies have investigated natural genetic resistance in North American white pines (Pinus subgenus Strobus) against pine blister rust Cronartium ribicola that has caused devastated damage in many pine populations since it was introduced to North America a century ago (Geils et al. 2010). Resistant trees were observed at only low frequency in the North American pine populations and the heritability of susceptibility was estimated to be relatively low based on early progeny studies (Bingham et al. 1969). Still, through selection and crossing of nonsusceptible trees, it was possible to develop a resistance breeding programme based on a combination of dominant R-gene(s) conferring hypersensitive response and quantitative resistance expected to be of polygenic nature (King et al. 2010).
The genetic and allelic background of the apparent resistance in F. excelsior remains unknown. The presence of high levels of genetic variation in susceptibility to the disease is intriguing, because we assume that H. pseudoalbidus is a recent invasive species in the region. Genes responsible for genetic resistance may have been present in the natural gene pool as neutral variation (in mutation-drift balance; cf. Kimura 1983) made possible by high effective population numbers in the wind-pollinated species (Namkoong 1991). Alternatively, the genes involved in the resistance may have pleiotropic effects on traits under balancing selection. In a previous study, we found a surprisingly strong genetic correlation between susceptibility and phenology (McKinney et al. 2011). Correlations between susceptibility and phenology were also observed in Castanea susceptibility to Phytophthora cinnamomi (Miranda-Fontaina et al. 2007) and in case of Ulmus sp. against Ophiostoma novo-ulmi (Ghelardini 2007). An alternative hypothesis is that selection against the pathogen has already been ongoing in parts of the natural distribution area of F. excelsior prior to the recent outbreak of the disease. A recent investigation of herbarium specimens has suggested that H. pseudoalbidus has been present in the southern part of the F. excelsior distribution area for at least 30 years, and before the first observations of symptoms (Queloz et al. 2010). Further, H. pseudoalbidus is closely related to H. albidus, which has been known as a harmless decomposer of ash foliage since 1896 (Kowalski and Holdenrieder 2009; Queloz et al. 2010). When and how H. pseudoalbidus emerged remains unknown.
Evolutionary significance
The resistance of the native F. excelsior populations must be regarded as being low. Of the 101 tested mother trees, no trees are expected to produce completely healthy offspring, only a single tree is expected to produce almost healthy offspring (estimated PDS-breeding value below 10%) and only four trees are expected to produce fairly healthy offspring (estimated PDS-breeding value below 20%) based on the 2010 assessment. This is in agreement with the findings in the clonal trials (McKinney et al. 2011), where only one out of 39 tested clones had PDS below 10% (and 3 clones below 20%) based on assessments in 2009. Unfortunately, both studies thus point towards a scenario where the majority of F. excelsior trees will become increasingly damaged, and ecosystems with F. excelsior as the dominating or keystone species risk temporary collapse. This process is accelerated by the present forest practice in Denmark where mature Ash trees are being intensively logged to harvest the valuable trees before their wood is damaged by the fungal infection.
However, the high level of additive genetic variance in susceptibility (with estimated PDS-breeding values from 8% to 100%), the high narrow-sense heritability and the low level of genotype by environment interactions suggest that the potential for recovery is present in the surveyed natural populations. Strong natural selection is expected to take place in favour of trees with low susceptibility. Given the high level of additive variation, the response in fitness can be substantial, if trees with low PDS-breeding values contribute more offspring to the next generation than average for mature trees in the population (Walsh 2008). The actual effects are difficult to predict, because the relationship between PDS and the expected relative contribution to next generation remains unknown. We did observe many dying trees (total accumulated mortality during the first 6 years in the present trial was 40%, where offspring from some mother trees had experiences as high as 84% mortality). This leads us to expect a strong positive relation between PDS and fitness, which we expect to trigger rapid response towards increased fitness of the subsequent generation. The trees we have studied are still not seeding, and we are therefore reluctant to estimate the expected contemporary quantitative evolution. This must await empirical data that allows us to infer on the relations between PDS-breeding values and fertility and mortality over time.
The importance of the observed genetic variation should be analysed in a landscape genetic context. Our study suggests that low-susceptible trees are relatively rare and distributed randomly across the landscape. Strong selection in favour of trees with low susceptibility may therefore lead to significant spatial genetic structures, where seed and pollen flow distances, compared to the distance between future surviving trees, will determine the size of effective breeding populations in future landscapes. Inbreeding depression is a potential risk if the effective population number becomes very low (Lienert 2004). This alone may decrease future fitness of the species, and thereby render the species more susceptible to other biotic stresses and further reduce the ability of F. excelsior to compete with other species for its ecological niches. Fortunately, F. excelsior trees have been reported to connect even in a fragmented landscape through pollen flow (Bacles and Ennos 2008). The overall effect on future population structure will probably vary substantially between landscapes depending on a priori species density and type of landscape management.
Our results suggest that future restoration of F. excelsior forests based on identification and breeding of hyposensitive trees is an important option. The expected effects of artificial selection and breeding for trees with low level of susceptibility can, in principle, be estimated based on the breeders equation Gain = i*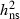 √Vp (where i denoted the selection intensity and Vp the phenotypic variance; Falconer and Mackay 1996). The idealized infinite allele model behind the breeder's equation (Walsh 2008) may be a crude approximation in the present case, but the estimated high level of
√Vp (where i denoted the selection intensity and Vp the phenotypic variance; Falconer and Mackay 1996). The idealized infinite allele model behind the breeder's equation (Walsh 2008) may be a crude approximation in the present case, but the estimated high level of 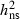 and Vp suggests that substantial progress can be obtained from selection and breeding. Given the low expected frequency of hyposensitive trees, substantial progress will depend on precise selection made by effectively screening and testing of a large number (thousands) of trees, because high selection intensity will otherwise result is an unacceptable low number of selected trees left to form the basis for future restoration programs.
and Vp suggests that substantial progress can be obtained from selection and breeding. Given the low expected frequency of hyposensitive trees, substantial progress will depend on precise selection made by effectively screening and testing of a large number (thousands) of trees, because high selection intensity will otherwise result is an unacceptable low number of selected trees left to form the basis for future restoration programs.
The robustness of the observed partial resistance needs to be studied further before more decisive conclusions are drawn regarding the potential of selection and breeding for conservation of F. excelsior forests. The observed continuous distribution in PDS-breeding values combined with high levels of additive genetic variation and narrow-sense heritability observed in the present study, suggests presence of a partial resistance system based on multiple loci. Results from controlled inoculations in a recent work (McKinney et al. in press) support that resistance is likely to be of a partial and quantitative nature. This observation is important, because yet another unknown risk is that the pathogen evolves new virulent strains. Recent studies by Rytkönen et al. (2011) and Kraj et al. (in press) have identified relative high levels of genetic variation between isolates of C. fraxinea (the asexual form of H. pseudoalbidus), but at present, the origin and evolution of the pathogen is basically unknown. Nevertheless, our general observations so far (cf. above) point towards a partial host resistance of predominantly quantitative nature against an apparent necrotrophic pathogen, factors that would normally indicate a resistance system that does not rely on simple R alleles to which the pathogen can easily adapt (Collinge et al. 2008).
Acknowledgments
The work was supported by Godfred Birkedal Hartmanns Familiefond, the Danish Forest and Nature Agency (Praksisnære forsøg) and Nordic Forest Research Co-operation Committee. Viggo Jensen provided valuable technical assistance in the field. David B. Collinge and Iben M. Thomsen are acknowledged for their valuable discussion and comments on the manuscript. Three anonymous reviewers also provided important comments. The original data for this study will be stored at Dryad – doi: 10.5061/dryad.62v0p8q2.
Conflict of interest
The authors declare no conflict of interest.
Literature cited
- Aitken SN, Yeaman S, Holliday JA, Wang TL, Curtis-McLane S. Adaptation, migration or extirpation: climate change outcomes for tree populations. Evolutionary Application. 2008;1:95–111. doi: 10.1111/j.1752-4571.2007.00013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacles CFE, Ennos RA. Paternity analysis of pollen-mediated gene flow for Fraxinus excelsior L. in a chronically fragmented landscape. Heredity. 2008;101:368–380. doi: 10.1038/hdy.2008.66. [DOI] [PubMed] [Google Scholar]
- Bakys R, Vasaitis R, Barklund P, Thomsen I, Stenlid J. Occurrence and pathogenicity of fungi in necrotic and nonsymptomatic shoots of declining common ash (Fraxinus excelsior) in Sweden. European Journal of Forest Research. 2009a;128:51–60. [Google Scholar]
- Bakys R, Vasaitis R, Barklund P, Ihrmark K, Stenlid J. Investigations concerning the role of Chalara fraxinea in declining Fraxinus excelsior. Plant Pathology. 2009b;58:284–292. [Google Scholar]
- Barrett LG, Thrall PH, Burdon JJ, Linde CC. Life history determines genetic structure and evolutionary potential of host-parasite interactions. Trends in Ecology & Evolution. 2008;23:678–685. doi: 10.1016/j.tree.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham RT, Olson F-J, Becker WA, Marsden MA. Breeding blister rust resistance western white pine. V. Estimates of heritability, combining ability, and genetic advance based on tester matings. Silvae Genetica. 1969;18:28–33. [Google Scholar]
- Burdon RD. Genetic correlation as a concept for studying genotype–environment interaction in forest tree breeding. Silvae Genetica. 1977;26:168–175. [Google Scholar]
- Burdon RD. Genetic diversity and disease resistance: some considerations for research, breeding, and deployment. Canadian Journal of Forest Research. 2001;31:596–606. [Google Scholar]
- Burdon JJ, Thrall PH. Coevolution of plants and their pathogens in natural habitats. Science. 2009;324:755–756. doi: 10.1126/science.1171663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdon JJ, Thrall PH, Ericson L. The current and future dynamics of disease in plant communities. Annual Review of Phytopathology. 2006;44:19–39. doi: 10.1146/annurev.phyto.43.040204.140238. [DOI] [PubMed] [Google Scholar]
- Collinge D, Lund O, Thordal-Christensen H. What are the prospects for genetically engineered, disease resistant plants? European Journal of Plant Pathology. 2008;121:217–231. [Google Scholar]
- Dieters MJ, Hodge GR, White TL. Genetic parameter estimates for resistance to rust (Cronartium quercuum) infection from full-sib tests of slash pine (Pinus elliottii), modelled as functions of rust incidence. Silvae Genetica. 1996;45:235–242. [Google Scholar]
- Dowkiw A, Bastien C. Presence of defeated qualitative resistance genes frequently has major impact on quantitative resistance to Melampsora larici-populina; leaf rust in P. interamericana hybrid poplars. Tree Genetics & Genomes. 2007;3:261–274. [Google Scholar]
- Eriksson G, Namkoong G, Roberds JH. Dynamic gene conservation for uncertain futures. Forest Ecology and Management. 1993;62:15–37. [Google Scholar]
- Ersoz ES, Wright MH, Gonzalez-Martinez SC, Langley CH, Neale DB. Evolution of disease response genes in Loblolly Pine: insights from candidate genes. PLoS ONE. 2010;5:e14234. doi: 10.1371/journal.pone.0014234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Commission. Interpretation Manual of European Union habitats. EUR27. Bruxelles, Belgium: DG Environment, Nature and Biodiversity, The European Commission; 2007. p. 142. [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4th edn. Harlow, UK: Longman Scientific and Technical; 1996. p. 438. [Google Scholar]
- FRAXIGEN. Ash Species in Europe: Biological Characteristics and Practical Guidelines for Sustainable Use. Oxford, UK: Department of Plant Sciences, University of Oxford; 2005. p. 128. [Google Scholar]
- Funda T, Lstiburek M, Lachout P, Klapste J, El-Kassaby YA. Optimization of combined genetic gain and diversity for collection and deployment of seed orchard crops. Tree Genetics & Genomes. 2009;5:583–593. [Google Scholar]
- Geils BW, Hummer KE, Hunt RS. White pines, Ribes, and blister rust: a review and synthesis. Forest Pathology. 2010;40:147–185. [Google Scholar]
- Gerard PR, Fernandez-Manjarres JF, Frascaria-Lacoste N. Temporal cline in a hybrid zone population between Fraxinus excelsior L and Fraxinus angustifolia Vahl. Molecular Ecology. 2006;15:3655–3667. doi: 10.1111/j.1365-294X.2006.03032.x. [DOI] [PubMed] [Google Scholar]
- Ghelardini L. Bud burst phenology, dormancy release and susceptibility to Dutch elm disease in elms (Ulmus spp.) Acta Universitatis Agriculturae Sueciae. 2007;2007:134. [Google Scholar]
- Gilmour AR, Gogel BJ, Cullis BR, Thompson R. ASReml User Guide Release 3.0. Hemel Hempstead, UK: VSN International Ltd; 2009. [Google Scholar]
- Griffin GJ. Blight control and restoration of the American chestnut. Journal of Forestry. 2000;98:22–27. [Google Scholar]
- Hansen JK, Wellendorf H, Kjaer ED. Low cost improvement of Coastal Douglas-fir (Pseudotsuga menziesii var. menziesii (Mirb.) franco) by application of the breeding seed orchard approach in Denmark. Silvae Genetica. 2005;54:218–227. [Google Scholar]
- Houle D. Comparing evolvability and variability of quantitative traits. Genetics. 1992;130:195–204. doi: 10.1093/genetics/130.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husson C, Scala B, Caël O, Frey P, Feau N, Ioos R, Marçais B. Chalara fraxinea is an invasive pathogen in France. European Journal of Plant Pathology. 2011;130:311–324. [Google Scholar]
- Jump AS, Marchant R, Penuelas J. Environmental change and the option value of genetic diversity. Trends in Plant Science. 2009;14:51–58. doi: 10.1016/j.tplants.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press; 1983. [Google Scholar]
- King JN, David A, Noshad D, Smith J. A review of genetic approaches to the management of blister rust in white pines. Forest Pathology. 2010;40:292–313. [Google Scholar]
- Kowalski T. Chalara fraxinea sp nov associated with dieback of ash (Fraxinus excelsior) in Poland. Forest Pathology. 2006;36:264–270. [Google Scholar]
- Kowalski T, Holdenrieder O. The teleomorph of Chalara fraxinea, the causal agent of ash dieback. Forest Pathology. 2009;39:304–308. [Google Scholar]
- Kraj W, Zarek M, Kowalski T. Genetic variability of Chalara fraxinea, dieback cause of European ash (Fraxinus excelsior L. Mycological Progress. in press. doi: 10.1007/s11557-010-0724-z. Available from: http://www.springerlink.com/content/h838779388401575/ (accessed on 23 November 2011) [Google Scholar]
- Kuparinen A, Savolainen O, Schurr FM. Increased mortality can promote evolutionary adaptation of forest trees to climate change. Forest Ecology and Management. 2010;259:1003–1008. [Google Scholar]
- Lefort F, Brachet S, Frascaria-Lacoste N, Edwards KJ, Douglas GC. Identification and characterization of microsatellite loci in ash (Fraxinus excelsior L) and their conservation in the olive family (Oleaceae) Molecular Ecology. 1999;8:1088–1090. [Google Scholar]
- Lienert J. Habitat fragmentation effects on fitness of plant populations – a review. Journal for Nature Conservation. 2004;12:53–72. [Google Scholar]
- McKinney LV, Nielsen LR, Hansen JK, Kjaer ED. Presence of natural genetic resistance in Fraxinus excelsior (Oleraceae) to Chalara fraxinea (Ascomycota): an emerging infectious disease. Heredity. 2011;106:788–797. doi: 10.1038/hdy.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney LV, Thomsen IM, D Kjær E, Nielsen LN. Genetic resistance to Chalara fraxinea limits fungal growth and symptom occurrence in Fraxinus excelsior. Forest Pathology. in press. doi: 10.1111/j.1439-0329.2011.00725.x. Available from: http://onlinelibrary.wiley.com/doi/10.1111/j.1439-0329.2011.00725.x/abstract (accessed on 23 November 2011) [Google Scholar]
- Milgate AW, Potts BM, Joyce K, Mohammed C, Vaillancourt RE. Genetic variation in Eucalyptus globulus for susceptibility to Mycosphaerella nubilosa and its association with tree growth. Australasian Plant Pathology. 2005;34:11–18. [Google Scholar]
- Miranda-Fontaina ME, Fernandez-Lopez J, Vettraino AM, Vannini A. Resistance of Castanea clones to Phytophthora cinnamomi: testing and genetic control. Silvae Genetica. 2007;56:11–21. [Google Scholar]
- Namkoong G. Maintaining genetic diversity in breeding for resistance in forest trees. Annual Review of Phytopathology. 1991;29:325–342. [Google Scholar]
- Nielsen LR, Kjaer ED. Gene flow and mating patterns in individuals of wych elm (Ulmus glabra) in forest and open land after the influence of Dutch elm disease. Conservation Genetics. 2010;11:257–268. [Google Scholar]
- Nord-Larsen T, Johannsen VK, Vesterdal L, Jørgensen BB, Bastrup-Birk A. Skove og Plantager 2008 [Forests and Plantations 2008. In Danish] Copenhagen, Denmark: University of Copenhagen; 2009. ISBN 978-87-7903-448-8 (internet) [Google Scholar]
- Pautasso M, Dehnen-Schmutz K, Holdenrieder O, Pietravalle S, Salama N, Jeger MJ, Lange E, et al. Plant health and global change – some implications for landscape management. Biological Reviews. 2010;85:729–755. doi: 10.1111/j.1469-185X.2010.00123.x. [DOI] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit RJ, Hampe A. Some evolutionary consequences of being a tree, Annu. Annual review of ecology, evolution, and systematics. 2006;37:187–214. [Google Scholar]
- Pluiura A, Lygis V, Suchockas Y, Bartevicius E. Performance of twenty-four European excelsior populations in three Lithuanian progeny trials with a special emphasis on resistance to Chalara fraxinea. Baltic Forestry. 2011;17:17–33. [Google Scholar]
- Queloz V, Grünig CR, Berndt R, Kowalski T, Sieber TN, Holdenrieder O. Cryptic speciation in Hymenoscyphus albidus. Forest Pathology. 2010;41:133–142. [Google Scholar]
- Ritland K. Extensions of models for the estimation of mating systems using n independent loci. Heredity. 2002;88:221–228. doi: 10.1038/sj.hdy.6800029. [DOI] [PubMed] [Google Scholar]
- Roberds JH, Bishir JW. Risk analyses in clonal forestry. Canadian Journal of Forest Research. 1997;27:425–432. [Google Scholar]
- Rytkönen A, Lilja A, Drenkhan R, Gaitnieks T, Hantula J. First record of Chalara fraxinea in Finland and genetic variation among isolates sampled from Aland, mainland Finland Estonia and Latvia. Forest Pathology. 2011;41:169–174. [Google Scholar]
- SAS Institute Inc. SAS/STAT User Guide. Gary, IN, USA: SAS Institute Inc; 2008. [Google Scholar]
- Skovsgaard JP, Thomsen IM, Skovgaard IM, Martinussen T. Associations among symptoms of dieback in evenaged stands of ash (Fraxinus excelsior L) Forest Pathology. 2009;40:7–18. [Google Scholar]
- Solla A, Bohnens J, Collin E, Diamandis S, Franke A, Gil L, Buron M, et al. Screening European elms for resistance to Ophiostoma novo-ulmi. Forest Science. 2005;51:134–141. [Google Scholar]
- Spitze K. Population structure in daphnia obtusa: quantitative genetic and allozymic variation. Genetics. 1993;135:367–374. doi: 10.1093/genetics/135.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stener L-G. Studie av klonskillnader i känslighet för askskottsjuka[A Study on clonal variation in susceptibility to Ash die-back] Sweden: SkogForsk; 2007. Arbetsrapport 648. ISSN 1404-305x (in Swedish) [Google Scholar]
- Stukenbrock EH, Mcdonald BA. The origins of plant pathogens in agro-ecosystems. Annual review of Phytopathology. 2008;46:75–100. doi: 10.1146/annurev.phyto.010708.154114. [DOI] [PubMed] [Google Scholar]
- Valen VL. A new evolutionary law. Evolutionary Theory. 1973;1:1–30. [Google Scholar]
- Walsh B. Handbook of Statistical Genetics. Chichester, England: John Wiley & Sons, Ltd; 2008. Evolutionary quantitative genetics. [Google Scholar]
- White TL, Hodge GR. Predicting Breeding Values: with Applications in Forest Tree Improvement. Boston, MA, USA: Kluwer Academic Pub; 1989. p. 388. [Google Scholar]