

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a condition in which excess fat accumulates in the liver of a patient without a history of alcohol abuse. Nonalcoholic steatohepatitis (NASH), a severe form of NAFLD, can progress to liver cirrhosis and hepatocellular carcinoma. NAFLD is regarded as a hepatic manifestation of metabolic syndrome and incidence has been increasing worldwide in line with the increased prevalence of obesity, type 2 diabetes, and hyperlipemia. Animal models of NAFLD/NASH give crucial information, not only in elucidating pathogenesis of NAFLD/NASH but also in examining therapeutic effects of various agents. An ideal model of NAFLD/NASH should correctly reflect both hepatic histopathology and pathophysiology of human NAFLD/NASH. Animal models of NAFLD/NASH are divided into genetic, dietary, and combination models. In this paper, we review commonly used animal models of NAFLD/NASH referring to their advantages and disadvantages.
Keywords: Animal model, Nonalcoholic fatty liver disease, Nonalcoholic steatohepatitis, Metabolic syndrome, Histopathology
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a condition in which excess fat accumulates in the liver of a patient without a history of alcohol abuse[1]. NAFLD is classified into simple steatosis and nonalcoholic steatohepatitis (NASH). In NASH, not only steatosis but also intralobular inflammation and hepatocellular ballooning are present, often accompanied by progressive fibrosis[2]. Long-standing NASH may progress to liver cirrhosis, and hepatocellular carcinoma (HCC) may be an outcome[3-5].
NAFLD is regarded as a hepatic manifestation of metabolic syndrome[6,7]. NAFLD has been increasing worldwide over recent decades in line with the increased prevalence of obesity, type 2 diabetes, and hyperlipemia. NAFLD/NASH is currently regarded as the most common chronic liver disease worldwide. It is estimated that about 20% of all adults have NAFLD and 2%-3% of adults have NASH[8].
The pathogenesis of NASH has not been completely elucidated, and treatments for NASH other than lifestyle modification by diet and exercise have not been fully established[5]. Studies of NAFLD/NASH using human materials have limitations, because the occurrence and progression of NAFLD/NASH require a long period of several decades, and ethical limitations exist in administering drugs to patients or collecting liver tissues from patients. Animal models of NAFLD/NASH give crucial information, not only in elucidating the pathogenesis of NAFLD/NASH but also in examining therapeutic effects of various agents. These animal models need to reflect correctly both the histopathology and pathophysiology of human NAFLD/NASH. Recently, several review articles on animal models of NAFLD/NASH have been published[9,10]. In this paper, we firstly review the histopathology and pathogenesis of NAFLD/NASH, and thereafter, we review current animal models of NAFLD/NASH referring to their advantages and disadvantages, with emphasis on a fructose-enriched diet model that was established by us.
HISTOPATHOLOGY OF NAFLD
The most important histological characteristic of NAFLD is steatosis of hepatocytes. Steatosis of hepatocytes is classified into macrovesicular and microvesicular. Macrovesicular steatosis is characterized by large vacuoles occupying almost the entire cytoplasm and pushing the nucleus to the periphery of the cell. Microvesicular steatosis is characterized by multiple small lipid vacuoles, and the nucleus is located at the center of the cell. Typically, steatosis in NAFLD is centrilobular and macrovesicular. However, steatosis may be present throughout the lobule, and microvesicular steatosis may also be present. Steatosis in more than 5% of hepatocytes is necessary for a diagnosis of NAFLD[11].
In NASH, not only steatosis but also intralobular inflammation and hepatocellular ballooning are present, and this is usually accompanied by fibrosis. Mallory’s hyaline (MH) may also be present. Intralobular inflammation in NASH is typically mild and of a mixed type and includes a small number of lymphocytes, macrophages, and neutrophils. Neutrophils tend to infiltrate in the area of marked steatosis and around MH. Portal inflammation is usually mild; however, relatively intense chronic inflammation may be present in the portal area.
Hepatocellular ballooning is a form of liver cell injury recognized as a swollen hepatocyte with a rarefied cytoplasm. It is most apparent near steatotic liver cells, typically in zone 3.
MH is an eosinophilic and amorphous structure in the cytoplasm of hepatocytes and is observed in alcoholic hepatitis, NASH, chronic cholestasis, and HCC[2]. MH in NASH is most frequently found in the areas of perisinusoidal fibrosis in zone 3 and may be accompanied by neutrophilic infiltration. MH in NASH is usually less distinct than that in alcoholic hepatitis. Immunohistochemically, MH is positive for cytokeratin (CK) 8, CK 18, p62, and ubiquitin[2].
Other pathological features that are often observed in NASH include fat cysts and lipogranulomas, and glycogenated nuclei frequently occur in hepatocytes in zone 1. Furthermore, megamitochondria and iron deposition may be observed in NASH.
Fibrosis usually originates in the perisinusoidal regions of zone 3 (perisinusoidal fibrosis) and may also be present in the periportal area. As the disease progresses, bridging fibrosis and liver cirrhosis may develop. In the process, steatosis and lesion activity may resolve, resulting in a diagnosis of “cryptogenic cirrhosis” (burn-out NASH).
Brunt et al[12] proposed the grading and staging system of NASH, and the NASH Clinical Research Network designed and validated the NAFLD activity score[13]. These systems are frequently used in both clinical studies and animal experiments. Intralobular changes are milder and portal inflammation is more severe in pediatric NAFLD than in adult NAFLD[14,15]. Furthermore, steatosis in pediatric NAFLD is not necessarily predominant in zone 3[15,16].
PATHOGENESIS OF NAFLD/NASH
The “two-hit” hypothesis proposed by Day et al[17] is widely accepted as the pathogenesis of NAFLD/NASH; the first hit causes fat accumulation in hepatocytes, and the second hit causes inflammation and fibrosis. Fat accumulation in the liver is closely associated with metabolic derangements that are related to central obesity and insulin resistance[18]. The most reproducible risk factors for NAFLD/NASH are central obesity, insulin resistance, fasting hyperglycemia, and hypertriglyceridemia[19], and NAFLD/NASH is regarded as a hepatic manifestation of metabolic syndrome[6,7].
Hepatic steatosis occurs when the rate of import or synthesis of fatty acids by hepatocytes exceeds the rate of export or catabolism[20,21]. Accordingly, the following 4 mechanisms are possible causes of lipid accumulation within the liver: (1) increased delivery and uptake into hepatocytes of long-chain fatty acids (LCFA) due to excess dietary intake or release from adipose tissue; (2) increased de novo hepatic LCFA and triglyceride synthesis; (3) failure of very low-density lipoprotein (VLDL) synthesis and triglyceride export; and (4) failure of LCFA elimination due to impaired hepatic mitochondrial β-oxidation[22].
Once steatosis has developed, the liver is sensitized; thus, an inflammatory response may be precipitated by a variety of stimuli[22]. Oxidative stress, pro-inflammatory cytokine [e.g., tumor necrosis factor (TNF)-α]-mediated hepatocyte injury, altered lipid partitioning and hepatotoxicity mediated by free fatty acids, abnormal intrahepatic cholesterol loading, hyperinsulinemia, hyperleptinemia, hypoadiponectinemia, and apoptosis are all thought to be important second hits causing NASH[22-24].
ANIMAL MODELS OF NAFLD/NASH
An ideal animal model of NAFLD/NASH should reflect hepatic histopathology and pathophysiology of human NAFLD/NASH. Accordingly, the liver of the animal model of NASH should show steatosis, intralobular inflammation, hepatocellular ballooning, and, ideally, perisinusoidal fibrosis in zone 3 and susceptibility to liver tumors. Furthermore, the animal should show metabolic abnormalities such as obesity, insulin resistance, fasting hyperglycemia, dyslipidemia, and altered adipokine profile. It is questionable whether the results of a study using an animal model that does not completely fulfill these conditions can be extrapolated to human disease. Animal models of NAFLD/NASH are classified into genetic models, nutritional models, and combination models of genetic and nutritional factors. Numerous animal models of NAFLD/NASH have been reported to date; however, no animal model completely reflects hepatic histopathology and pathophysiology of human NAFLD/NASH. It is, therefore, important to select the animal model that best conforms to the aim of the study. Biochemical and pathological characteristics of animal models described in this paper are summarized in Table 1. As described below, in many of the genetic models of NASH [e.g., sterol regulatory element binding protein (SREBP)-1c transgenic mice and phosphatase and tensin homologue deleted on chromosome 10 (PTEN) null mice], hepatic steatosis occurs first, and steatohepatitis develops later. Ob/ob, db/db, and KK-Ay mice do not progress to steatohepatitis spontaneously. On the other hand, in a dietary model induced by methionine and choline deficiency, steatohepatitis occurs very quickly.
Table 1.
Biochemical and pathological characteristics of animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis
| Model | Obesity | Insulin resistance | Steatosis | Steatohepatitis | Fibrosis |
| SREBP-1c transgenic mice | No (decreased adiposity) | Yes | Yes | Yes | Yes |
| Ob/ob mice | Yes | Yes | Yes | No (does not develop spontaneously) | No (resistant to fibrosis) |
| Db/db mice | Yes | Yes | Yes | No (does not develop spontaneously) | No (does not develop spontaneously) |
| KK-Ay mice | Yes | Yes | Yes | No (does not develop spontaneously) | No (does not develop spontaneously) |
| PTEN null mice | No | No | Yes | Yes | Yes |
| PPAR-α knockout mice | No | No | No (steatosis occurs in the starved state) | No | No |
| AOX null mice | No | No | Yes | Yes | No |
| MAT1A null mice | No | No | Yes | Yes | Yes |
| Methionine and choline deficiency | No (decreased weight and adiposity) | Hepatic insulin resistance | Yes | Yes (severe) | Yes |
| High fat | Yes | Yes | Yes | Yes (mild) | Yes |
| Cholesterol and cholate (atherogenic diet) | No (decreased weight) | Hepatic insulin resistance | Yes | Yes | Yes |
| Fructose | No | Yes | Yes | No/Yes | No |
SREBP: Sterol regulatory element binding protein; PTEN: Phosphatase and tensin homologue deleted on chromosome 10; AOX: Acyl-coenzyme A oxidase; MAT1A: Methionine adenosyltransferase-1A; PPAR: Peroxisome proliferator-activated receptor.
GENETIC MODELS
SREBP-1c transgenic mice
In the fat tissue of these mice, SREBP-1c, a lipogenic transcription factor, is overexpressed. This creates a model of congenital lipodystrophy in which severe insulin resistance and diabetes develop secondary to impaired adipose differentiation[25]. In these mice, decreased fat tissue with lipid accumulation in the liver is observed, and marked hepatic steatosis occurs by 8 d of age. When fed a standard diet, steatosis, lobular inflammation, and perivenular and pericellular fibrosis develop at 20 wk[26]; ballooned hepatocytes and MH are also observed. These mice appear to be appropriate for the lipodystrophy-associated steatohepatitis model. However, it is questionable whether they can be used as a model for typical NAFLD/NASH, because visceral fat characteristically increases in human NAFLD/NASH.
Ob/ob mice
Ob/ob mice possess a spontaneous mutation in the leptin gene (leptin-deficient). Leptin is an adipokine produced by white adipose tissue and operates on the hypothalamic ventral median nucleus exerting a marked anorexic effect[22]. Ob/ob mice are hyperphagic, inactive, and extremely obese, and show hyperglycemia, insulin resistance, and hyperinsulinemia[27]. Ob/ob mice develop spontaneous hepatic steatosis[28]; this does not, however, progress to steatohepatitis spontaneously. Secondary insults such as a methionine- and choline-deficient (MCD) diet, a high fat (HF) diet, or low-dose lipopolysaccharide (endotoxin) are needed to trigger steatohepatitis in ob/ob mice[29,30]. Another feature of ob/ob mice is that they are protected against fibrosis[31], a phenomenon which led to the characterization of leptin as an essential mediator of hepatic fibrogenesis[31,32]. Mutations in the ob gene are not prevalent in obese subjects or NASH patients, and leptin levels correlate poorly with the development of NASH[33].
Db/db mice
Db/db mice possess a natural mutation in the leptin receptor (Ob-Rb) gene[34] and, therefore, show normal or elevated levels of leptin but are resistant to the effects of leptin. These mice are obese, insulin resistant, and diabetic, and develop macrovesicular hepatic steatosis. They develop NASH when a second hit such as an MCD diet is added[35]. When db/db mice are fed an MCD diet, significant liver fibrosis is observed as compared to that in ob/ob mice[36]. The advantage of ob/ob and db/db mice is that the phenotype of these mice simulates the human condition of metabolic syndrome in many aspects. However, these mice have the disadvantage that they do not spontaneously develop steatohepatitis or liver fibrosis.
KK-Ay mice
A heterozygous mutation of the agouti gene (KK-Ay/a) results in a loss of melanocortin and an obese phenotype due to hyperphagia from impaired hypothalamic appetite suppression[9]. Although these mice develop hepatic steatosis in conjunction with obesity and insulin resistance, significant steatohepatitis does not occur spontaneously. It was reported that KK-Ay mice exhibited increased susceptibility to MCD diet-induced steatohepatitis, where hypoadiponectinemia most likely played a key role in exacerbation of both inflammatory and profibrogenic responses[37].
PTEN 10 null mice
PTEN is a tumor suppressor gene encoding a lipid phosphatase whose major substrate is phosphatidylinositol-3,4,5-triphosphate (PIP3). PTEN is a negative regulator of several signaling pathways such as phosphatidylinositol 3-kinase and serine-threonine protein kinase B (PKB, or Akt)[38]. These pathways regulate apoptosis, cell proliferation, and tumor formation[38]. Liver-specific Pten knockout mice (AlbCrePTEN flox/flox mice) show extensive hepatomegaly and steatohepatitis, and the histopathology is similar to that in human NASH[39]. Steatosis is observed at 10 wk of age, and steatohepatitis with fibrosis is observed at 40 wk. Hepatocellular adenomas occur with an incidence of 47% by 44 wk, and by 74-78 wk, all of the livers show adenomas, whereas 66% develop HCCs[40]. Pten knockout mice are hypersensitive to insulin, and Pten null hepatocytes have high proliferative activity in vitro[40]. The advantage of this model is that the histological phenotype resembles that of human NASH. The disadvantage of this model is that it is hypersensitive to insulin.
Peroxisome proliferator-activated receptor-α knockout mice
Peroxisome proliferator-activated receptor-α (PPAR-α) is a key regulator of genes involved in peroxisomal, mitochondrial, and microsomal fatty acid oxidation systems in the liver, and a significant decrease in PPAR-α is observed in the HF diet model[41]. Fat does not accumulate in the liver of mice with a homozygous mutation of the PPAR-α gene under conditions of normal feeding, but in the starved state, hepatic steatosis occurs because fatty acid oxidation is inhibited[42].
Acyl-coenzyme A oxidase null mice
Acyl-coenzyme A oxidase (AOX) is the rate-limiting enzyme of peroxisomal β-oxidation of LCFA. AOX null (AOX -/-) mice have defective peroxisomal β-oxidation of LCFA, which accumulate in the liver and lead to steatohepatitis. AOX -/- mice begin to exhibit microvesicular fatty change of hepatocytes in zones 2 and 3 of liver lobules at 7 d of age[43]. By 30 d of age, the AOX -/- mouse liver shows increased severity of steatosis of liver parenchymal cells and concomitant occurrence of focal inflammatory cell infiltrate[43]. At 2 mo of age, clusters of hepatocytes with peroxisome-rich eosinophilic granular cytoplasm are observed in periportal areas[43]. By 4-5 mo of age, increased expressions of PPAR-α, cytochrome P450 (Cyp) 4a10 and Cyp 4a14, and increased levels of H2O2 are observed[44]. However, compensative increase of fatty acid oxidation is observed by 6-7 mo of age, and hepatic steatosis recovers by regeneration of hepatocytes. AOX -/- mice develop hepatocellular adenomas and HCCs by 15 mo of age[44].
Methionine adenosyltransferase-1A null mice
Methionine adenosyltransferase-1A (MAT1A) is a liver-specific rate-limiting enzyme of methionine metabolism and catalyzes the formation of S-adenosylmethionine. MAT1A null mice have decreased levels of antioxidants, including glutathione, and decreased expressions of genes involved in lipid oxidation such as Cyp 4a10 and Cyp 4a14[44]. MAT1A null mice spontaneously develop steatohepatitis after 8 mo of age, and proliferation of hepatocytes increases, resulting in tumor development[45,46]. The mice are susceptible to choline-deficient diet-induced fatty liver at 3 mo of age[45]. Although MAT1A null mice are hyperglycemic, their insulin levels are normal and they do not appear to develop other features of metabolic syndrome[46].
DIETARY MODELS OF NASH
Methionine and choline deficiency
The MCD diet contains high sucrose and fat (40% sucrose, 10% fat) but lacks methionine and choline, which are essential for hepatic β-oxidation and production of VLDL[22]. In addition, choline deficiency impairs hepatic VLDL secretion[47]. As a result, lipid is deposited in the liver. Furthermore, oxidative stress[48,49] and changes in cytokines and adipocytokines[50] occur, contributing to the liver injury. Antagonizing oxidative stress by increasing antioxidant capacities attenuates the degree of steatohepatitis and stresses the importance of reactive oxygen species in this model[51].
Serum alanine aminotransferase (ALT) level is consistently increased after MCD-diet feeding in mice[52]. Steatohepatitis occurs at day 10[52], and perisinusoidal fibrosis is observed by 8-10 wk in mice[48,53]. After 10 wk of MCD feeding, extensive macrovesicular steatosis is observed in all areas except for the periportal region, and many necroinflammatory foci containing lymphocytes and neutrophils are observed in mice[54]. Although the MCD model causes more severe inflammation, oxidative stress, mitochondrial damage, apoptosis, and fibrogenesis than other animal nutritional models of NASH[55], the severity of NASH in rodents fed the MCD diet may depend on the species, gender, and strain of the animal[56]. Kirsch et al[57] compared the effects of MCD diet using male and female Wistar, Long-Evans, and Sprague-Dawley rats, and C57BL/6 mice. As a result, the Wistar strain and the male sex were associated with the greatest degree of steatosis in rats. Of the groups studied, male C57BL/6 mice developed the most inflammation and necrosis, and best approximated the histological features of NASH.
The main disadvantage of the MCD model is that the metabolic profile of the model is opposite to that of typical human NASH. Namely, mice fed the MCD diet show significant weight loss (often, more than 20% weight loss after 3 wk), low fasting blood sugar, peripheral insulin sensitivity, low serum insulin and leptin levels, and unchanged or increased serum adiponectin levels[50,58-61]. To improve these problems, genetically obese mice, such as ob/ob and db/db mice, are occasionally used as the MCD-fed animal. The main advantages of the MCD diet are that it is easy to obtain and use.
HF diet
Lieber et al[62] reported a diet model of NASH by using an HF diet (71% of energy from fat, 11% from carbohydrates, and 18% from proteins). Rats fed this diet ad libitum for 3 wk showed elevated plasma insulin levels reflecting insulin resistance. Rats fed the HF diet developed marked panlobular steatosis, and the hepatic lipid concentrations of these rats were approximately twice those of control rats fed the standard Lieber-DeCarli diet (35% fat, 47% carbohydrates, and 18% protein). Like human NASH, the rats fed the HF diet developed oxidative damage in the liver. When dietary consumption was restricted, steatosis and inflammation in the liver, oxidative stress, and plasma insulin levels were decreased. Feeding of an HF emulsion to Sprague-Dawley rats also induced changes closely resembling human NASH[63]. In the longitudinal study by Ito et al[64], chronic administration of an HF diet (60% of calories from fat) caused steatohepatitis in male C57BL/6J mice.
Intragastric overfeeding of mice with an HF diet up to 85% in excess of standard intake for 9 wk has been reported to replicate the histopathological and pathogenic features of NASH[65]. Overfed C57BL/6 mice became obese (71% higher body weight), had increased visceral fat (white adipose tissue: WAT), and showed hyperglycemia, hyperinsulinemia, hyperleptinemia, glucose intolerance, and insulin resistance. Almost half (46%) of the animals developed NASH, and their plasma ALT levels showed 9- to 10-fold increases. Neutrophilic infiltration and perisinusoidal fibrosis reminiscent of human NASH were observed. The WAT exhibited increased TNF-α and leptin expressions and reduced adiponectin expression.
An HF diet is widely used to cause hepatic steatosis and NASH in experimental animals. However, it seems that the HF diet model produces variable results with regard to the degree of steatosis, inflammation, and fibrosis, and the results depend on rodent species and strain, the fat content in the diet, the composition of dietary fat, and the duration of treatment. For example, Sprague-Dawley rats appear susceptible to steatohepatitis development when fed an HF diet, and this is likely associated with their susceptibility to diet-induced obesity[54]. On the other hand, it was reported that long term high saturated fat feeding did not induce hepatic steatosis and NASH in Wistar rats[66]. In mice, it was reported that BALB/c male mice accumulated more hepatic lipid than C57BL/6J male mice when fed an HF diet[67]. In general, compared with the MCD model, the degree of liver injury in the HF model is less severe[10]. Among the HF models, the histopathology and pathophysiology of the intragastric overfeeding method most resemble those of human NASH. However, this method is difficult to implement, because it requires specific equipment and expertise. The optimization of the composition of the HF diet to reliably cause NASH in animals by ad libitum administration warrants future investigation. Recently, Ogasawara et al[68] reported a combined model of HF diet and gold thioglucose (GTG) administration. GTG is known to induce lesions in the ventromedial hypothalamus, leading to hyperphagia and obesity. They administered GTG intraperitoneally to C57BL/6 mice, and thereafter, fed an HF diet to the mice for 12 wk. As a result, obesity with increased abdominal adiposity, glucose intolerance, insulin resistance, and steatohepatitis with hepatocyte ballooning, MH, and pericellular fibrosis were induced.
Cholesterol and cholate
Matsuzawa et al[69] fed mice an atherogenic diet containing 1.25% cholesterol and 0.5% cholate and observed the progressive formation of steatosis, inflammation, and fibrosis in a time-dependent manner over 6-24 wk. In the model, hepatocellular ballooning, characteristic of human NASH, was observed at 24 wk. When 60% fat (cocoa butter) was added to the diet, development of these histopathological features was accelerated, and hepatocellular ballooning was observed at 12 wk. Furthermore, the atherogenic diet induced oxidative stress. Thus, it is conceivable that a combination diet of HF, cholesterol, and cholate in animals would cause histological features reminiscent of human NASH. However, the mice fed this diet were systematically insulin sensitive, albeit they showed hepatic insulin resistance. In fact, the mice lost 9% body weight during the experiment and had small epididymal fat pads and low plasma triglyceride levels compared with those in control mice. Therefore, this model appears to differ from human NASH in metabolic status.
Fructose
NAFLD/NASH is regarded as a hepatic manifestation of metabolic syndrome. Experimental animals fed a fructose-enriched diet are recognized as good models of metabolic syndrome. We examined livers of Wistar rats fed a high-fructose (70%) diet for 5 wk and found significantly higher macrovesicular steatosis (Figure 1A) and intralobular inflammation (Figure 1B) grades, liver:body weight ratios, and hepatic triglyceride concentrations than those in control rats[70]. In this study, the distribution of steatosis in the rats fed a high-fructose diet was characteristically predominant in zone 1. This pattern differs from that of human adult NAFLD, in which steatosis is usually predominant in zone 3. Rats fed a high-fructose diet showed significantly higher expressions of interleukin (IL)-6 protein and TNF-α protein in the liver compared with those in control rats (unpublished data). By adding plant leaf extract to the high-fructose diet, hepatic fatty change (Figure 2) and expression of IL-6 protein in the liver were completely suppressed (unpublished data).
Figure 1.
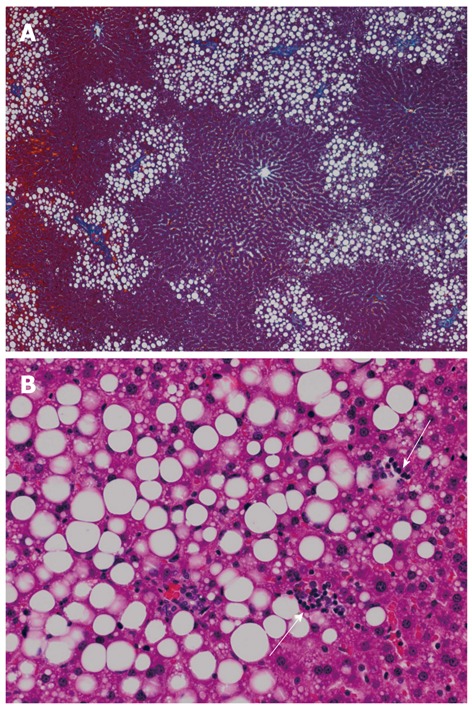
Liver histology of rats fed a high-fructose diet for 5 wk. A: Hepatic steatosis, mainly distributed in zone 1, is observed (azan stain, × 40); B: Both macrovesicular and microvesicular steatosis are evident as well as scattered necroinflammatory foci (arrows) (hematoxylin and eosin stain, × 200).
Figure 2.
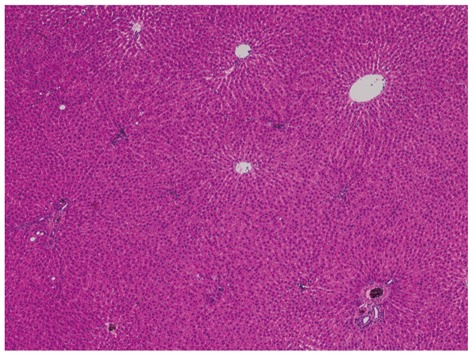
Liver histology of rats fed a high-fructose diet + plant leaf extract. Hepatic fatty change is completely suppressed by the plant leaf extract (hematoxylin and eosin stain, × 40).
Other groups also reported induction of NAFLD/NASH by fructose in experimental animals. Ackerman et al[71] found that male Sprague-Dawley rats fed a fructose-enriched (60%) diet developed macrovesicular and microvesicular steatosis in the liver. Armutcu et al[72] reported that male Wistar albino rats provided with drinking water containing 10% fructose for 10 d developed macrovesicular and microvesicular steatosis but did not develop inflammation in the liver. In mice, by adding 30% fructose to the drinking water, 3- to 4-fold increases in hepatic triglyceride levels and marked increases in hepatic steatosis and weight were observed at 8 wk[73]. Recently, it was reported that C57BL/6 mice fed a high-fat high-carbohydrate diet and provided with drinking water containing 55% fructose for 16 wk developed a NASH-like phenotype with significant fibrosis as well as obesity[74]. Inflammation caused by endogenous toxins of fructose metabolites is suggested as one of the mechanisms of the second hit in the pathogenesis of NASH[75].
COMBINED MODEL OF GENETIC MODIFICATION AND NUTRITIONAL/DIETARY CHALLENGES
Many animal models combine naturally occurring genetic mutations or targeted gene modifications with dietary or chemical challenges so that the histopathology and pathophysiology of the models more closely resemble those of human NAFLD. Sahai et al[36] fed an MCD diet to ob/ob and db/db mice and observed that db/db mice had significantly higher serum ALT levels and more severe hepatic inflammation and fibrosis than those in ob/ob and wild-type mice. PPAR-α null mice fed an MCD diet show more severe steatohepatitis than that in wild-type mice fed the same diet[76,77]. Many other models combining genetic abnormalities with nutritional challenges have been reported[78-80].
CONCLUSION
As reviewed in this paper, many animal models of NASH have been developed to date. These animal models do not replicate the full spectrum of the disease in humans; however, they can be used in verifying hypotheses on the pathogenesis of NASH and in performing interventional studies. We hope that animal models which more closely reflect the histopathology and pathophysiology of human NASH will be developed in the future, and information on pathogenesis and treatment of NASH will increase by using these models.
Footnotes
Peer reviewer: Kazuaki Takabe, MD, PhD, Surgical Oncology, Virginia Commonwealth University, Surg Onc/VCU, PO Box 980011, West Hospital 7-402, 1200 East Broad Street, Richmond, VA 23298-0011, United States
S- Editor Gou SX L- Editor Logan S E- Editor Zhang DN
References
- 1.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi Y, Fukusato T. Pathology of nonalcoholic steatohepatitis. In: Current Research in Hepatology 2., editor. Trivandrum: Research Media; 2008. pp. 99–112. [Google Scholar]
- 3.Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11:74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 4.Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol. 2003;98:2042–2047. doi: 10.1111/j.1572-0241.2003.07659.x. [DOI] [PubMed] [Google Scholar]
- 5.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 7.Machado M, Cortez-Pinto H. Non-alcoholic steatohepatitis and metabolic syndrome. Curr Opin Clin Nutr Metab Care. 2006;9:637–642. doi: 10.1097/01.mco.0000241677.40170.17. [DOI] [PubMed] [Google Scholar]
- 8.Neuschwander-Tetri BA. Nonalcoholic steatohepatitis and the metabolic syndrome. Am J Med Sci. 2005;330:326–335. doi: 10.1097/00000441-200512000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Schattenberg JM, Galle PR. Animal models of non-alcoholic steatohepatitis: of mice and man. Dig Dis. 2010;28:247–254. doi: 10.1159/000282097. [DOI] [PubMed] [Google Scholar]
- 10.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 11.Brunt EM, Tiniakos DG. Alcoholic and non-alcoholic fatty liver disease. In: Odze RD, Goldblum JR, Crawford JM, editors. Pathology of the GI Tract, Liver, Biliary Tract and Pancreas. Philadelphia: Saunders; 2009. pp. 1087–1114. [Google Scholar]
- 12.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 13.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 14.Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, Lavine JE. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology. 2005;42:641–649. doi: 10.1002/hep.20842. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi Y, Inui A, Fujisawa T, Takikawa H, Fukusato T. Histopathological characteristics of non-alcoholic fatty liver disease in children: Comparison with adult cases. Hepatol Res. 2011;41:1066–1074. doi: 10.1111/j.1872-034X.2011.00855.x. [DOI] [PubMed] [Google Scholar]
- 16.Carter-Kent C, Brunt EM, Yerian LM, Alkhouri N, Angulo P, Kohli R, Ling SC, Xanthakos SA, Whitington PF, Charatcharoenwitthaya P, et al. Relations of steatosis type, grade, and zonality to histological features in pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2011;52:190–197. doi: 10.1097/MPG.0b013e3181fb47d3. [DOI] [PubMed] [Google Scholar]
- 17.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi Y, Fukusato T. Pediatric nonalcoholic fatty liver disease: overview with emphasis on histology. World J Gastroenterol. 2010;16:5280–5285. doi: 10.3748/wjg.v16.i42.5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 20.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- 21.Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis. 2004;8:639–671. doi: 10.1016/j.cld.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day CP, Saksena S. Non-alcoholic steatohepatitis: definitions and pathogenesis. J Gastroenterol Hepatol. 2002;17 Suppl 3:S377–S384. doi: 10.1046/j.1440-1746.17.s3.31.x. [DOI] [PubMed] [Google Scholar]
- 24.Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL, Brown MS. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. 1998;12:3182–3194. doi: 10.1101/gad.12.20.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakayama H, Otabe S, Ueno T, Hirota N, Yuan X, Fukutani T, Hashinaga T, Wada N, Yamada K. Transgenic mice expressing nuclear sterol regulatory element-binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism. 2007;56:470–475. doi: 10.1016/j.metabol.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev. 1979;59:719–809. doi: 10.1152/physrev.1979.59.3.719. [DOI] [PubMed] [Google Scholar]
- 28.Diehl AM. Lessons from animal models of NASH. Hepatol Res. 2005;33:138–144. doi: 10.1016/j.hepres.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 29.Brix AE, Elgavish A, Nagy TR, Gower BA, Rhead WJ, Wood PA. Evaluation of liver fatty acid oxidation in the leptin-deficient obese mouse. Mol Genet Metab. 2002;75:219–226. doi: 10.1006/mgme.2002.3298. [DOI] [PubMed] [Google Scholar]
- 30.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206–213. doi: 10.1016/s0168-8278(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 32.Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y, Sato N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology. 2001;34:288–297. doi: 10.1053/jhep.2001.26518. [DOI] [PubMed] [Google Scholar]
- 33.Chalasani N, Crabb DW, Cummings OW, Kwo PY, Asghar A, Pandya PK, Considine RV. Does leptin play a role in the pathogenesis of human nonalcoholic steatohepatitis? Am J Gastroenterol. 2003;98:2771–2776. doi: 10.1111/j.1572-0241.2003.08767.x. [DOI] [PubMed] [Google Scholar]
- 34.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 35.Wortham M, He L, Gyamfi M, Copple BL, Wan YJ. The transition from fatty liver to NASH associates with SAMe depletion in db/db mice fed a methionine choline-deficient diet. Dig Dis Sci. 2008;53:2761–2774. doi: 10.1007/s10620-007-0193-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, Whitington PF. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1035–G1043. doi: 10.1152/ajpgi.00199.2004. [DOI] [PubMed] [Google Scholar]
- 37.Okumura K, Ikejima K, Kon K, Abe W, Yamashina S, Enomoto N, Takei Y, Sato N. Exacerbation of dietary steatohepatitis and fibrosis in obese, diabetic KK-A(y) mice. Hepatol Res. 2006;36:217–228. doi: 10.1016/j.hepres.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, Sherwin R, Devaskar S, Lesche R, Magnuson MA, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci USA. 2004;101:2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato W, Horie Y, Kataoka E, Ohshima S, Dohmen T, Iizuka M, Sasaki J, Sasaki T, Hamada K, Kishimoto H, et al. Hepatic gene expression in hepatocyte-specific Pten deficient mice showing steatohepatitis without ethanol challenge. Hepatol Res. 2006;34:256–265. doi: 10.1016/j.hepres.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A, et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol. 2006;169:846–860. doi: 10.2353/ajpath.2006.050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cook WS, Jain S, Jia Y, Cao WQ, Yeldandi AV, Reddy JK, Rao MS. Peroxisome proliferator-activated receptor alpha-responsive genes induced in the newborn but not prenatal liver of peroxisomal fatty acyl-CoA oxidase null mice. Exp Cell Res. 2001;268:70–76. doi: 10.1006/excr.2001.5266. [DOI] [PubMed] [Google Scholar]
- 44.London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis. 2007;11:55–74, viii. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 45.Lu SC, Alvarez L, Huang ZZ, Chen L, An W, Corrales FJ, Avila MA, Kanel G, Mato JM. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci USA. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martínez-Chantar ML, Corrales FJ, Martínez-Cruz LA, García-Trevijano ER, Huang ZZ, Chen L, Kanel G, Avila MA, Mato JM, Lu SC. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- 47.Yao ZM, Vance DE. Reduction in VLDL, but not HDL, in plasma of rats deficient in choline. Biochem Cell Biol. 1990;68:552–558. doi: 10.1139/o90-079. [DOI] [PubMed] [Google Scholar]
- 48.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, Walsh SV, Tsujita T, Dillon JF, Ashford ML, Hayes JD. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic Biol Med. 2010;48:357–371. doi: 10.1016/j.freeradbiomed.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 50.Larter CZ, Yeh MM, Williams J, Bell-Anderson KS, Farrell GC. MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. J Hepatol. 2008;49:407–416. doi: 10.1016/j.jhep.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 51.Oz HS, Im HJ, Chen TS, de Villiers WJ, McClain CJ. Glutathione-enhancing agents protect against steatohepatitis in a dietary model. J Biochem Mol Toxicol. 2006;20:39–47. doi: 10.1002/jbt.20109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dela Peña A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663–1674. doi: 10.1053/j.gastro.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 53.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 54.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 55.Gao D, Wei C, Chen L, Huang J, Yang S, Diehl AM. Oxidative DNA damage and DNA repair enzyme expression are inversely related in murine models of fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1070–G1077. doi: 10.1152/ajpgi.00228.2004. [DOI] [PubMed] [Google Scholar]
- 56.Fan JG, Qiao L. Commonly used animal models of non-alcoholic steatohepatitis. Hepatobiliary Pancreat Dis Int. 2009;8:233–240. [PubMed] [Google Scholar]
- 57.Kirsch R, Clarkson V, Shephard EG, Marais DA, Jaffer MA, Woodburne VE, Kirsch RE, Hall Pde L. Rodent nutritional model of non-alcoholic steatohepatitis: species, strain and sex difference studies. J Gastroenterol Hepatol. 2003;18:1272–1282. doi: 10.1046/j.1440-1746.2003.03198.x. [DOI] [PubMed] [Google Scholar]
- 58.Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 59.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 60.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 61.Leclercq IA, Lebrun VA, Stärkel P, Horsmans YJ. Intrahepatic insulin resistance in a murine model of steatohepatitis: effect of PPARgamma agonist pioglitazone. Lab Invest. 2007;87:56–65. doi: 10.1038/labinvest.3700489. [DOI] [PubMed] [Google Scholar]
- 62.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–509. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 63.Zou Y, Li J, Lu C, Wang J, Ge J, Huang Y, Zhang L, Wang Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006;79:1100–1107. doi: 10.1016/j.lfs.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 64.Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, Hirose H, Ito M, Ishihara A, Iwaasa H, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. 2007;37:50–57. doi: 10.1111/j.1872-034X.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 65.Deng QG, She H, Cheng JH, French SW, Koop DR, Xiong S, Tsukamoto H. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology. 2005;42:905–914. doi: 10.1002/hep.20877. [DOI] [PubMed] [Google Scholar]
- 66.Romestaing C, Piquet MA, Bedu E, Rouleau V, Dautresme M, Hourmand-Ollivier I, Filippi C, Duchamp C, Sibille B. Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr Metab ( Lond) 2007;4:4. doi: 10.1186/1743-7075-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nishikawa S, Yasoshima A, Doi K, Nakayama H, Uetsuka K. Involvement of sex, strain and age factors in high fat diet-induced obesity in C57BL/6J and BALB/cA mice. Exp Anim. 2007;56:263–272. doi: 10.1538/expanim.56.263. [DOI] [PubMed] [Google Scholar]
- 68.Ogasawara M, Hirose A, Ono M, Aritake K, Nozaki Y, Takahashi M, Okamoto N, Sakamoto S, Iwasaki S, Asanuma T, et al. A novel and comprehensive mouse model of human non-alcoholic steatohepatitis with the full range of dysmetabolic and histological abnormalities induced by gold thioglucose and a high-fat diet. Liver Int. 2011;31:542–551. doi: 10.1111/j.1478-3231.2010.02443.x. [DOI] [PubMed] [Google Scholar]
- 69.Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakanuma Y, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–1403. doi: 10.1002/hep.21874. [DOI] [PubMed] [Google Scholar]
- 70.Kawasaki T, Igarashi K, Koeda T, Sugimoto K, Nakagawa K, Hayashi S, Yamaji R, Inui H, Fukusato T, Yamanouchi T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J Nutr. 2009;139:2067–2071. doi: 10.3945/jn.109.105858. [DOI] [PubMed] [Google Scholar]
- 71.Ackerman Z, Oron-Herman M, Grozovski M, Rosenthal T, Pappo O, Link G, Sela BA. Fructose-induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension. 2005;45:1012–1018. doi: 10.1161/01.HYP.0000164570.20420.67. [DOI] [PubMed] [Google Scholar]
- 72.Armutcu F, Coskun O, Gürel A, Kanter M, Can M, Ucar F, Unalacak M. Thymosin alpha 1 attenuates lipid peroxidation and improves fructose-induced steatohepatitis in rats. Clin Biochem. 2005;38:540–547. doi: 10.1016/j.clinbiochem.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 73.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–1104. doi: 10.1002/hep.23122. [DOI] [PubMed] [Google Scholar]
- 74.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, Balistreri WF, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology. 2010;52:934–944. doi: 10.1002/hep.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nomura K, Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem. 2012;23:203–208. doi: 10.1016/j.jnutbio.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 76.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38:123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 77.Kashireddy PV, Rao MS. Lack of peroxisome proliferator-activated receptor alpha in mice enhances methionine and choline deficient diet-induced steatohepatitis. Hepatol Res. 2004;30:104–110. doi: 10.1016/j.hepres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 78.Carmiel-Haggai M, Cederbaum AI, Nieto N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005;19:136–138. doi: 10.1096/fj.04-2291fje. [DOI] [PubMed] [Google Scholar]
- 79.Arsov T, Larter CZ, Nolan CJ, Petrovsky N, Goodnow CC, Teoh NC, Yeh MM, Farrell GC. Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochem Biophys Res Commun. 2006;342:1152–1159. doi: 10.1016/j.bbrc.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 80.Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lütjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–486. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]