

Abstract
There is increasing evidence supporting a causal role of oxidatively damaged DNA in neurodegeneration during the natural aging process and neurodegenerative diseases such as Parkinson’s and Alzheimer’s. The presence of redox-active catecholamine neurotransmitters coupled with the localization of catalytic copper to DNA suggests a plausible role for these agents in the induction of oxidatively generated DNA damage. In this study we have investigated the role of Cu(II)-catalyzed oxidation of several catecholamine neurotransmitters and related neurotoxins to induce oxidatively generated DNA damage. Auto-oxidation of all catechol neurotransmitters and related congeners tested resulted in the formation of nearly a dozen oxidation DNA products resulting in a decomposition pattern that was essentially identical for all agents tested. The presence of Cu(II), and to a lesser extent Fe(III), had no effect on the decomposition pattern but substantially enhanced the DNA product levels by up to 75 fold, with dopamine producing the highest levels of unidentified oxidation DNA products (383 ± 46 adducts/106 nucleotides), comparable to 8-oxo-7,8-dihydro-2′-deoxyguanosine levels under the same conditions (122 ± 19 adducts/106 nucleotides). The addition of sodium azide, 2,2,6,6-tetramethyl-4-piperidone, tiron, catalase, bathocuproine or methional to the dopamine/Cu(II) reaction mixture resulted in a substantial decrease (>90%) in oxidation DNA product levels, indicating a role of singlet oxygen, superoxide, H2O2, Cu(I) and Cu(I)OOH in their formation. While the addition of N-tert-butyl-α-phenylnitrone significantly decreased (67%) dopamine-mediated oxidatively damaged DNA, three other hydroxyl radical scavengers, ascorbic acid, sodium benzoate and mannitol, had little to no effect on these oxidation DNA product levels, suggesting that free hydroxyl radicals may have limited involvement in this dopamine/Cu(II)-mediated oxidatively generated DNA damage. These studies suggest a possible contributory role of oxidatively generated DNA damage by dopamine and related catechol neurotransmitters/neurotoxins in neurodegeneration and cell death. We also found that a naturally occurring broad spectrum antioxidant, ellagic acid, was substantially effective (nearly 50% inhibition) at low doses (1 μM) at preventing this dopamine/Cu(II)-mediated oxidatively generated DNA damage. Since dietary ellagic acid has been found to reduce oxidative stress in rat brains, a neuro-protective role of this polyphenol is plausible.
Keywords: Neurotransmitters, Dopamine, Catecholamines, Oxidative DNA adducts, 32P-Postlabeling
Introduction
Significant evidence indicates that oxidative stress plays a central role in the aging brain [1–3] and neurodegenerative diseases such as Alzheimer’s (AD)1 [1, 3–4] and Parkinson’s disease (PD) [5–7]. Post-mortem studies of patients with neurologic degeneration have demonstrated increased levels of oxidatively damaged DNA in AD [4] and dementia with Lewy body brains [8] and increased indices of oxidative stress in PD brains, including increased levels of lipid peroxidation and iron, decreased mitochondrial complex I activity and levels of glutathione [9] as compared to age-matched control subjects. A common trait shared between AD and PD is the degradation of neurons in specific regions of the brain which are believed to be mediated, at least in part, by reactive oxygen species (ROS) [10]. Post-mitotic cells, such as neurons, are believed to be particularly sensitive to the deleterious effects of ROS since damaged cells cannot be replaced. Moreover, the high oxygen metabolism taking place in the brain also increases neuronal vulnerability to oxidatively generated damage [11]. ROS have been shown to cause, in addition to neuronal cell death [12–13], damage to a wide range of neuronal cellular components including, membrane lipids [14–16], proteins [15, 17] and DNA [5, 7].
Both PD and AD are considered diseases of the ageing brain since the most important risk factor for these diseases is advancing age [11, 18]. Not only have increased levels of oxidatively damaged DNA in brain tissue been shown to be inversely proportional with life span [2–3] and correlate with increasing age of the human brain [3], but they have also been shown to play a key role in selective neuronal loss in both PD [6] and AD [3]. Nuclear DNA (nDNA), and mitochondrial DNA (mtDNA) in particular, appear to be important targets for the progression of neurodegeneration in the natural aging process as well as several diseases including AD and PD [11, 18]. Mitochondria provide the primary source of energy in the cell. mtDNA is exceptionally susceptible to oxidatively generated damage as it lacks protective histones and is located in close proximity to the respiratory chain, the cell’s main source or ROS [11, 18]. One line of evidence supporting mtDNA’s increased vulnerability to oxidative stress is apparent in that elevated levels of oxidatively damaged DNA in both nDNA and mtDNA have been found in AD brains [19–20] however, 3- to 10-fold higher levels of oxidized bases were identified in mtDNA [4, 21]. Also noteworthy, is that mtDNA has no non-coding sequences [22] thus potentially resulting in impaired functional consequences [4]. Impairment in mitochondrial function resulting from mutations in nDNA and mtDNA may result in neuronal cell death via defects in oxidative phosphorylation [4]. It should also be recognized that damage to either nDNA or mtDNA can indirectly affect the other. Damage to mtDNA can lead to impaired energy generation and potential leaks of ROS during respiration which can in turn result in oxidatively generated to nDNA. Conversely, nDNA encodes the majority of mitochondrial proteins and thus its integrity has a direct impact on mitochondrial respiration and function [18].
A growing body of evidence indicates that dopamine (DA) [23–26] an essential neurotransmitter, and possibly other catecholamine neurotransmitters such as norepinephrine (NE) [27], contribute to neuronal cell death and the development of neurodegenerative diseases such as PD [28] and AD [29]. One common trait and plausible mechanism shared by these catechol neurotransmitters is their ability to redox cycle producing cellular and DNA damaging ROS [23, 26–27, 30]. The oxidation of these catecholamines to DNA-reactive species has been shown to occur non-enzymatically, in the presence of transition metals such as copper and iron, and has been proposed to proceed via quinone and semi-quinone intermediates [30–31]. Both iron and copper are prevalent in human tissues, including the brain and altered levels of these essential metals have been found in brain tissues of patients with varied neurodegenerative diseases [31–33]. Since approximately 20% of the total copper is stored in the nucleus [34], DNA is a major target for copper-catalyzed oxidations. In the current study we investigated oxidatively generated DNA damage formed by copper-mediated oxidation of dopamine and its catecholinergic analogs by employing a new 32P-postlabeling TLC system for the detection of oxidative DNA adducts. The specific ROS involved in this DNA adduction were also studied by employing a variety of known free radical scavengers.
Experimental Procedures
Chemicals
All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO) or Aldrich (Milwaukee, WI) unless stated otherwise. Sources of chemicals used for 32P-postlabeling analysis were as described previously [35].
Oxidation DNA Products
Salmon testis (ST) DNA (300 μg/ml), freed from RNA by treatment with RNases and solvent extractions [35], was incubated (37°C, 4 h) with either vehicle (1% DMSO), catecholamine neurotransmitters or dopamine analogs (30 μM) in the presence or absence of a metal catalyst (30 μM), CuCl2, CuCl, FeCl3 or FeSO4 in 10 mM Tris HCl, pH 7.4. Intervention with ROS scavengers was carried out as described above using dopamine (30 μM) as the substrate in the presence of CuCl2 (30 μM) and 200 U/ml superoxide dismutase (SOD), 200 U/ml catalase, 200 U/ml tyrosinase, 10 mM tiron, 10 and 100 mM sodium azide, 10 mM 2,2,6,6-tetramethyl-4-piperidone (TMP), 150 μM bathocuproine, 10 mM 3-(methylthio)propionaldehyde (methional), 10 mM sodium benzoate, 10 mM mannitol, 10 mM N-tert-butyl-α-phenylnitrone (TBP), 150 μM ascorbic acid or 1, 6, 30 or 150 μM ellagic acid. All ROS modifiers were dissolved in HPLC water except bathocuproine, TBP, methional and ellagic acid which were dissolved in DMSO. Modified DNA was purified by solvent extractions and ethanol precipitation of DNA as previously described [35].
In order to generate readily detectable oxidation DNA products and to obtain reliable quantitative data in the presence of potent ROS scavengers, significantly higher levels of copper were used in these studies compared to known biological levels.
32P-Postlabeling Analysis
Analyses of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG) and the unidentified oxidation DNA products were performed separately as described elsewhere [36–37], respectively. Briefly, DNA (6 μg) was hydrolyzed to nucleoside 3′-monophosphates with micrococcal nuclease and spleen phosphodiesterase and adducts were enriched by treatment with nuclease P1 (unidentified DNA products) or 1-directiuonal PEI-cellulose TLC. The DNA products were 5′ −32P-labeled in the presence of molar excess of [γ −32P]ATP (<3 μM; 550 Ci/mmol) and T4 polynucleotide kinase. The labeled nucleotides were treated with a mixture of nuclease P1 and potato apyrase to convert labeled bisphosphates of the residual normal nucleotides to monophosphates and unused [γ −32P]ATP to 32Pi, respectively. The labeled 3′,5′-bisphosphate of 8-oxodG were converted to 5′-monophosphate by treatment with nuclease P1. The unidentified oxidation DNA products were resolved by two-directional polyethyleneimine (PEI)-cellulose TLC (D1 = 45 mM sodium phosphate, pH 5.8/1 M formic acid onto a 6 cm Whatman no. 17 paper wick; D2 = 100 mM sodium phosphate, pH 6.0/10% acetonitrile (v/v). The 8-oxodG were separated by two-directional PEI-cellulose TLC (D1 = 0.6 M formic acid onto a 5 cm Whatman no. 17 paper wick; and D 2 = 3 M sodium phosphate, pH 6.0).
Total nucleotides were analyzed by labeling dilute DNA digest (2 ng) in parallel with adducts followed by one-directional PEI-cellulose TLC in 0.5 M acetic acid/2 M formic acid [36]. The decomposition pattern was visualized and quantified by a Packard InstantImager. Oxidation DNA product levels were determined by relative adduct labeling (RAL), i.e., RAL = [cpm of the oxidative products/cpm of normal nucleotides] × 1/dilution factor. Levels are expressed as oxidation DNA products/106 nucleotides.
Co-chromatography of oxidation DNA products of Cu(II)-catalyzed reaction of dopamine and H2O2 were performed by applying similar amount of radioactivity in the DNA products in the two reaction mixtures individually and in combination onto PEI-cellulose thin layers and resolving the DNA products in two different solvents as indicated in the figure legend (see Figure 3).
Figure 3.

Autoradiographs representing co-chromatography of 32P-labeled DNA products resulting from Cu2+ (30 μM)-mediated activation of dopamine (30 μM), H2O2 (30 μM) and their mixture. Similar amounts of radioactivity were of the DNA products, individually and in combination, were applied onto PEI-cellulose thin layers and resolved using two different solvent systems: system 1) D1 = 35 mM sodium phosphate, pH 6 onto a 6 cm Whatman no. 17 paper wick; D2 = 80 mM sodium phosphate, pH 6/10% acetonitrile (v/v) and system 2) D1 = 35 mM sodium phosphate, pH 6 onto a 6 cm Whatman no. 17 paper wick; D2 = 0.9:1 (v/v) isopropyl alcohol:4M ammonium hydroxide.
Statistical Analysis
Statistical comparisons were made using the two-way ANOVA followed by Dunnett’s multiple comparison test. Values were considered significantly different when P ≤ 0.05.
Results
Detection of Oxidatively Damaged DNA
All catecholamine neurotransmitters and their congeners (30 μM) tested (Figure 1), in the presence of Cu(II) (30 μM CuCl2) and ST-DNA (300 μg/ml), resulted, in addition to 8-oxodG, the formation of a DNA decomposition pattern containing nearly a dozen unidentified oxidation DNA products (Figure 2) as detected by 32P-postlabeling coupled with a PEI-cellulose TLC [38]. The decomposition pattern was chromatographically identical for all neurotransmitters and their congeners tested (not shown). The characterization of these DNA decomposition products as oxidation DNA products is based on their ability to migrate under low-salt conditions, while DNA adducts formed from the covalent binding of quinone metabolites of catecholamine neurotransmitters, such as dopamine, to DNA require high-salt and high-urea concentrations to displace them from the origin during thin-layer chromatography [30]. An identical DNA decomposition pattern (Figure 3) was also found upon comparison with a Fenton-type reaction known to generate hydroxyl radicals from H2O2 (30 μM) with CuCl2 (30 μM) in the presence of ST-DNA (300 μg/ml) further supporting oxidative based characterization of these decomposition products.
Figure 1.

Structure of catecholamine neurotransmitters and their congeners.
Figure 2.

Representative autoradiographs of 32P-labeled DNA products resulting from auto-oxidation of dopamine (30 μM) and Cu2+ (30 μM)-mediated activation of dopamine (30 μM) and H2O2 (30 μM). Specific conditions are described in text. An identical DNA decomposition pattern following auto-oxidation and Cu(II)-mediated oxidation of dopamine was found and visualized with longer exposure times. Oxidation DNA products were resolved by two-directional polyethyleneimine (PEI)-cellulose TLC (D1 = 45 mM sodium phosphate, pH 5.8/1 M formic acid onto a 6 cm Whatman no. 17 paper wick; D2 = 100 mM sodium phosphate, pH 6.0/10% acetonitrile (v/v). OR, origin.
Most of these oxidation DNA products detected were found to be oxidatively modified dinucleotides since these DNA products co-migrated with oxidation DNA products formed by reaction of CuCl2 and individual dinucleotides (for example dApG, dGpT, etc.). Chromatographic identity of the DNA-derived and the dinucleotide-derived products was established in two different solvent systems (data not shown).
Unidentified oxidation DNA product levels mediated by Cu(II)-activation of the catechol neurotransmitters and their congeners ranged from 80 to 383 oxidative products/106 nucleotides with dopamine resulting in the highest levels (Figure 4). For comparison purposes 8-oxodG levels were also measured and ranged from 37 to 172 per 106 nucleotides with epinephrine resulting in the highest levels following Cu(II)-mediated catalysis. The Fenton-type reaction of H2O2 (30 μM) with CuCl2 (30 μM) resulted in 20-fold lower oxidation DNA product levels than dopamine with CuCl2 under the same conditions. In the absence of copper, unidentified oxidation DNA product levels ranged from <5 to 30 oxidative products/106 nucleotides for the individual neurotransmitters and their analogs while 8-oxodG levels ranged from 6 to 13 adducts/106 nucleotides (data not shown), indicating their potential for auto-oxidation.
Figure 4.

Oxidation DNA product levels of catecholamine neurotransmitters and their congeners (30 μM) following incubation with DNA (300 μg/ml) in the presence and absence (auto-oxidation) of CuCl2 (30 μM). Vehicle used was 1% DMSO. DNA product levels were determined as the mean of 3 to 4 replicates ± SE
In order to determine if copper was the only transition metal which could catalyze formation of these oxidation DNA products from catecholic neurotransmitters, dopamine was reacted with either Cu(I) (CuCl), Cu(II) (CuCl2), Fe(II) (FeSO4), or Fe(I) (FeCl3) (30 μM) in the presence of DNA (300 μg/ml) and 10 mM Tris HCl, pH 7.4. While both copper and iron-mediated activation of dopamine resulted in a chromatographically-identical DNA decomposition patterns, Cu (I), Fe(II) and Fe(I) resulted in oxidation DNA product levels 2, 6 and 15-fold lower, respectively than found in the presence of Cu(II) (data not shown).
Effect of ROS Modifiers on Dopamine-Mediated Oxidatively Damaged DNA
In order to determine the specific ROS which may be involved in copper-catalyzed dopamine oxidative DNA decomposition several ROS modifiers - ascorbic acid, mannitol, SOD, sodium benzoate, TBP, tyrosinase, TMP, sodium azide, catalase, tiron, bathocuproine and methional - used in previous studies at similar concentrations [30, 38], were added to the standard reaction mixture. No differences in the unidentified oxidative DNA decomposition patterns were observed following intervention with the modifiers tested (data not shown). However, significant differences in oxidation DNA product levels were observed (Figure 5). Bathocuproine, a Cu(I)-specific chelator, and methional, a CuOOH and hydroxyl radical scavenger, were two of the most potent inhibitors (>97%) of dopamine-mediated oxidative DNA decomposition tested. Also, substantially effective inhibitors of this oxidatively damaged DNA are catalase (97% inhibition), an enzyme which catalyzes the decomposition of H2O2 to water and oxygen, sodium azide (93% inhibition) a singlet oxygen and hydroxyl radical scavenger, at the high dose (100 mM) but not the lower dose (10 mM), and TMP (91% inhibition). Tiron, a superoxide scavenger, also substantially inhibited (97%) this oxidatively generated damage while SOD, an enzyme that catalyzes the dismutation of superoxide into oxygen and hydrogen peroxide, was largely ineffective (20% inhibition). The hydroxyl radical scavenger TBP significantly prevented (67%) dopamine-mediated oxidative DNA decomposition, however, the classic hydroxyl radical scavengers, mannitol and sodium benzoate were ineffective modulators (3% and 36%, respectively) of this oxidatively generated DNA damage as was ascorbic acid which, in fact, increased these oxidation DNA product levels by 36%, although was not statistically significant.
Figure 5.

Effect of ROS modifiers or vehicle (HPLC water) on Cu2+ (30 μM)-mediated oxidatively generated DNA damage by dopamine (30 μM). Levels of the oxidation DNA products were determined as the mean of 3 to 4 replicates ± SE. Asterick (*) indicates values significantly different from vehicle at P ≤ 0.05. Specific conditions are described in text. Modifiers and target ROS: bathocuproine, Cu(I); methional, Cu(I)OOH and hydroxyl radical; catalase, hydrogen peroxide; SOD and tiron, superoxide; sodium azide and TMP, singlet oxygen; ascorbic acid, mannitol and sodium benzoate, hydroxyl radical and TBP, hydroxyl radical, singlet oxygen and hydroperoxyl radical.
A similar pattern was observed for the ROS scavengers tested on 8-oxodG (Figure 5). TBP, TMP, sodium azide, catalase, tiron, bathocuproine and methional were all significant inhibitors of 8-oxodG formation (67–98%) while mannitol and SOD were ineffective.
Ellagic acid, a broad spectrum antioxidant and direct scavenger of a variety of free radicals, was highly effective at inhibiting dopamine/Cu(II)-mediated oxidatively generated DNA damage by 44%, 52%, 74% and 99% at low concentrations of 1, 6, 30 and 150 μM, respectively (Figure 6). Interestingly, tyrosinase, an enzyme which catalyzes the oxidation of phenols such as tyrosine and dopamine, also greatly inhibited dopamine-mediated oxidatively damaged DNA (84%).
Figure 6.
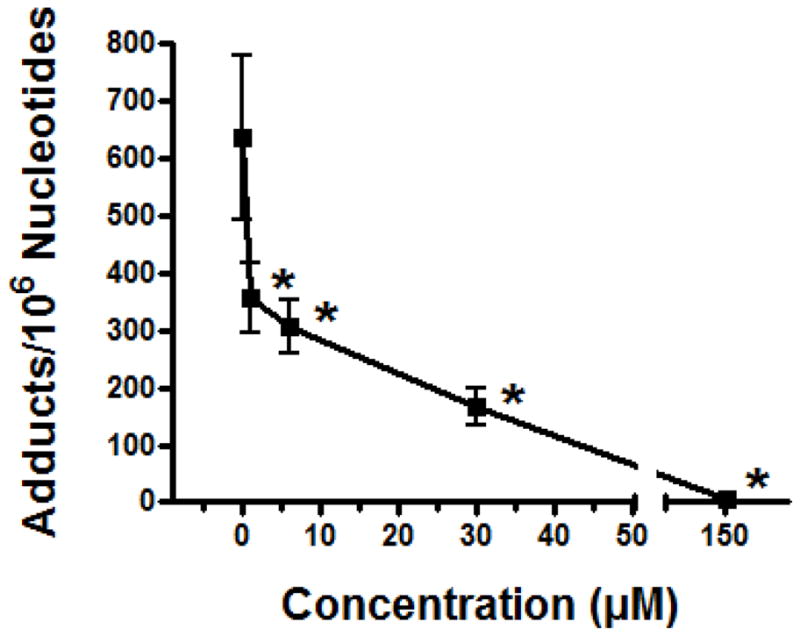
Effect of ellagic acid (1–150 μM) on Cu2+ (30 μM)-mediated oxidatively generated DNA damage by dopamine (30 μM). Oxidation DNA product levels were determined as the mean of 3 to 4 replicates ± SE. Asterick (*) indicates values significantly different from vehicle at P ≤ 0.05. Specific conditions are described in text.
Discussion
A growing body of evidence implicates a causal role of ROS and their subsequent damage to cellular macromolecules, such as DNA, in the neurodegeneration of the aging brain and in the development of neurodegenerative diseases such as PD. Loss of dopaminergic neurons in the substantia nigra is the hallmark of PD, however, significant degeneration of noradrenergic cells in the locus coeruleus has also been documented in PD as well other neurodegenerative diseases, including AD and Down Syndrome [27, 39–40]. The loss of these critical dopaminergic and noradrenergic neurons has been linked to oxidation, via quinone formation and subsequent redox cycling and ROS generation, of the catecholamine neurotransmitters dopamine [23, 30] and norepinephrine [27], respectively. We have demonstrated, in this study, that the endogenous catecholamine neurotransmitters dopamine and epinephrine can auto-oxidize resulting in the formation of numerous oxidation DNA products. The dopamine analogs, 6-OH dopamine, 5-OH dopamine and L-DOPA were also found to auto-oxidize in a similar fashion yielding an identical DNA decomposition pattern for all catecholamines tested.
Although the chemical structure of these oxidative products has not been identified, several observations indicate that they are oxidatively derived. The thin-layer chromatography system employed for separating these adducts is based on low-salt conditions, where as separation of aromatic adducts derived from the covalent binding of the quinone/semi-quinone derivatives of catecholamines, such as dopamine, to DNA requires high-salt high-urea conditions [30]. Further, the migration pattern of these oxidation products, under these low-salt conditions, is also in proximity to known oxidative markers, including 5-hydroxy-2′-deoxyuridine, thymidine glycol, and 8-oxodG [37]. Importantly, chromatographic comparison with DNA decomposition products generated from typical Fenton chemistry, reduction of H2O2 in the presence of reduced transition metals Fe(II) or Cu(I), resulted in a chromatographically identical DNA decomposition pattern as that generated by the catecholamines in this study. Further, these oxidation products were also essentially chromatographically identical to those of numerous structurally diverse redox-active agents, including estrogen catechols (unpublished data), PAH diols [41] and dihydroxylated PCBs [38]. Finally, the DNA decomposition pattern generated by these catecholamine neurotransmitters, as well as those generated by other redox-active agents exhibiting an identical decomposition pattern, has been found to be inhibited by antioxidants [38, 42].
Oxidation of the catecholamines in the presence of the transition metal copper, and to a lesser extent iron, resulted in an identical DNA decomposition pattern but substantially enhanced the DNA oxidation product levels by 4 to 75 fold. This observation of increased oxidation DNA products by catecholamine neurotransmitters in the presence of copper is supported by a recent study in which the auto-oxidation of a neurodegenerative dopamine analog, 6-OH dopamine, was found to be accelerated in the presence of Cu(II) and Fe(III)/EDTA but at a much faster rate for Cu(II) than Fe(III)/EDTA [43]. Further, in the same study, Cu(II)-mediated catalysis of 6-OH dopamine also resulted in higher 8-oxodG levels than Fe(III)/EDTA in a cellular system. Although all catecholamines tested in the current study resulted in significantly increased levels of unidentified oxidation DNA products in the presence of Cu(II), dopamine, followed by 6-OH dopamine, produced the highest amounts resulting in oxidative product levels of 378 adducts/106 nucleotides. For comparison purposes, 8-oxodG levels were measured using 32P-postlabeling [36] and were found to be three fold lower (122 adducts/106 nucleotides) for Cu(II)-mediated activation of dopamine (data not shown) under the same reaction conditions.
Copper is an essential and biologically relevant metal required for normal enzymatic function and found throughout the human body including the brain. In a recent study, levels of copper were found to approximate 16 and 31 ng/g in the substantia nigra and the locus coeruleus, respectively, of normal brain tissue [44]. Copper and its abnormal homeostasis have been linked to the pathogenesis of numerous neurological diseases including neuronal cell death in PD [45]. DNA is believed to be an especially sensitive site within the cell for copper-mediated damage, as these ions have a high affinity for nucleic acids and bind to DNA both at intrastrand and interstrand levels [34]. In a recent study, copper-mediated catalysis of 6-hydroxydopamine, a neurotoxin used to produce PD in animal models, increased the formation of 8-oxodG followed by a subsequent initiation of apoptosis of human neuroblastoma cells [43]. This study further supports the potential role of copper-catalyzed catecholamine induced oxidatively damaged DNA and neuronal cell death in PD.
In order to investigate the mechanism(s) and identify the potential ROS involved in this catecholamine-mediated oxidatively generated DNA damage, several ROS modifiers were studied for their effects on Cu(II)-catalyzed dopamine oxidation and subsequent DNA decomposition. Catalase, an enzyme which catalyzes the decomposition of H2O2 to water and oxygen, was one of the most effective inhibitors of this oxidative polar DNA adduction implicating H2O2 as an important mediator in this DNA damage. The significance of H2O2 in the Cu(II)-catalyzed oxidation of dopamine to form these oxidation DNA products directly supports our observation of an identical DNA decompositiont pattern as that of the reaction of Cu(II) with H2O2. Studies using the catechol neurotoxin 6-OH dopamine, also showed a significant in the presence of Cu(II) in neuronal cells [43]. Cu(I) was also important for production of H2O2 the dopamine/Cu(II)-mediated oxidatively generated DNA damage as bathocuproine, a Cu(I)-specific chelator, substantially inhibited its formation. Tiron, a superoxide scavenger, but not SOD, also significantly inhibited this oxidatively generated DNA damage. In fact, higher levels of SOD actually increased the oxidative DNA decomposition in this dopamine/Cu(II) system (data not shown). SOD’s effect was not unexpected as H2O2 is generated during the dismutation of superoxide. The singlet oxygen scavengers, sodium azide and TMP, were also effective inhibitors of oxidatively damaged DNA indicating a contributory role of singlet oxygen in mediating dopamine/Cu(II)-induced oxidative DNA degradation. Studies have shown that oxidation of double-stranded DNA in aqueous solutions by singlet oxygen result predominantly in the formation of 8-oxodG, particularly when the extent of guanine oxidation remains low (<1%) [46]. However, oxidation of 2′-deoxyguanosine by singlet oxygen has also been shown to yield other oxidative products including 5-hydroxy-8-oxo-7,8-dihydro-2′-deoxyguanosine (5-OH-8-oxodG), and upon rearrangement, spiroiminodihydantoin nucleosides (dSP) [46]. It should be noted, however, that the diastereomers of dSP were not found to be generated in DNA duplexes under conditions where the extent of guanine oxidation was low. Further, decreased specificity of the ROS scavengers tested for particular radical species at somewhat high doses cannot be ruled out as a potential confounding factor in these studies.
Classical hydroxyl radical scavengers [47], mannitol, sodium benzoate and ascorbic acid were ineffective inhibitors of dopamine-mediated oxidatively damaged DNA. However, TBP, a well known hydroxyl radical scavenger, with recently identified singlet oxygen [48] and hydroperoxyl [49] scavenging properties as well, was a moderate inhibitor (37%) of this oxidatively damaged DNA suggesting that singlet oxygen and/or the hydroperoxyl radical, but not the hydroxyl radical, may mediate dopamine/Cu(II) oxidatively generated DNA damage. Methional, a hydroxyl radical scavenger and inhibitor of metal-oxygen complexes [50], substantially inhibited dopamine/Cu(II) oxidative product levels. The lack of efficacy of the classical hydroxyl radical scavengers tested suggests that methional is most likely scavenging a copper-peroxide complex which may be an important mediator of this oxidatively damaged DNA. Several studies have implicated a copper-peroxide complex in the involvement of oxidatively generated DNA damage, including the formation of 8-oxodG and DNA strand breaks, by redox cycling of catechols [43, 51–52]. Although still being debated, site-specific DNA-Cu(I)OOH complex is believed to be the primary intermediate in DNA damage. This complex is hypothesized to release hydroxyl radicals that can immediately react with adjacent DNA sites but are essentially unavailable to radical scavengers in the medium due to their juxtapose proximity to DNA and their high reaction rates [51]. Alternatively, singlet oxygen has been implicated as the primary species mediating the formation of 8-oxodG, and now based on this study, additional oxidation DNA products, from copper-catalyzed oxidation of catechols and H2O2 [47, 53]. The formation of a copper-hydroperoxide complex has also been suggested to be the source of the singlet oxygen in these studies.
Based on these findings we propose the following mechanism and potential ROS involved for the formation of these unidentified oxidation DNA products and subsequent neuronal cell death from Cu(II)-catalyzed oxidation of dopamine and related catecholamine neurotransmitters and neurotoxins (Figure 7). The auto-oxidation of catecholamine neurotransmitters, such as dopamine, and related catecholamines is substantially enhanced by the presence of transitions metals, especially copper, in their oxidized form. In the presence of O2, these catechols can redox cycle producing quinone metabolites via semi-quinone intermediates while simultaneously reducing the metal. Superoxide is then generated by the reduction of molecular oxygen followed by the re-generation of the metal to its oxidized state. Superoxide can also react with Cu(I) to produce H2O2 which then complexes with either Cu(I) or Cu(II) bound to DNA to form DNA-Cu(I)OOH. Following the formation of this DNA-Cu(I)OOH a variety of oxidative DNA lesions, such as 8-oxodG and the unidentified oxidation DNA products in this study, result due to the production of hydroxyl radicals at site-specific regions of the DNA. Singlet oxygen may also be produced from this complex yielding predominantly 8-oxodG and potentially other minor DNA degradation products either from oxidation of 2′-deoxyguanosine or possibly further oxidation of 8-oxodG [54]. Recent studies strongly support the role of one-electron oxidation as the predominant pathway in the degradation of DNA by Cu(II) in the presence of hydrogen peroxide as opposed to the generation of free hydroxyl radicals by the Fenton reaction [53]. One-electron oxidation of DNA leads to the predominant degradation of guanine since it exhibits the lowest oxidation potential among DNA components. Our data support this mechanism in dopamine/Cu(II)-mediated DNA oxidation as the majority of DNA degradation products detected appear to be guanine derived. However, a favored degradation of guanine could also be accounted for by the preferential complexation of Cu(I) to guanine resulting in the generation of site-specfic nonscavengeable hydroxyl radicals which may also account for low levels of oxidatively generated damage to other DNA bases adjacent to copper-bound guanine [53].
Figure 7.

Proposed role of ROS in dopamine-mediated oxidatively generated DNA damage and subsequent neuronal cell death. The auto-oxidation of catecholamine neurotransmitters, such as dopamine, and related catecholamines, is substantially enhanced by the presence of transitions metals, especially copper, in their oxidized form. In the presence of O2, these catechols can redox cycle producing quinone metabolites, which can bind directly to DNA forming covalent DNA adducts, via semi-quinone intermediates while simultaneously reducing the metal. Superoxide is then generated by the reduction of molecular oxygen followed by the re-generation of the metal to its oxidized state. Superoxide can also react with Cu(I) to produce H2O2 which then can complex with either Cu(I) or Cu(II) bound to DNA to form DNA-Cu(I)OOH. Following the formation of this DNA-Cu(I)OOH a variety of oxidative DNA lesions, including the unidentified oxidation DNA products studied here, result due to the production of hydroxyl radicals at site-specific regions of the DNA. Singlet oxygen may also be produced from this complex yielding 8-oxodG.
In order to assess the potential of a broad spectrum antioxidant to mediate dopamine/Cu(II)-induced oxidative DNA adduction, ellagic acid was evaluated for its efficacy in this model system. Ellagic acid is a naturally occurring polyphenol found in a wide variety of fruits, nuts and particularly berries, including raspberries, strawberries, and black currants and is the major phenolic constituent present in distilled beverages [55]. An average daily intake of ellagic acid, through consumption of fruits and vegetables, has been estimated to be 5.2 mg [56]. Ellagic acid exhibits potent antioxidant activity in addition to its antimutagenic and anti-inflammatory properties [57]. The primary antioxidant mechanism of ellagic acid has been attributed to its direct scavenging of free radicals, nitrogen reactive species and ROS, including hydroxyl radicals, peroxyl radicals, NO2 radicals and peroxynitrite [57–58]. Other potential protective mechanisms of ellagic acid include shielding of DNA from attack by its direct association with this macromolecule [59], inhibition of ROS production and chelation of metal ions such as copper [57, 60]. In the current study, ellagic acid substantially inhibited dopamine/Cu(II)-mediated oxidative DNA decomposition at all doses tested, by nearly 50% at doses as low as 1 μM. These results coupled with those of a recent study which indicated that dietary ellagic acid significantly protected brain tissue of rats from tetrachlrodibenzodioxin (TCDD)-induced superoxide anion production, lipid peroxidation and DNA strand breaks [61], support the potential role of ellagic acid in neuroprotection of oxidatively generated DNA damage.
Also of interest, tyrosinase, a copper monooxygenase which catalyzes the oxidation of catechols, such as dopamine, to their corresponding quinones, substantially inhibited dopamine/Cu(II)-mediated oxidative DNA adduction. In a previous study tyrosinase was shown to increase the covalent incorporation of [H3]-dopamine into DNA resulting in the formation of covalent DNA adducts, presumably via a quinone-related pathway [62]. Based on these previous results, tyrosinase was included in the current study and was hypothesized to increase the formation of these dopamine/Cu(II)-mediated oxidation DNA products by increasing the rate of redox cycling between dopamine and its corresponding quinone. The unexpected decrease observed in the oxidation DNA product levels may be attributed to its catalase activity [63] or possibly by sequestering, otherwise available Cu(II), in its active site.
In conclusion, dopamine and other catecholamine neurotransmitters and neurotoxins were found to produce significant levels of numerous oxidation DNA products which were substantially elevated in the presence of transition metals, especially Cu(II). The formation of these products is mediated by Cu(I), singlet oxygen, superoxide and H2O2 and potentially results from one-electron oxidation and/or the site-specific attack of hydroxyl radicals via a DNA-Cu(I)OOH complex. Since accumulation of oxidative DNA base modifications has been identified as a major contributing factor to genomic instability and mitochondrial dysfunction in aging and neurodegenerative disorders such as PD and AD [11, 64], catecholamine-mediated oxidatively damaged DNA in nuclear, and particularly mitochondrial, DNA likely contributes to neuronal death associated with these degenerative processes. Naturally occurring antioxidants such as ellagic acid may attenuate these neurodegenerative processes and should be further evaluated towards this role in vivo.
Acknowledgments
This work was supported by USPHS grant CA-90892 and Agnes Brown Duggan Endowment. Dr. Ramesh Gupta is Agnes Brown Duggan Chair in Oncological Research.
Footnotes
Some part of this work was performed at the Department of Preventive Medicine and Environmental Health, and Graduate Center for Toxicology, University of Kentucky, Lexington, Kentucky 40536
Abbreviations: AD, Alzheimer’s disease; PD, Parkinson’s disease; ROS, reactive oxygen species; nDNA, nuclear DNA; mtDNA, mitochondrial DNA; DA, dopamine; NE, norepinephrine; ST-DNA, salmon testis-DNA; SOD, superoxide dismutase; TBP, N-tert-butyl-α-phenylnitrone; TMP, 2,2,6,6-tetramethyl-4-piperidone; PEI, polyethyleneimine; 8-oxodG, 8-oxo-7,8-dihydro-2′-deoxyguanosine; TCDD, tetrachlrodibenzodioxin; 5-OH-8-oxodG, 5-hydroxy-8-oxo-7,8-dihydro-2′-deoxyguanosine; dSP, spiroiminodihydantoin nucleosides
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- 3.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 6.Kikuchi A, Takeda A, Onodera H, Kimpara T, Hisanaga K, Sato N, Nunomura A, Castellani RJ, Perry G, Smith MA, Itoyama Y. Systemic increase of oxidative nucleic acid damage in Parkinson’s disease and multiple system atrophy. Neurobiol Dis. 2002;9:244–248. doi: 10.1006/nbdi.2002.0466. [DOI] [PubMed] [Google Scholar]
- 7.Rao KS. Free radical induced oxidative damage to DNA: relation to brain aging and neurological disorders. Indian J Biochem Biophys. 2009;46:9–15. [PubMed] [Google Scholar]
- 8.Lyras L, Perry RH, Perry EK, Ince PG, Jenner A, Jenner P, Halliwell B. Oxidative damage to proteins, lipids, and DNA in cortical brain regions from patients with dementia with Lewy bodies. J Neurochem. 1998;71:302–312. doi: 10.1046/j.1471-4159.1998.71010302.x. [DOI] [PubMed] [Google Scholar]
- 9.Jenner P, Olanow CW. Understanding cell death in Parkinson’s disease. Ann Neurol. 1998;44:S72–84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- 10.Dorszewska J, Florczak J, Rozycka A, Kempisty B, Jaroszewska-Kolecka J, Chojnacka K, Trzeciak WH, Kozubski W. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer’s and Parkinson’s diseases. Acta Neurobiol Exp (Wars) 2007;67:113–129. doi: 10.55782/ane-2007-1639. [DOI] [PubMed] [Google Scholar]
- 11.Weissman L, de Souza-Pinto NC, Stevnsner T, Bohr VA. DNA repair, mitochondria, and neurodegeneration. Neuroscience. 2007;145:1318–1329. doi: 10.1016/j.neuroscience.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 12.Lotharius J, Falsig J, van Beek J, Payne S, Dringen R, Brundin P, Leist M. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25:6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J Neurochem. 2004;88:657–667. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- 14.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 15.Markesbery WR, Carney JM. Oxidative alterations in Alzheimer’s disease. Brain Pathol. 1999;9: 133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bayir H, Kapralov AA, Jiang J, Huang Z, Tyurina YY, Tyurin VA, Zhao Q, Belikova NA, Vlasova, Maeda A, Zhu J, Na HM, Mastroberardino PG, Sparvero LJ, Amoscato AA, Chu CT, Greenamyre JT, Kagan VE. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C: protection against apoptosis versus delayed oxidative stress in Parkinson disease. J Biol Chem. 2009;284:15951–15969. doi: 10.1074/jbc.M900418200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson’s disease: proximal triggers, distal effectors, and final steps. Apoptosis. 2009;14:478–500. doi: 10.1007/s10495-008-0309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 19.de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab Invest. 2000;80:1323–1335. doi: 10.1038/labinvest.3780140. [DOI] [PubMed] [Google Scholar]
- 20.Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J. Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol Aging. 1990;11:169–173. doi: 10.1016/0197-4580(90)90542-8. [DOI] [PubMed] [Google Scholar]
- 21.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 22.Wallace DC. Diseases of the mitochondrial DNA. Annu Rev Biochem. 1992;61:1175–1212. doi: 10.1146/annurev.bi.61.070192.005523. [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki I, Asanuma M. Dopaminergic neuron-specific oxidative stress caused by dopamine itself. Acta Med Okayama. 2008;62:141–150. doi: 10.18926/AMO/30942. [DOI] [PubMed] [Google Scholar]
- 24.Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- 25.Luo Y, Roth GS. The roles of dopamine oxidative stress and dopamine receptor signaling in aging and age-related neurodegeneration. Antioxid Redox Signal. 2000;2:449–460. doi: 10.1089/15230860050192224. [DOI] [PubMed] [Google Scholar]
- 26.Masserano JM, Baker I, Venable D, Gong L, Zullo SJ, Merril CR, Wyatt RJ. Dopamine induces cell death, lipid peroxidation and DNA base damage in a catecholaminergic cell line derived from the central nervous system. Neurotox Res. 2000;1:171–179. doi: 10.1007/BF03033288. [DOI] [PubMed] [Google Scholar]
- 27.Manini P, Panzella L, Napolitano A, d’Ischia M. Oxidation chemistry of norepinephrine: partitioning of the O-quinone between competing cyclization and chain breakdown pathways and their roles in melanin formation. Chem Res Toxicol. 2007;20:1549–1555. doi: 10.1021/tx700254q. [DOI] [PubMed] [Google Scholar]
- 28.Michel PP, Ruberg M, Hirsch E. Dopaminergic neurons reduced to silence by oxidative stress: an early step in the death cascade in Parkinson’s disease? Sci STKE. 2006;2006:pe19. doi: 10.1126/stke.3322006pe19. [DOI] [PubMed] [Google Scholar]
- 29.da Silva GF, Ming LJ. Metallo-ROS in Alzheimer’s disease: oxidation of neurotransmitters by CuII-beta-amyloid and neuropathology of the disease. Angew Chem Int Ed Engl. 2007;46:3337–3341. doi: 10.1002/anie.200604421. [DOI] [PubMed] [Google Scholar]
- 30.Levay G, Ye Q, Bodell WJ. Formation of DNA adducts and oxidative base damage by copper mediated oxidation of dopamine and 6-hydroxydopamine. Exp Neurol. 1997;146:570–574. doi: 10.1006/exnr.1997.6560. [DOI] [PubMed] [Google Scholar]
- 31.Spencer JP, Jenner A, Aruoma OI, Evans PJ, Kaur H, Dexter DT, Jenner P, Lees AJ, Marsden DC, Halliwell B. Intense oxidative DNA damage promoted by L-dopa and its metabolites. Implications for neurodegenerative disease. FEBS Lett. 1994;353:246–250. doi: 10.1016/0014-5793(94)01056-0. [DOI] [PubMed] [Google Scholar]
- 32.Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain. 1991;114(Pt 4):1953–1975. doi: 10.1093/brain/114.4.1953. [DOI] [PubMed] [Google Scholar]
- 33.Dexter DT, Ward RJ, Wells FR, Daniel SE, Lees AJ, Peters TJ, Jenner P, Marsden CD. Alpha-tocopherol levels in brain are not altered in Parkinson’s disease. Ann Neurol. 1992;32:591–593. doi: 10.1002/ana.410320420. [DOI] [PubMed] [Google Scholar]
- 34.Agarwal K, Sharma A, Talukder G. Effects of copper on mammalian cell components. Chem Biol Interact. 1989;69:1–16. doi: 10.1016/0009-2797(89)90094-x. [DOI] [PubMed] [Google Scholar]
- 35.Gupta R. In: Technologies for Detection of DNA Damage and Mutations32P-Postlabeling for detection of DNA adducts. Pfeifer G, editor. New York: Plenum Press; 1996. pp. 45–61. [Google Scholar]
- 36.Gupta RC, Arif JM. An improved (32)P-postlabeling assay for the sensitive detection of 8-oxodeoxyguanosine in tissue DNA. Chem Res Toxicol. 2001;14:951–957. doi: 10.1021/tx000131d. [DOI] [PubMed] [Google Scholar]
- 37.Ravoori S, Vadhanam MV, Davey DD, Srinivasan C, Nagarajan B, Gupta RC. Modulation of novel DNA adducts during human uterine cervix cancer progression. Int J Oncol. 2006;29:1437–1443. [PubMed] [Google Scholar]
- 38.Spencer WA, Lehmler HJ, Robertson LW, Gupta RC. Oxidative DNA adducts after Cu(2+)-mediated activation of dihydroxy PCBs: role of reactive oxygen species. Free Radic Biol Med. 2009;46:1346–1352. doi: 10.1016/j.freeradbiomed.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gesi M, Soldani P, Giorgi FS, Santinami A, Bonaccorsi I, Fornai F. The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci Biobehav Rev. 2000;24:655–668. doi: 10.1016/s0149-7634(00)00028-2. [DOI] [PubMed] [Google Scholar]
- 40.Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- 41.Vadhanam MV, Krzeminski J, Amin S, Gupta RC. Cu2+-mediated oxidative DNA adducts from aromatic hydrocarbon catechols and diones. Proc Natl Acad Sci U S A. 2002;43:abstract 4289. [Google Scholar]
- 42.Aiyer HS, Vadhanam MV, Gupta RC. Efficacy of cancer chemopreventive agents in protecting against oxidative DNA damage from Cu2+-mediated activation of 4-hydroxy estradiol. Proc Natl Acad Sci U S A. 2002;43:abstract 5682. [Google Scholar]
- 43.Kobayashi H, Oikawa S, Umemura S, Hirosawa I, Kawanishi S. Mechanism of metal-mediated DNA damage and apoptosis induced by 6-hydroxydopamine in neuroblastoma SH-SY5Y cells. Free Radic Res. 2008;42:651–660. doi: 10.1080/10715760802270334. [DOI] [PubMed] [Google Scholar]
- 44.Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, Giaveri G, Arosio P, Santambrogio P, Fariello RG, Karatekin E, Kleinman MH, Turro N, Hornykiewicz O, Zucca FA. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A. 2004;101:9843–9848. doi: 10.1073/pnas.0403495101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Desai V, Kaler SG. Role of copper in human neurological disorders. Am J Clin Nutr. 2008;88:855S–858S. doi: 10.1093/ajcn/88.3.855S. [DOI] [PubMed] [Google Scholar]
- 46.Cadet J, Ravanat JL, Martinez GR, Medeiros MH, Di Mascio P. Singlet oxygen oxidation of isolated and cellular DNA: product formation and mechanistic insights. Photochem Photobiol. 2006;82:1219–1225. doi: 10.1562/2006-06-09-IR-914. [DOI] [PubMed] [Google Scholar]
- 47.Park JH, Gopishetty S, Szewczuk LM, Troxel AB, Harvey RG, Penning TM. Formation of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo) by PAH o-quinones: involvement of reactive oxygen species and copper(II)/copper(I) redox cycling. Chem Res Toxicol. 2005;18:1026–1037. doi: 10.1021/tx050001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawai A, Nishinaka Y, Arai T, Hirota K, Mori H, Endo N, Miyoshi T, Yamashita K, Sasada M. Alpha-phenyl-N-tert-butyl nitrone has scavenging activity against singlet oxygen ((1)O(2)) and attenuates (1)O(2)-induced neuronal cell death. J Pharmacol Sci. 2008;108:545–549. doi: 10.1254/jphs.08233sc. [DOI] [PubMed] [Google Scholar]
- 49.Durand G, Choteau F, Pucci B, Villamena FA. Reactivity of superoxide radical anion and hydroperoxyl radical with alpha-phenyl-N-tert-butylnitrone (PBN) derivatives. J Phys Chem A. 2008;112:12498–12509. doi: 10.1021/jp804929d. [DOI] [PubMed] [Google Scholar]
- 50.Pryor WA, Tang RH. Ethylene formation from methional. Biochem Biophys Res Commun. 1978;81:498–503. doi: 10.1016/0006-291x(78)91562-0. [DOI] [PubMed] [Google Scholar]
- 51.Schweigert N, Acero JL, von Gunten U, Canonica S, Zehnder AJ, Eggen RI. DNA degradation by the mixture of copper and catechol is caused by DNA-copper-hydroperoxo complexes, probably DNA-Cu(I)OOH. Environ Mol Mutagen. 2000;36:5–12. doi: 10.1002/1098-2280(2000)36:1<5::aid-em2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 52.Oikawa S, Kawanishi S. Site-specific DNA damage induced by NADH in the presence of copper(II): role of active oxygen species. Biochemistry. 1996;35:4584–4590. doi: 10.1021/bi9527000. [DOI] [PubMed] [Google Scholar]
- 53.Frelon S, Douki T, Favier A, Cadet J. Hydroxyl radical is not the main reactive species involved in the degradation of DNA bases by copper in the presence of hydrogen peroxide. Chem Res Toxicol. 2003;16:191–197. doi: 10.1021/tx025650q. [DOI] [PubMed] [Google Scholar]
- 54.Shao J, Geacintov NE, Shafirovich V. Oxidation of 8-oxo-7,8-dihydro-2′-deoxyguanosine by oxyl radicals produced by photolysis of azo compounds. Chem Res Toxicol. 2010;23:933–938. doi: 10.1021/tx100022x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldberg DM, Hoffman B, Yang J, Soleas GJ. Phenolic constituents, furans, and total antioxidant status of distilled spirits. J Agric Food Chem. 1999;47:3978–3985. doi: 10.1021/jf9811626. [DOI] [PubMed] [Google Scholar]
- 56.Radtke J, Linseisen J, Wolfram G. Phenolic acid intake of adults in a Bavarian subgroup of the national food consumption survey. Z Ernahrungswiss. 1998;37:190–197. doi: 10.1007/s003940050016. [DOI] [PubMed] [Google Scholar]
- 57.Priyadarsini KI, Khopde SM, Kumar SS, Mohan H. Free radical studies of ellagic acid, a natural phenolic antioxidant. J Agric Food Chem. 2002;50:2200–2206. doi: 10.1021/jf011275g. [DOI] [PubMed] [Google Scholar]
- 58.Gerhauser C, Klimo K, Heiss E, Neumann I, Gamal-Eldeen A, Knauft J, Liu GY, Sitthimonchai S, Frank N. Mechanism-based in vitro screening of potential cancer chemopreventive agents. Mutat Res. 2003;523–524:163–172. doi: 10.1016/s0027-5107(02)00332-9. [DOI] [PubMed] [Google Scholar]
- 59.Thulstrup PW, Thormann T, Spanget-Larsen J, Bisgaard HC. Interaction between ellagic acid and calf thymus DNA studied with flow linear dichroism UV-VIS spectroscopy. Biochem Biophys Res Commun. 1999;265:416–421. doi: 10.1006/bbrc.1999.1694. [DOI] [PubMed] [Google Scholar]
- 60.Bock PE, Srinivasan KR, Shore JD. Activation of intrinsic blood coagulation by ellagic acid: insoluble ellagic acid-metal ion complexes are the activating species. Biochemistry. 1981;20:7258–7266. doi: 10.1021/bi00528a032. [DOI] [PubMed] [Google Scholar]
- 61.Hassoun EA, Vodhanel J, Abushaban A. The modulatory effects of ellagic acid and vitamin E succinate on TCDD-induced oxidative stress in different brain regions of rats after subchronic exposure. J Biochem Mol Toxicol. 2004;18:196–203. doi: 10.1002/jbt.20030. [DOI] [PubMed] [Google Scholar]
- 62.Stokes AH, Brown BG, Lee CK, Doolittle DJ, Vrana KE. Tyrosinase enhances the covalent modification of DNA by dopamine. Brain Res Mol Brain Res. 1996;42:167–170. doi: 10.1016/s0169-328x(96)00164-7. [DOI] [PubMed] [Google Scholar]
- 63.Yamazaki S, Morioka C, Itoh S. Kinetic evaluation of catalase and peroxygenase activities of tyrosinase. Biochemistry. 2004;43:11546–11553. doi: 10.1021/bi048908f. [DOI] [PubMed] [Google Scholar]
- 64.Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K. Oxidative damage in nucleic acids and Parkinson’s disease. J Neurosci Res. 2007;85:919–934. doi: 10.1002/jnr.21191. [DOI] [PubMed] [Google Scholar]