

Abstract
Campylobacter jejuni is the most common bacterial cause of foodborne disease in the developed world. Its general physiology and biochemistry, as well as the mechanisms enabling it to colonize and cause disease in various hosts, are not well understood, and new approaches are required to understand its basic biology. High-throughput sequencing technologies provide unprecedented opportunities for functional genomic research. Recent studies have shown that direct Illumina sequencing of cDNA (RNA-seq) is a useful technique for the quantitative and qualitative examination of transcriptomes. In this study we report RNA-seq analyses of the transcriptomes of C. jejuni (NCTC11168) and its rpoN mutant. This has allowed the identification of hitherto unknown transcriptional units, and further defines the regulon that is dependent on rpoN for expression. The analysis of the NCTC11168 transcriptome was supplemented by additional proteomic analysis using liquid chromatography-MS. The transcriptomic and proteomic datasets represent an important resource for the Campylobacter research community.
Introduction
Campylobacter jejuni is the most common bacterial cause of foodborne disease in the developed world, with an estimated one in every 100 individuals in the UK developing a Campylobacter-related illness each year, resulting in an economic burden of up to £500 million per annum (Humphrey et al., 2007). Although most cases are self-limiting, C. jejuni has been associated with severe post-infection complications, including polyneuropathies such as Guillain–Barré syndrome (Goodyear et al., 1999). The bacterium is a common gut commensal of animals destined for human consumption, and faecal contamination of meat during processing is a recognized route of transmission to humans (Corry & Atabay, 2001; Herman et al., 2003). C. jejuni (NCTC11168) was the first food-borne pathogen to be sequenced (Parkhill et al., 2000) due to its importance as a pathogen and the fact that it has a relatively small genome (1.6 Mb). Many other Campylobacter species have been sequenced, and these genome sequences have facilitated many functional genomics-based approaches to study the roles of individual genes and proteins in colonization, environmental survival and virulence.
An important step in the control of bacterial gene expression is the initiation of transcription, which is dependent on the DNA-dependent RNA polymerase core enzyme binding a sigma factor (Polyakov et al., 1995). Unlike Escherichia coli, which has seven sigma factors, the genome sequence of C. jejuni NCTC11168 revealed the presence of only three (Parkhill et al., 2000): RpoD (σ70), responsible for the transcription of most genes (Petersen et al., 2003), and RpoN (σ54) and RpoF/FliA (σ28), which control the expression of genes required for flagellum biosynthesis and chemotaxis (Hendrixson et al., 2001; Jagannathan et al., 2001; Wassenaar et al., 1994). The lack of sigma factors in C. jejuni suggests that alternative regulatory mechanisms must exist and/or that the two alternative sigma factors play wider regulatory roles than those characterized in other species. Although σ54 is known to be involved in the transcription of flagellar genes including those encoding the hook, basal body and minor flagellin (Carrillo et al., 2004; Hendrixson & DiRita, 2003; Wösten et al., 2004), the complete RpoN regulon in C. jejuni has not been fully defined.
C. jejuni remains a poorly understood pathogen and many of the paradigms that have been established with ‘model’ bacterial species such as E. coli and Salmonella enterica do not apply to it. Consequently, new approaches are required to understand this pathogen. Recently, several eukaryotes (Cloonan et al., 2008; Mortazavi et al., 2008; Wilhelm et al., 2008) and prokaryotes (Perkins et al., 2009; Yoder-Himes et al., 2009) have been profiled at the transcriptome level using direct high-throughput Illumina sequencing of cDNA. This technique offers the ability to survey the entire transcriptome of an organism in a high-throughput and quantitative manner (Marioni et al., 2008; Wang et al., 2009). Since a large number of sequencing reads can be readily obtained, the method is sensitive and offers a large dynamic range. Thus RNA-seq can be used to detect and quantify RNA expressed at very low levels, in contrast with what can be found using DNA microarrays (Nagalakshmi et al., 2008).
In this study we have used Illumina high-throughput DNA sequencing to study mRNA expression levels (i.e. the transcriptome) of C. jejuni NCTC11168, and have compared the transcriptome data with protein expression data. In addition we have used RNA-seq to map and quantify the transcriptome of an rpoN mutant. We have compared RNA-seq with existing microarray technologies for their abilities to identify differentially expressed genes, and we have identified putative small non-coding RNAs (ncRNAs) that would not have been apparent using microarray technology.
Methods
Bacteria.
E. coli strain DH5α was grown at 37 °C on Luria–Bertani (LB) agar or in LB broth. Preparation of electrocompetent E. coli cells and transformations were performed as described previously (Dower et al., 1988). C. jejuni was routinely cultured on Mueller–Hinton (MH) agar (Oxoid) supplemented with 5 % defibrinated horse blood (hereafter referred to as MH blood agar plates) at 42 °C under microaerobic conditions (5 % O2, 5 % CO2, 5 % H2, 85 % N2) in a MACS VA500 variable atmosphere workstation (Don Whitley Scientific). Liquid cultures of C. jejuni were grown in brain heart infusion (BHI) broth (Oxoid) at 42 °C under microaerobic conditions with agitation at 150 r.p.m. Where necessary for selection, media were supplemented with chloramphenicol (10 µg ml−1), ampicillin (100 µg ml−1) or trimethoprim (10 µg ml−1).
Generation of C. jejuni NCTC11168 rpoN : : cat mutant.
The C. jejuni rpoN mutant was generated by insertional inactivation of the gene using a chloramphenicol acetyltransferase (cat) cassette. Standard methods were used for molecular cloning (Sambrook & Russell, 2001). Chromosomal and plasmid DNA purifications were performed using commercial kits following the manufacturers’ instructions (Qiagen). Routine DNA modifications including restriction endonuclease digestion of DNA, modifications of DNA and ligations were carried out according to the manufacturers, instructions (Promega, Invitrogen, Roche, New England Bioloabs). PCR primers were designed using Primer3 (http://frodo.wi.mit.edu/primer3/) and purchased from Sigma. All PCRs were performed in 50 µl reaction volumes in 0.2 ml Eppendorf tubes in an Applied Biosystems GeneAmp PCR Systems 9700 (Applied Biosystems). Reactions contained 200 µM dNTPs, 2 mM Mg2+, 0.01 vols Proof Start DNA polymerase (Qiagen; 2.5 U µl−1), 0.1 vols polymerase buffer (10×), 1 µM forward and reverse primers, and template DNA (~50 ng plasmid DNA or ~100 ng chromosomal DNA). Typical thermal cycler conditions were 94 °C for 4 min, 30 cycles of 94 °C for 1 min, 55 °C for 1 min and 72 °C for 1 min, followed by a final extension of 72 °C for 7 min. DNA and RNA concentration and purity were measured using a Nanodrop ND-1000 spectrophotometer. The rpoN gene was amplified by PCR from C. jejuni NCTC11168 chromosomal DNA using primers ajg285 (5′-GGGGGGATCCATGTTAAAGCAAAAAATCACC-3′) and ajg286 (5′-GGGGGGATCCTTATCCTTCAAGTTCATATAA-3′); the resulting fragment was cloned as a BamHI fragment into BamHI-digested pUC19 to generate the plasmid pAK1. To facilitate subsequent selections a chloramphenicol resistance gene with its promoter was amplified by PCR from pEnterprise2 (Hendrixson et al., 2001) using primers ajg289 (5′-GGATCCCTTAAGCTCGGCGGTGTTCCTTTCCAA-3′) and ajg290 (5′-GGATCCCTTAAGCGCTTTAGTTCCTAAAGGG-3′) and inserted as an AflII fragment into AflII-digested pAK1 (rpoN contained a unique AflII restriction site), generating pAK2. pAK2, with a cat cassette in the forward orientation relative to rpoN, was introduced into C. jejuni by natural transformation using a plate biphasic method adapted from Van Vliet et al. (1998). The structure of a representative isolate, with a chromosomally located rpoN : : cat insertion, was confirmed by PCR and Southern hybridization (data not shown).
Motility assays.
Motility assays were performed essentially as described by Silverman & Simon (1973). Briefly, a platinum wire was dipped into a single colony and used to stab MH motility medium containing 0.4 % agar. Motility was assessed by measuring colony diameter after incubation at 42 °C for 16 h.
RNA preparation for RNA-seq.
C. jejuni strains were cultured for 48 h on MH blood agar plates with antibiotics added as appropriate. Bacterial lawns were harvested in 1 ml BHI broth, 50 µl of which was inoculated into 10 ml BHI broth in a 50 ml Falcon centrifuge tube with a loosened cap, and grown for 16 h at 42 °C under microaerobic conditions with agitation at 150 r.p.m. to generate a starter culture. The OD600 of starter cultures was measured by using a 6305 UV/Visible Spectrophotometer (Jenway). Starter cultures were diluted appropriately in BHI broth and used to inoculate 10 ml BHI broth containing 10 µg trimethoprim ml−1 in a 50 ml falcon tube (previously equilibrated at 42 °C in microaerobic conditions for 16 h) to a calculated OD600 of 0.00002. Cultures were grown at 42 °C under microaerobic conditions with shaking at 150 r.p.m. for 24 h. Growth was monitored by recording OD600 and by determining viable c.f.u. ml−1 by serial 10-fold dilution of cultures in BHI broth and plating onto MH agar. Plates were incubated microaerobically at 42 °C for 48 h before colonies were counted. C. jejuni cultures were fixed with 2 vols RNA protect bacteria (Qiagen) and harvested. RNA was isolated from the pellet using the SV RNA isolation kit (Promega) according to the manufacturer’s instructions. 23S and 16S rRNA was depleted using a MicrobExpress kit (Ambion). Genomic DNA was removed with two digestions, using amplification-grade DNase I (Invitrogen), to below levels detectable by PCR. RNA was reverse-transcribed using random primers (Invitrogen) and Superscript III (Invitrogen) at 45 °C for 3 h and denatured at 70 °C for 15 min. leuB (using primers QJAW084 5′-GCAAGTATAGATGCTTATGGAGTG-3′ and QJAW085 5′-CTCTTTCAGGTCTTTGATCTATGG-3′), aroA (QJAW092 5′-GCTTTGGCTAAGGGTAAATCTAGT-3′ and QJAW093 5′-ATCAAGTTCTCTAGCTTCAACACC-3′), lpxD (QJAW104 5′-GGAGCTTATATAGGCGATAATGTC-3′ and QJAW105 5′-CAAAACCGTCACTTCCTATTACAC-3′) and guaB (QJAW108 5′-GGGTGTTGATGTTGTTGTGC-3′ and QJAW109 5′-GGCGATATTTCCTGCGATAA-3′) were used as targets for a PCR as a positive control for reverse transcription.
Library construction and sequencing.
Library construction and sequencing were carried out as described previously (Perkins et al., 2009).
Read mapping and visualization.
Reads were mapped to the C. jejuni NCTC11168 genome sequence (GenBank accession no. AL111168) using Novoalign version 2.05 (Novocraft Technologies; http://www.novocraft.com) with default settings. Reads that mapped to more than one position in the genome were randomly distributed amongst the matching regions. Mapped reads were visualized using the version of BamView (Carver et al., 2010) integrated into Artemis (Rutherford et al., 2000). All subsequent analyses were performed using R version 2.8.0.
Quantification of transcript levels.
To estimate the level of transcription for each gene, the number of reads that mapped within each annotated coding sequence (CDS) was determined. To account for the possibility of unannotated ncRNA genes, the number of reads that mapped within each intergenic region of >150 bp was also determined. Reads that mapped within 50 bp of the adjacent genes were discarded to prevent the presence of 5′ and 3′ untranslated regions of adjacent transcripts from influencing the read count for the intergenic region. To enable comparison of expression levels, between both different RNA-seq experiments and different genes within the same experiment, it is necessary to normalize the read counts. The number of reads per kb of transcript per million mapped reads (RPKM) has been proposed as a useful metric that normalizes for variation in transcript length and sequence yield (Mortazavi et al., 2008). RPKM values for each gene and intergenic region were calculated for the data from each Illumina lane, and mean RPKM values were determined for the wild-type and rpoN mutant samples. As RPKM values are log-normally distributed, it is convenient to express them as log2(RPKM+1). The complete dataset from this study has been deposited in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) with the accession no. E-MTAB-706.
Analysis of differential expression.
Differential expression was assessed using DESeq (Anders & Huber, 2010). The total read count was determined for each gene and intergenic region by combining data from the two technical replicate sequencing runs. Read counts for the wild-type and the rpoN mutant were compared to determine the log2-fold change in abundance of each transcript. DESeq allows a P-value to be determined in the absence of any available biological replicates, by treating the two conditions as replicates, under the assumption that only a small proportion of transcripts is differentially expressed. P-values were calculated under this assumption, and adjusted for multiple testing using the false discovery rate controlling procedure (Benjamini & Hochberg, 1995).
Microarray analysis.
Raw data from an earlier rpoN mutant microarray study (Kamal et al., 2007) were retrieved from BµG@Sbase and reanalysed by using Limma (Smyth, 2005). Background signals were subtracted from the foreground, and any resulting negative signals were assigned an arbitrary value of 0.5, to prevent problems arising from taking the log of a negative number. Within-array Loess normalization was performed for each array, and signals were scaled between arrays to have the same median-absolute-deviation (Smyth & Speed, 2003). Within-array replicate spot signals were averaged, and correlation of the signals for technical replicate arrays was assessed using the duplicate Correlation function (Smyth et al., 2005). Differential expression was assessed by using a linear modelling approach with empirical Bayes statistics (Smyth, 2004), and the resultant P-values were adjusted for multiple testing using the false discovery rate controlling procedure (Benjamini & Hochberg, 1995).
Cellular fractionation and protein sequencing.
Bacteria were harvested from 5 ml culture (bacteria were grown as described above for RNA preparation for RNA-seq) by centrifugation in a micro-centrifuge (Hettich Mikro 22R) at 13 000 r.p.m. and 4 °C for 2 min. The supernatant was removed by aspiration, and the protein samples were resuspended in 2× final sample buffer (FSB) [0.125 M Tris/HCl (pH 6.8), 4 % SDS, 20 % glycerol, 10 % 2-mercaptoethanol, 0.05 % Bromophenol Blue], reduced with DTT and alkylated with iodoacetamide prior to separation in a 4–12 % NuPAGE Bistris gel (Invitrogen). Gels were stained with colloidal Coomassie Blue (Sigma) and bands were excised and in-gel digested with trypsin (sequencing grade; Roche). The extracted peptides were analysed with online nano liquid chromatography (LC)-MS/MS on an Ultimate 3000 Nano/Capillary LC System (Dionex) coupled to a Q-Tof Premier (Waters) or LTQ FT Ultra (ThermoElectron) mass spectrometer equipped with a nanoelectrospray source. Samples were first loaded and desalted on a PepMap C18 trap (0.3 or 0.5 mm id × 5 mm, Dionex) then separated on a C18 analytical column [Atlantis 100 µm id × 22 cm (Waters), PepMap 75 µm id × 25 cm (Dionex) or BEH 75 µm id x 10 cm (Waters)] over a 45, 60 or 90 min linear gradient of 4–32 % CH3CN/0.1 % FA. The Q-Tof Premier mass spectrometer was operated in the standard data-dependent acquisition (DDA) mode at a resolution of 10 000 (V mode) controlled by Masslynx 4.1. The survey scans were acquired over m/z 400–1500, and the four most abundant multiply charged ions (2+, 3+ and 4+) with a minimal intensity at 20 or 30 counts s−1 were subject to MS/MS for 3 s with dynamic exclusion for 120 s. The instrument was externally calibrated. The Raw files were processed by Proteinlynx Global Server (PLGS) 2.2.5 with and without deconvolution of MS/MS spectra by MaxEnt3. The LTQ FT Ultra mass spectrometer was operated in a similar DDA mode to the Q-Tof premier with a resolution at 100 000 at m/z 400, as described previously (Pickard et al., 2010). The raw files were processed with BioWorks 3.3 (ThermoElectron). The data were subjected to a database search with Mascot Server 2.2 (Matrix Science) against an in-house-built Campylobacter genomic six-frame translated database using the following parameters: trypsin/P with maximum three missed cleavage sites; peptide mass tolerance at ±20 p.p.m. (FT) or 50 p.p.m. (Q-Tof Premier); MS/MS fragment mass tolerance at ±0.49 Da (FT) or 0.2 Da (Q-Tof Premier); and variable modifications, Acetyl (Protein N-term), Carbamidomethyl (C), Deamidated (NQ), Dioxidation (M), Formyl (N-term), Gln→pyro-Glu (N-term Q), Methyl (E), Oxidation (M). The Mascot result was processed with the in-house Percolator (Brosch et al., 2009), and then filtered with the following rules: peptides with less than 7 amino acids or a posterior error probability >0.05 were removed. Proteins with two or more non-redundant peptides were kept. For those proteins with only one peptide identified, the peptide must be found in both datasets (Q-Tof Premier and FT) and with a PEP <0.01 and ≥10 amino acids. If the peptide was found in only one of the datasets, then the peptide must also have a Mascot score above the identification threshold. This resulted in an overall false discovery rate <0.1 %. The complete dataset from this study has been deposited in the PRIDE database (http://www.ebi.ac.uk/pride/q.do) with the accession nos 17661 and 17684.
Secondary structure and conservation analysis for C. jejuni non-coding candidates.
To identify potential unannotated ncRNA genes, large intergenic regions (>150 bp) from the complete genome sequence of C. jejuni NCTC11168 were searched against RFAMSEQ (a subset of the EMBL nucleotide database) using a combination of wu-blast filters and covariance models (Altschul et al., 1990; Eddy & Durbin, 1994). Reliable matches were subsequently aligned to the reference sequence, and a consensus secondary structure was predicted using the WAR package (Torarinsson & Lindgreen, 2008); this formed a ‘seed’ alignment. Covariance models were built from the resulting alignment and secondary structure; these were searched against RFAMSEQ10 using the Rfam pipeline (Eddy & Durbin, 1994; Gardner et al., 2009). New and reliable hits were added to the seed, and a new CM was built and researched. This procedure was iterated until there were no new reliable hits. The subsequent alignments were analysed using the structured RNA gene prediction methods RNAz (Washietl et al., 2005) and Alifoldz (Washietl & Hofacker, 2004). These were computed using a sliding window across alignments generated from the seed sequences using clustal w (Thompson et al., 1994). The window size used was 100 columns and a step-size of 10 columns for both methods. Each prediction method was run on both the forward and reverse complement strands.
Results and Discussion
Mapping DNA sequence reads generated by Illumina-based RNA-seq to the annotated C. jejuni NCTC11168 genome – global interpretations
In order to characterize the C. jejuni transcriptome using RNA-seq, RNA was prepared from a pool of five independent cultures of C. jejuni NCTC11168 grown for 24 h to an OD600 1.0 (log10 10 c.f.u. ml−1) in BHI broth. The 16S and 23S rRNA species were depleted prior to sequencing using selective capture and magnetic separation. The resulting RNA was reverse-transcribed into cDNA which was then processed into a library of molecules that could be sequenced on the Illumina Genome Analyser. The procedure was repeated for RNA derived from a C. jejuni rpoN : : cat mutant. To assess technical variance, we sequenced each biological sample on two Illumina lanes. The sequence reads were mapped to the genome sequence of NCTC11168 and were displayed using the genome browser Artemis and Bamview (Fig. 1). The total number of reads obtained and mapped for each sample is detailed in Supplementary Table S1 (available with the online version of this paper). As previously reported for RNA-seq analysis of eukaryotic (Wilhelm et al., 2008) and prokaryotic (Perkins et al., 2009) RNA, the sequence coverage was not uniform across each CDS; however, our technical replicates were in close agreement (Supplementary Fig. S1), with the log2(RPKM+1) values for each gene and intergenic region giving Pearson correlation coefficients of 0.984 and 0.982 between replicates for the wild-type and rpoN : : cat samples, respectively (P<2.2×10−16 in both cases). Full details of the number of reads mapping to each gene and intergenic region together with log2(RPKM+1) values are available in Supplementary Table S2.
Fig. 1.

RNA-seq sequence data mapped to the C. jejuni NCTC11168 genome and visualized using Artemis and Bamview. The top section shows the position of reads derived from the wild-type (red) and reads from the rpoN : : cat mutant (green). The middle section shows a plot of sequence coverage, using the same colour scheme. The lower section shows a representation of the DNA strands and the six possible reading frames, and indicates the positions of annotated features. The highlighted gene, Cj1242, is downregulated in the rpoN mutant. This gene encodes the invasion antigen CiaC (Christensen et al., 2009).
Protein-coding genes
The distribution of log2(RPKM+1) values for the 1624 protein-coding genes annotated in the C. jejuni NCTC11168 genome (Gundogdu et al., 2007) is shown in Fig. 2. The distribution is approximately normal, with a mean±sd of 7.44±2.14. Only three protein-coding genes did not have any reads that mapped within their coding regions from either the wild-type or rpoN : : cat RNA-seq: Cj0344, Cj0877c and Cj0973. These genes are all annotated as encoding hypothetical proteins; Cj0877c is described as ‘very hypothetical’. Two more genes, annotated as encoding the hypothetical protein Cj0971 and the RepA homologue Cj1667c, had no mapped reads in the wild-type sample but did have mapped reads derived from the rpoN : : cat sample.
Fig. 2.
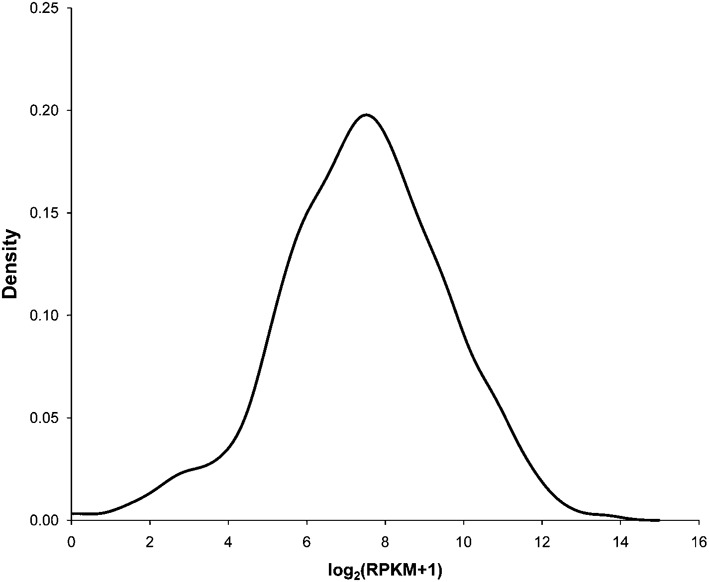
Density plot showing the distribution of log2(RPKM+1) values obtained for the protein-coding genes in wild-type C. jejuni NCTC11168.
Pseudogenes
The original annotation of C. jejuni NCTC11168 identified 20 pseudogenes (Parkhill et al., 2000), and reanalysis of the genome revised this total to 19 (Gundogdu et al., 2007). We were able to detect reads for all of the pseudogenes (Supplementary Table S3, available with the online version of this paper) although only four exhibited a log2(RPKM+1) value above the first quartile of the distribution obtained from the protein-coding genes (Cj0501, Cj0565, Cj1064 and Cj1528). Closer inspection of the data indicates that most of the pseudogenes had a strong transcriptional profile at the start of the CDS with just a few isolated reads mapping to the remainder of the CDS (Fig. 3). Two of the pseudogenes (Cj1064 and Cj0501) had strong transcriptional profiles throughout the entire CDS, suggesting that transcription may be maintained to express functional domains or peptides, or that the inactivation of these genes was evolutionarily recent. The sequences of our reads matched the published genome sequences for these two genes.
Fig. 3.

Artemis plots showing RNA-seq data obtained for pseudogenes (as for Fig. 1). (a) Cj0654c showed an expression pattern typical of most pseudogenes in the genome, with a high level of expression at the 5′ end of the gene that diminishes towards the 3′ end. (b) Cj1064 showed a high level of expression across the length of the pseudogene.
tRNA
There are 43 tRNA-encoding genes annotated in the C. jejuni NCTC11168 genome (Gundogdu et al., 2007), with 33 distinct anticodons. The log2(RPKM+1) values of the tRNA genes show a similar mean to the protein-coding genes (7.67; Student’s t-test, P = 0.3188), but a reduced variance (Fisher’s F test, P = 0.0040). It is a common observation that the use of alternative synonymous codons is non-random (Sharp et al., 2010); in C. jejuni this has been attributed to the low GC-content of the genome, with a preference for AT-rich synonyms (Fuglsang, 2003). It might be expected that the expression levels of the tRNAs would reflect the codon usage bias, but this does not seem to be the case (Supplementary Tables S4 and S5, available with the online version of this paper). For example, the tRNA gene with the highest log2(RPKM+1) value (12.12) encodes tRNA-Arg with an anticodon CCU. However, the codon recognized by this tRNA, AGG, is used only 1340 times in the genome, whereas the synonym AGA is used 8195 times, yet its cognate tRNA with the anticodon UCU has the lowest log2(RPKM+1) value of all the arginine tRNAs (7.88). Leucine is the most frequently used amino acid in the C. jejuni proteome, yet the four tRNA-Leu genes all have average log2(RPKM+1) values. The levels of tRNA transcription seem to be dependent on genomic context, for example, the CCU anticodon tRNA-Arg is encoded downstream of the highly expressed fusA gene, and the three tRNA-Ile genes are located within rRNA operons.
ncRNA sequences
A limitation of expression microarrays is that they only provide information on specific genomic regions to which probe sequences have been designed. RNA-seq allows investigation of transcription across the whole genome, and hence enables the discovery of novel ncRNAs (Yoder-Himes et al., 2009). Four ncRNAs (excluding rRNA and tRNA) are currently annotated in the C. jejuni NCTC11168 genome: the signal recognition particle RNA, the TPP riboswitch, the RNA component of RNaseP and 10Sa RNA (Gundogdu et al., 2007). Transcripts were identified in the RNA-seq data for all of these. Additionally, we performed a screen of all large intergenic regions for potential unannotated ncRNA genes based on evolutionary conservation and structural prediction using the Rfam alignment pipeline (Gardner et al., 2009), RNAz (Washietl et al., 2005) and Alifoldz (Washietl & Hofacker, 2004), and identified five candidate regions which may be worth investigating further (intergenic_671549–671895, intergenic_722652–722740, intergenic_906748–907066, intergenic_1127982–1128192 and intergenic_1575021–1575288) (Table 1 and Fig. 4), although one of these (intergenic_906748–907066) did not show a significant level of expression in our data.
Table 1. Secondary structure and conservation analysis for C. jejuni ncRNA candidates.
ID, unique identifier containing genomic location; maxRnazP, the maximal RNAz probability on the forward or reverse strand – values greater than 0.5 were classified as putative ncRNA genes; minAlifoldZ, the lowest (best) Z-score from the RNAalifoldZ predictions on the forward or reverse strand; numSeqs, no. of homologous sequences used in the Rfam seed alignment; aveLen, average length of the sequences in the Rfam seed alignment; GC content, average G+C content for all the sequences in the Rfam seed alignment; GC enrichment, enrichment of G+C nucleotides relative to the genomic background [computed as log2(GC-content/0.3055)]. One homologue was identified in the C. jejuni genome (EMBL ACC:AL111168.1) for each ncRNA candidate.
| ID no. | MaxRnazP | MinAlifoldZ | numSeqs | aveLen | aveID | GC | |||
| Forward | Reverse | Forward | Reverse | Content | Enrichment | ||||
| Intergenic_671549–671895 | 0.957647 | 0.796566 | −5.9 | −5.1 | 8 | 342.9 | 74 | 0.285 | −0.1001 |
| Intergenic_722652–722740 | 0.995352 | 0.995194 | −4.6 | −4.4 | 5 | 89.4 | 89 | 0.295 | −0.0504 |
| Intergenic_906748–907066 | 0.932765 | 0.89473 | −1.1 | −6.6 | 5 | 311 | 89 | 0.232 | −0.397 |
| Intergenic_1127982–1128192 | 0.999921 | 0.559228 | −8.6 | −2.4 | 5 | 206.8 | 78 | 0.281 | −0.1205 |
| Intergenic_1575021–1575288 | 0.999869 | 0.999657 | −7.7 | −8.2 | 5 | 262.6 | 88 | 0.221 | −0.4671 |
Fig. 4.

Consensus secondary structure predictions for the forward (left) and reverse (right) complement intergenic_671549–671895 (a), intergenic_722652–722740 (b), intergenic_906748–907966 (c), intergenic_1127982–1128192 (d) and intergenic_1575021–1575288 (e). The colour markup indicates the sequence conservation.
RNA-seq and proteome analysis of C. jejuni hypothetical genes and pseudogenes
mRNA transcript levels do not necessarily correlate with protein levels, or indeed whether the transcript is translated. We carried out a comprehensive proteomic analysis using LC-MS of peptides from whole-cell lysates of C. jejuni NCTC11168. We obtained good coverage with 11462 peptides mapping to 1041 predicted genes. We did not obtain any peptides that mapped to any of the pseudogenes. This might suggest that, even though we obtained transcripts for a number of the pseudogenes, they are not translated. Alternatively, any translated products may be below the level of detection or below our ~6 kDa molecular mass cut-off. Fig. 5 shows the number of peptides obtained for each protein-coding gene plotted against the log2(RPKM+1) value. Fitting a log-linear model with quasi-Poisson errors indicates a significant relationship between mRNA expression level and the observed peptide count (P<2×1016). For the gene pnp (Cj1253), a peptide sequence was obtained that included one amino acid encoded upstream of the annotated start codon. This suggests that the annotated pnp start codon is incorrect; the most plausible alternative is the TTG codon at position 1183537.
Fig. 5.

Plot of the observed number of peptides from wild-type C. jejuni NCTC11168 against log2(RPKM+1) as a measure of the mRNA expression level. The blue line indicates the predicted protein level according to the fitted log-linear regression with quasi-Poisson errors. Red points indicate mRNAs predicted to encode peptides below the 6 kDa threshold of detection.
RNA-seq and microarray analysis of a C. jejuni rpoN mutant
We generated a C. jejuni rpoN : : cat mutant and compared the growth of the mutant with the wild-type. Bacteria were cultured in MH broth with shaking in microaerobic conditions at 42 °C. The doubling time for each strain was determined for exponential phase growth: wild-type, 1.40±0.12 generations per hour; mutant 1.41±0.01 generations per hour. The difference between the growth rates was not statistically significant (Student’s t-test, P>0.05) based on viable counts. Motility assays were performed on the wild-type and rpoN : : cat mutant. Following 72 h incubation at 42 °C in microaerobic conditions, the mutant was completely non-motile (data not shown). Genes that were expressed in the rpoN : : cat mutant were identified from the RNA-seq data using DEseq (Anders & Huber, 2010). The log2 (fold changes) and estimated P-values obtained from the data are available in Supplementary Table S2. Twenty-five genes and two intergenic regions showed significantly altered expression in the rpoN : : cat mutant compared with the wild-type (P<0.1; see Table 2). Seventeen protein-coding genes showed significant downregulation in the rpoN mutant, including 12 that are annotated as encoding flagellar proteins and two others (Cj0040 and Cj1465) that are co-located with flagellar gene clusters. The other three downregulated genes (Cj0243c, Cj1242 and Cj1650) are all annotated as encoding hypothetical proteins, although Cj1242 has recently been identified as the invasion antigen CiaC, which is secreted by the C. jejuni flagellar type III secretion system and is potentially important in virulence (Christensen et al., 2009). Cj1650 is an orthologue of the Helicobacter pylori protein HP1076, which has been identified as a flagellar export co-chaperone (Lam et al., 2010). Two intergenic regions, located between tpx–napA (positions 731983–732128) and luxS–Cj1199 (positions 1127982–1128192), were also identified as significantly downregulated. The latter region was also identified in the screen for potential ncRNA genes, suggesting the possibility of a novel ncRNA involved in flagellar regulation. Genes identified as being upregulated in the rpoN mutant include two gene clusters: Cj0423–Cj0425, which is annotated as encoding a putative integral membrane protein and two putative periplasmic proteins; and metAB, which encode enzymes involved in amino acid metabolism and are located close to flgE2. An additional membrane protein gene, cstA, and another amino acid metabolism gene, cysM, are also upregulated in the rpoN mutant, together with Cj0667, Cj0898 and Cj1650, all of which are of unknown function. Interestingly, Cj0667 is the first gene in the operon containing rpoN.
Table 2. RNA-seq data for genes and intergenic regions identified as differentially expressed in the rpoN : : cat mutant relative to the wild-type.
The log2(fold change) (logFC) values for the RNA-seq and microarray, and the wild-type log2(RPKM+1) values are coloured by value (red >yellow >blue). RNaseq and microarray P-values of <0.05 are in bold type.

We compared differentially expressed genes identified in the RNA-seq data with those identified in a reanalysis of data derived from an earlier microarray-based study (Kamal et al., 2007). Both studies compare wild-type C. jejuni NCTC11168 with an rpoN mutant, although it should be noted that different growth conditions were employed. (Kamal et al. used bacteria from a 24 h plate culture, resuspended in MH broth to OD600 ~0.1, and grown at 37 °C for 18 h with shaking at 75 r.p.m.) All but one of the genes identified as being significantly downregulated in the rpoN mutant by RNA-seq were also significantly downregulated in the microarray data (Fig. 6 and Supplementary Table S2). Conversely, for the upregulated genes, only one (metA) was significant in both datasets. The microarray data detected considerably more significantly downregulated genes than were identified using RNA-seq. This is in part due to the additional biological replicates available for the microarray data, which add statistical power. However, most of the genes in question show no evidence of downregulation in the RNA-seq data irrespective of P-value, which would be unexpected were they genuinely part of the rpoN regulon. It is possible that the assignment of these genes as differentially expressed is an artefact of the microarray hybridization or analysis procedure. Alternatively, the differences between the two datasets could reflect the different culture conditions employed in the two studies, although we consider this unlikely. As RNA-seq becomes increasingly commonplace, it will be possible to resolve this issue by characterizing the effect of growth conditions on the rpoN regulon.
Fig. 6.

log2(fold change) values obtained using RNA-seq, plotted against the equivalent values from the microarray study (Kamal et al., 2007). Genes showing significant differential expression in the RNA-seq data (P<0.05) are highlighted in red, genes showing significant differential expression in the microarray data (P<0.05) are shown as triangles. (Red triangles, differential expression in the RNA-seq and microarray data; red circles, differential expression in the RNA-seq data; black triangles, differential expression in the microarray data; black circles, no differential expression in either data set.)
Concluding remarks
In this study we have used direct high-throughput Illumina sequencing of cDNA (RNA-seq) to analyse the transcriptome of C. jejuni (NCTC11168). Our studies demonstrate the efficacy of high-throughput sequencing for defining mRNA expression levels and identifying differentially expressed genes and novel transcribed regions of the genome (for example, identification of potential unannotated ncRNA genes). We hope that the transcriptome and proteome datasets will be a useful resource to the Campylobacter research community.
Acknowledgements
This work was supported by the Department for Environment, Food and Rural Affairs (Defra) Senior Fellowship in Veterinary Microbiology to D. J. M. A. J. G. was supported by Defra/Higher Education Funding Council of England under the Veterinary Training and Research Initiative (grant no. VT0105), and by a Medical Research Council grant (G0801161) awarded to A. J. G. P. P. G. and T. T. P. were funded by the Wellcome Trust. L. Y. and J. C. were funded by the Wellcome Trust grant no. 079643/Z/06/Z.
Abbreviations:
- CDS
coding sequence
- LC
liquid chromatography
- ncRNA
non-coding RNA
- RNA-seq
transcriptome sequencing
- RPKM
reads per kb of transcript per million mapped reads
Footnotes
Five supplementary tables and a supplementary figure are available with the online version of this paper.
References
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J Mol Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate – a practical and powerful approach to multiple testing. J R Stat Soc B 57, 289–300. [Google Scholar]
- Brosch M., Yu L., Hubbard T., Choudhary J. (2009). Accurate and sensitive peptide identification with Mascot Percolator. J Proteome Res 8, 3176–3181. 10.1021/pr800982s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo C. D., Taboada E., Nash J. H., Lanthier P., Kelly J., Lau P. C., Verhulp R., Mykytczuk O., Sy J., et al. & other authors (2004). Genome-wide expression analyses of Campylobacter jejuni NCTC11168 reveals coordinate regulation of motility and virulence by flhA. J Biol Chem 279, 20327–20338. 10.1074/jbc.M401134200 [DOI] [PubMed] [Google Scholar]
- Carver T., Böhme U., Otto T. D., Parkhill J., Berriman M. (2010). BamView: viewing mapped read alignment data in the context of the reference sequence. Bioinformatics 26, 676–677. 10.1093/bioinformatics/btq010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J. E., Pacheco S. A., Konkel M. E. (2009). Identification of a Campylobacter jejuni-secreted protein required for maximal invasion of host cells. Mol Microbiol 73, 650–662. 10.1111/j.1365-2958.2009.06797.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan N., Forrest A. R., Kolle G., Gardiner B. B., Faulkner G. J., Brown M. K., Taylor D. F., Steptoe A. L., Wani S., et al. & other authors (2008). Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5, 613–619. 10.1038/nmeth.1223 [DOI] [PubMed] [Google Scholar]
- Corry J. E., Atabay H. I. (2001). Poultry as a source of Campylobacter and related organisms, Symposium series, 96S–114S. Society for Applied Microbiology. [DOI] [PubMed] [Google Scholar]
- Dower W. J., Miller J. F., Ragsdale C. W. (1988). High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16, 6127–6145. 10.1093/nar/16.13.6127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S. R., Durbin R. (1994). RNA sequence analysis using covariance models. Nucleic Acids Res 22, 2079–2088. 10.1093/nar/22.11.2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuglsang A. (2003). The genome of Campylobacter jejuni: codon and amino acid usage. APMIS 111, 605–618. 10.1034/j.1600-0463.2003.1110603.x [DOI] [PubMed] [Google Scholar]
- Gardner P. P., Daub J., Tate J. G., Nawrocki E. P., Kolbe D. L., Lindgreen S., Wilkinson A. C., Finn R. D., Griffiths-Jones S., et al. & other authors (2009). Rfam: updates to the RNA families database. Nucleic Acids Res 37 (Database issue), D136–D140. 10.1093/nar/gkn766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear C. S., O’Hanlon G. M., Plomp J. J., Wagner E. R., Morrison I., Veitch J., Cochrane L., Bullens R. W., Molenaar P. C., et al. & other authors (1999). Monoclonal antibodies raised against Guillain–Barré syndrome-associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyze muscle-nerve preparations. J Clin Invest 104, 697–708. 10.1172/JCI6837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundogdu O., Bentley S. D., Holden M. T., Parkhill J., Dorrell N., Wren B. W. (2007). Re-annotation and re-analysis of the Campylobacter jejuni NCTC11168 genome sequence. BMC Genomics 8, 162. 10.1186/1471-2164-8-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrixson D. R., DiRita V. J. (2003). Transcription of sigma54-dependent but not σ28-dependent flagellar genes in Campylobacter jejuni is associated with formation of the flagellar secretory apparatus. Mol Microbiol 50, 687–702. 10.1046/j.1365-2958.2003.03731.x [DOI] [PubMed] [Google Scholar]
- Hendrixson D. R., Akerley B. J., DiRita V. J. (2001). Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol Microbiol 40, 214–224. 10.1046/j.1365-2958.2001.02376.x [DOI] [PubMed] [Google Scholar]
- Herman L., Heyndrickx M., Grijspeerdt K., Vandekerchove D., Rollier I., De Zutter L. (2003). Routes for Campylobacter contamination of poultry meat: epidemiological study from hatchery to slaughterhouse. Epidemiol Infect 131, 1169–1180. 10.1017/S0950268803001183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey T., O’Brien S., Madsen M. (2007). Campylobacters as zoonotic pathogens: a food production perspective. Int J Food Microbiol 117, 237–257. 10.1016/j.ijfoodmicro.2007.01.006 [DOI] [PubMed] [Google Scholar]
- Jagannathan A., Constantinidou C., Penn C. W. (2001). Roles of rpoN, fliA, and flgR in expression of flagella in Campylobacter jejuni. J Bacteriol 183, 2937–2942. 10.1128/JB.183.9.2937-2942.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal N., Dorrell N., Jagannathan A., Turner S. M., Constantinidou C., Studholme D. J., Marsden G., Hinds J., Laing K. G., et al. & other authors (2007). Deletion of a previously uncharacterized flagellar-hook-length control gene fliK modulates the σ54-dependent regulon in Campylobacter jejuni. Microbiology 153, 3099–3111. 10.1099/mic.0.2007/007401-0 [DOI] [PubMed] [Google Scholar]
- Lam W. W., Woo E. J., Kotaka M., Tam W. K., Leung Y. C., Ling T. K., Au S. W. (2010). Molecular interaction of flagellar export chaperone FliS and cochaperone HP1076 in Helicobacter pylori. FASEB J 24, 4020–4032. 10.1096/fj.10-155242 [DOI] [PubMed] [Google Scholar]
- Marioni J. C., Mason C. E., Mane S. M., Stephens M., Gilad Y. (2008). RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18, 1509–1517. 10.1101/gr.079558.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M. (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. 10.1126/science.1158441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J., Wren B. W., Mungall K., Ketley J. M., Churcher C., Basham D., Chillingworth T., Davies R. M., Feltwell T., et al. & other authors (2000). The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668. 10.1038/35001088 [DOI] [PubMed] [Google Scholar]
- Perkins T. T., Kingsley R. A., Fookes M. C., Gardner P. P., James K. D., Yu L., Assefa S. A., He M., Croucher N. J., et al. & other authors (2009). A strand-specific RNA-Seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. PLoS Genet 5, e1000569. 10.1371/journal.pgen.1000569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen L., Larsen T. S., Ussery D. W., On S. L., Krogh A. (2003). RpoD promoters in Campylobacter jejuni exhibit a strong periodic signal instead of a -35 box. J Mol Biol 326, 1361–1372. 10.1016/S0022-2836(03)00034-2 [DOI] [PubMed] [Google Scholar]
- Pickard D., Toribio A. L., Petty N. K., van Tonder A., Yu L., Goulding D., Barrell B., Rance R., Harris D., et al. & other authors (2010). A conserved acetyl esterase domain targets diverse bacteriophages to the Vi capsular receptor of Salmonella enterica serovar Typhi. J Bacteriol 192, 5746–5754. 10.1128/JB.00659-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyakov A., Severinova E., Darst S. A. (1995). Three-dimensional structure of E. coli core RNA polymerase: promoter binding and elongation conformations of the enzyme. Cell 83, 365–373. 10.1016/0092-8674(95)90114-0 [DOI] [PubMed] [Google Scholar]
- Rutherford K., Parkhill J., Crook J., Horsnell T., Rice P., Rajandream M. A., Barrell B. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. 10.1093/bioinformatics/16.10.944 [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D. W. (2001). Molecular Cloning: a Laboratory Manual, 3rd edn Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Sharp P. M., Emery L. R., Zeng K. (2010). Forces that influence the evolution of codon bias. Philos Trans R Soc Lond B Biol Sci 365, 1203–1212. 10.1098/rstb.2009.0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman M., Simon M. (1973). Genetic analysis of flagellar mutants in Escherichia coli. J Bacteriol 113, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article 3. [DOI] [PubMed] [Google Scholar]
- Smyth G. K. (2005). Limma: linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor, pp. 397–420. Edited by Gentleman R., Carey V., Dudoit S., Irizarry R., Huber S. New York: Springer; 10.1007/0-387-29362-0_23 [DOI] [Google Scholar]
- Smyth G. K., Speed T. (2003). Normalization of cDNA microarray data. Methods 31, 265–273. 10.1016/S1046-2023(03)00155-5 [DOI] [PubMed] [Google Scholar]
- Smyth G. K., Michaud J., Scott H. S. (2005). Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21, 2067–2075. 10.1093/bioinformatics/bti270 [DOI] [PubMed] [Google Scholar]
- Thompson J. D., Higgins D. G., Gibson T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torarinsson E., Lindgreen S. (2008). WAR: Webserver for aligning structural RNAs. Nucleic Acids Res 36 (Web Server issue), W79–84. 10.1093/nar/gkn275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vliet A. H. M., Wood A. C., Henderson J., Wooldridge K., Ketley J. M. (1998). Genetic manipulation of enteric Campylobacter species. Methods in Microbiology 27, 407–419. 10.1016/S0580-9517(08)70301-5 [DOI] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63. 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S., Hofacker I. L. (2004). Consensus folding of aligned sequences as a new measure for the detection of functional RNAs by comparative genomics. J Mol Biol 342, 19–30. 10.1016/j.jmb.2004.07.018 [DOI] [PubMed] [Google Scholar]
- Washietl S., Hofacker I. L., Stadler P. F. (2005). Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci U S A 102, 2454–2459. 10.1073/pnas.0409169102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar T. M., Bleumink-Pluym N. M., Newell D. G., Nuijten P. J., van der Zeijst B. A. (1994). Differential flagellin expression in a flaA flaB+ mutant of Campylobacter jejuni. Infect Immun 62, 3901–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm B. T., Marguerat S., Watt S., Schubert F., Wood V., Goodhead I., Penkett C. J., Rogers J., Bähler J. (2008). Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453, 1239–1243. 10.1038/nature07002 [DOI] [PubMed] [Google Scholar]
- Wösten M. M., Wagenaar J. A., van Putten J. P. (2004). The FlgS/FlgR two-component signal transduction system regulates the fla regulon in Campylobacter jejuni. J Biol Chem 279, 16214–16222. 10.1074/jbc.M400357200 [DOI] [PubMed] [Google Scholar]
- Yoder-Himes D. R., Chain P. S., Zhu Y., Wurtzel O., Rubin E. M., Tiedje J. M., Sorek R. (2009). Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci U S A 106, 3976–3981. 10.1073/pnas.0813403106 [DOI] [PMC free article] [PubMed] [Google Scholar]