

Abstract
Natural killer (NK) cells exhibit a polarized phenotype with increased cytotoxicity and decreased IFN- γ production in chronic hepatitis C virus (HCV) infection. Here we asked whether this is due to type I interferon (IFN)-induced expression and phosphorylation levels of signal transducer and activator of transcription (STAT) molecules in NK cells and whether it affects the response and refractoriness of NK cells to IFN-α-based therapy of hepatitis C. STAT1 levels in NK cells were significantly higher in patients with chronic HCV infection than in uninfected controls. STAT1 levels and induction of phosphorylated STAT1 (pSTAT1) increased further during IFN-α-based therapy with preferential STAT1 over STAT4 phosphorylation. Induction of pSTAT1 correlated with increased NK cytotoxicity (TRAIL expression and degranulation) and decreased IFN-γ production. NK cells from patients with a greater than 2 log10 first phase HCV RNA decline to IFN-α-based therapy (>99% IFN effectiveness) displayed strong pSTAT1 induction in vivo and were refractory to further stimulation in vitro. In contrast, NK cells from patients with a less than 2 log10 first phase HCV RNA decline exhibited lower pSTAT1 induction in vivo (p=0.024) but retained greater IFN-α responsiveness in vitro (p=0.024). NK cells of all patients became refractory to in vivo and in vitro stimulation by IFN-α during the second phase virological response.
Conclusion
These data show that IFN-α-induced modulation of STAT1/4 phosphorylation underlies the polarization of NK cells towards increased cytotoxicity and decreased IFN-γ production in HCV infection, and that NK cell responsiveness and refractoriness correlate to the antiviral effectiveness of IFN-α-based therapy.
Keywords: interferon, innate, hepatitis C virus, treatment, natural killer
Introduction
Natural killer (NK) cells are innate immune cells that are best known for their immediate effector functions against virus-infected cells and tumor cells (1). These effector functions include the destruction of target cells via perforin/granzyme-mediated lysis or TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis, and the production of cytokines such as TNF-α, MIP-1β and IFN-γ (1). IFN-γ in particular has elicited great interest because it is abundantly produced, has direct antiviral activity and provides a link between innate and adaptive immunity by contributing to the priming of CD4+ and CD8+ T cells, and via induction of chemokines to T cell recruitment to the target organ (2).
Different effector functions have traditionally been associated with specific NK cell subsets, which can be distinguished based on CD56 expression. Approximately 90% of NK cells in the peripheral blood express low levels of CD56 on their cell surface. These CD56dim NK cells respond quickly to viral infection, exert cytotoxicity and produce chemokines and cytokines within hours (3–5). The remaining 10% of NK cells with high level of CD56 expression (CD56bright) respond slower and produce large amounts of IFN-γ and TRAIL with little perforin/granzyme-mediated cytotoxicity.
We and others have recently shown that patients with chronic HCV infection display a polarized NK cell phenotype with increased cytotoxicity and TRAIL production and decreased IFN-γ production (6–8). Induction of cytotoxicity and production of IFN-γ require differential STAT1/4 signaling as previously shown in a mouse model of lymphocytic choriomeningitis virus-induced hepatitis (9). In this model, virus-induced type I IFN results in increased expression of STAT1, which competes with STAT4 in the signaling events downstream of the IFN-α/β receptor (9). The result is preferential STAT1 over STAT4 phosphorylation, increased NK cell cytotoxicity and decreased IFN-γ production (9, 10). Interestingly, Miyagi et al. demonstrated increased STAT1 levels in NK cells of HCV-infected patients as compared to healthy controls and showed that in vitro stimulation with IFN-α resulted in preferential STAT1 over STAT4 phosphorylation (11). However, a demonstration that changes in IFN-signaling correlate with changes in NK cell function in HCV-infected patients has not yet been provided. Furthermore, the kinetics of the in vivo responsiveness of NK cells to IFN in humans are not known and may be very important for the therapeutic use of IFN-α, e.g. for the therapy of chronic HCV infection.
To address these points we performed a prospective analysis of STAT expression and phosphorylation in NK cells in chronic HCV infection and during the first 12 weeks of IFN-α-based therapy. This time period defines an early virological response (EVR), which is predictive of the ultimate treatment outcome (12). Changes in STAT signaling during this time period were correlated with changes in NK cell effector functions. In addition, the study included several time points during the first 48h of treatment, which allowed us to correlate changes in IFN-induced signaling in NK cells to the first phase decline in HCV titer (13). The results provide novel insights into the mechanisms of IFN-responsiveness and refractoriness of NK cells during viral infection and IFN-a-based therapy.
Materials and Methods
Study cohort
Peripheral blood NK cells were studied in 10 healthy subjects without HCV infection and 35 untreated patients with chronic HCV infection. Twenty-four patients with chronic HCV infection (Table 1) were prospectively studied during treatment with peginterferon (PegIFN) alfa-2a (180 μg/week s.c.) and weight-based ribavirin (RBV, 1000 mg for <75 kg bodyweight and 1200 mg for ≥75 kg bodyweight p.o daily for HCV genotypes 1 and 4; 800 mg p.o. daily for HCV genotypes 2 and 3) 4 weeks and 0h prior to treatment, and 6, 24 and 48h, and 1, 2, 4 and 12 weeks after treatment initiation. The week 1, 2, 4 and 12 samples were drawn prior to the weekly PegIFN injection. Two patients consented to an additional blood draw 6h after the week 12 PegIFN injection. All subjects gave written informed consent under protocols approved by the NIDDK Institutional Review Board conforming to the ethical guidelines of the 1975 Declaration of Helsinki.
Table 1.
Epidemiological and clinical data of HCV-infected patients
| Chronic HCV patients, not treated with PegIFN/RBV (n=35) | Chronic HCV patients, studied during PegIFN/RBV therapy (n=24) | |
|---|---|---|
| Early virological response, n (%) | n.a. a | 23 (100%) b |
| Gender (male/female) | 20/15 | 16/8 |
| Age at start of treatment, median (IQR c) years | 49.0 (44–55) | 53.5 (50–55) |
| Ethnicity (asian/african American/ caucasian/hispanic) | 7/10/18/0 | 3/5/15/1 |
| Body mass index, median (IQR) | 29.0 (23.7–34.1) | 28.9 (23.6–33.5) |
| IL-28B rs12979860 SNPd (CC/CT/TT) | n.d. e | 13/6/3 |
| HCV genotype (1/2/3/4/6) | 19/5/5/4/1/n.d. | 13/8/2/1 |
| Serum HCV RNA titer at start of treatment, mean (± SEM) log10 IU/mL | 6.04 (±0.11) | 6.30 (±0.15) |
| ALT level at start of treatment, median (IQR) U/mL | 74 (44–88) | 80 (45–112) |
n.a., not applicable
one patient did not yet reach the week 12 time point, thus was not evaluated for EVR.
IQR: interquartile range
SNP: single nucleotide polymorphism
n.d., not done
NK cell analysis
(i) IFN-α signaling
Expression of STAT1, pSTAT1 and pSTAT4 were assessed either directly in vivo or after in vitro stimulation of pre-warmed heparinized blood without or with 600 ng/mL consensus sequence IFN-α (IFN-α con1; InterMune Inc., Brisbane, CA) for 5 min at 37 °C. Cells were fixed and erythrocytes were lysed by incubation with 20-fold excess volume of Lyse/Fix buffer (BD Biosciences, San Jose, CA) for 10 min at 37 °C. After centrifugation, cells were permeabilized with Perm Buffer (BD Biosciences) for 20 min on ice, washed twice and resuspended in Staining Buffer (BD Biosciences).
All samples were stained with anti-CD56-PE (Beckman Coulter, Brea, CA) and anti-CD20-PerCP/Cy5.5 to identify NK cells and B cells, respectively, and with anti-CD3-FITC or anti-CD3-APC to exclude T cells. Cells were additionally stained with anti-STAT1-Alexa647, anti-pSTAT1-Alexa488 (which assesses tyrosine phosphorylation at Y701), or anti-pSTAT4-Alexa488 (assesses tyrosine phosphorylation at Y693) for 20 min at room temperature and analyzed on an LSRII with FacsDiva Version 6.1.3 (BD Biosciences) and FlowJo Version 8.8.2 (Tree Star, Ashland, OR) software.
(ii) Degranulation
Thawed PBMC were cultured overnight at 37°C, 5% CO2 in RPMI1640 with 10% fetal calf serum (Serum Source International, Charlotte, NC), 1% Penicillin/Streptomycin, 2 mM L-glutamine, 10 mM HEPES (Cellgro, Manassas, VA). The next day, PBMCs were counted and stimulated in the presence or absence of K562 cells (ATCC, Manassas, VA) to assess degranulation as described (6) but in the absence of additional cytokines. (iii) TRAIL expression: Thawed PBMC were stained with ethidium monoazide (EMA), anti-CD19-PeCy5 (BD Biosciences, San Jose, CA), anti-CD14-PeCy5 (Serotec, Raleigh, NC), anti-CD56-PeCy7, anti-CD3-AlexaFluor700 (BD Biosciences) and anti-TRAIL-PE (BD Biosciences).
(iii) IFN-γ production
Thawed PBMC were incubated with or without IL-12 (0.5 ng/ml; R&D Systems) and IL-15 (20 ng/ml R&D Systems) for 14h, followed by addition of brefeldin A for 4h and intracellular staining for IFN-γ as previously described (6).
Viral kinetics
HCV RNA levels were measured using Cobas TaqMan real-time PCR (Roche Diagnostics, Palo Alto, CA), with a lower limit of detection of 15 IU/mL. The first phase virological response was defined as the logarithmic decline in HCV RNA titer during the first 48h of therapy.
Genotyping
DNA samples were genotyped for the IL28B rs12979860 polymorphism with a TaqMan genotyping assay (Applied Biosystems Inc., Foster City, CA).
Statistical Analysis
GraphPad Prism Version 5.0 (GraphPad Software Inc, San Diego, CA) and JMP (SAS Inc. Cary, NC) was used to perform the (i) Mann-Whitney nonparametric two-sample rank test to compare NK cells from healthy subjects and patients, and from patients with strong and weak first phase responses, (ii) repeated measures ANOVA to assess changes in STAT1 and pSTAT1 expression during treatment, (iii) Wilcoxon signed rank test to determine changes in pSTAT1 and pSTAT4 levels and pSTAT1/pSTAT4 ratio from baseline to time points during treatment, and (iv) Spearman correlation analysis to study changes in pSTAT1 signaling in relation to NK cell function. A two-sided p-value of less than 0.05 was considered significant.
Results
Increased STAT1 expression in NK cells during HCV infection is further enhanced during PegIFN/RBV therapy
We have previously described that NK cells are activated in HCV infection but that activation does not result in equal stimulation of all effector functions (6). Specifically, NK cells of patients with chronic HCV infection display enhanced cytotoxicity as evidenced by increased degranulation and TRAIL production and decreased IFN-γ production as compared to uninfected controls (6). We demonstrated that this phenotype can be reproduced by in vitro stimulation of NK cells from healthy, uninfected controls with IFN-α (6).
To evaluate how IFN-α-based treatment of chronic hepatitis C modulates NK cell phenotype and function, we first studied the in vivo level of STAT1 in peripheral blood NK cells of chronically HCV-infected patients (Table 1) and healthy controls. As shown in figure 1A, the total NK cell population of HCV-infected patients as well as their CD56bright and CD56dim subsets showed increased levels of STAT1 when compared to healthy controls. This increase in STAT1 expression was not observed for T cells (Suppl. Fig. 1A). We then prospectively followed a group of HCV-infected patients during the first 12 weeks of IFN-based therapy for hepatitis C. All patients mounted an early virological response (i.e. serum HCV RNA levels at least 2 log10 lower at week 12 than prior to treatment). STAT1 expression increased significantly in the total NK cell population and the CD56bright and CD56dim subsets within 24h of therapy, and STAT1 levels increased further throughout the study period of 12 weeks (Fig. 1B-D, p<0.0001 for all populations). The same increase in STAT1 expression was observed in CD3+CD56- T cells (Suppl. Fig. 1B).
Fig. 1. Increased expression of STAT1 in NK cells during HCV infection is further enhanced by PegIFN/RBV therapy.
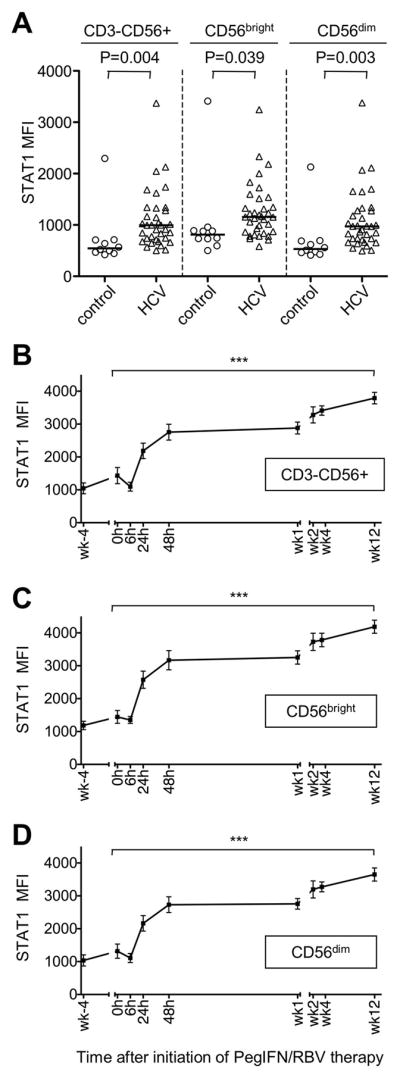
(A) In vivo STAT1 expression in CD3-CD56+ NK cells and their CD56bright and CD56dim subpopulations in HCV-infected patients and healthy, uninfected blood donors.
(B–D) In vivo STAT1 expression levels of all CD3-CD56+ NK cells (B) and their CD56bright (C) and CD56dim (D) subpopulations during therapy with PegIFN/RBV. Mean ± SEM are shown for 14 patients undergoing PegIFN/RBV therapy.h, hour; wk, week. *** P<0.001 by repeated measures ANOVA.
PegIFN/RBV therapy induces preferential phosphorylation of STAT1 over STAT4 in NK cells
To evaluate changes in signaling downstream of the IFN-α/β receptor we next studied the STAT1 and STAT4 phosphorylation at all treatment time points. Changes in pSTAT1 and pSTAT4 expression were greatest within the first 48h of therapy. In vivo pSTAT1 levels peaked in CD3-CD56+ NK cells and in their CD56bright and CD56dim subsets within 6h of therapy (MFI163±16 at baseline and 205±20 at maximum, p=0.005, p=0.018 and p=0.003, respectively, Fig. 2A).
Figure 2. PegIFN/RBV therapy results in preferential phosphorylation of STAT1 over STAT4 in NK cells.

(A–B) Maximal changes in pSTAT1 (A) and pSTAT4 (B) expression levels in chronic HCV patients of total CD3-CD56+ NK cells (left panel) and their CD56bright (middle panel) and CD56dim (right panel) subsets prior to and following PegIFN/RBV therapy initiation. (C) pSTAT1/pSTAT4 ratio (MFI) throughout the first 48h of therapy. *P<0.05, **P<0.01, **P<0.001 (comparing the indicated individual time points to the 0h time point).
In contrast, pSTAT4 levels decreased in the overall CD3-CD56+ NK cell population and in their CD56bright and CD56dim subsets in response to IFN-based therapy, reaching a minimum at the 48h time point (MFI 183±10 at 0h and 149±8 at 48h, p=0.011, p=0.023 and p=0.028, respectively, Fig. 2B). Since STAT1 and STAT4 signaling molecules both compete for phosphorylation at the IFN-α/β receptor (9) these data suggest that an increase in the expression of STAT1 (Fig. 1) results in preferential phosphorylation of STAT1 over STAT4 during IFN-based therapy (Fig. 2). Consistent with this interpretation, the pSTAT1/pSTAT4 ratio peaked 6h after initiation of therapy and remained increased up to 48h in the CD56dim NK cell subset (Fig. 2C).
PegIFN/RBV-induced STAT1 phosphorylation correlates to polarization of NK cell function
In a detailed prospective analysis we showed previously that NK cell effector functions are strongly induced in response to IFN-α (14). NK cells cytotoxicity as determined by TRAIL expression (Fig. 3A, left panel) and degranulation (Fig. 3B, left panel) peaked as early as 6h and 24h, respectively. Conversely, the frequency of IFN-γ producing NK cells reached its minimum 6h after treatment initiation (Fig. 3C, left panel) and never increased above pretreatment levels at later time points (14). Importantly, the increase in cytotoxicity as evidenced by TRAIL production directly correlated with the increase in pSTAT1 levels (r=0.586, p=0.014, Fig. 3A, right panel) and the increase in NK cell degranulation followed the same trend (r=0.453, p=0.078, Fig. 3B, right panel). In contrast, the change in IFN-γ production correlated inversely with the increase in pSTAT1 levels (r=0.549, p=0.015, Fig. 3C, right panel). These results support the interpretation that the polarization of NK cell function in patients with chronic hepatitis C is mediated by IFN-α since IFN-based therapy further drives this functional dichotomy by the induction of pSTAT1.
Figure 3. PegIFN/RBV-induced changes in pSTAT1 expression in NK cells correlate to changes in NK cell function.

Changes in TRAIL production (A, left graph), degranulation (B, left graph) and IFN-γ production (C, left graph) in response to PegIFN/RBV therapy initiation in correlation to changes in pSTAT1 expression level from 0h to 6h (A–C right graphs). r: Spearman correlation coefficient.
Prolonged PegIFN/RBV therapy results in refractoriness of NK cells to in vitro IFN-α stimulation
To evaluate whether NK cells are maximally stimulated by IFN-based therapy in vivo we isolated PBMCs at numerous time points within the first weeks of treatment, subjected them to in vitro stimulation with IFN-α and determined their pSTAT1 levels. As shown in figure 4A, in vitro induced pSTAT1 levels decreased after the initial 6h of PegIFN/RBV treatment, reached their minimum after the first week of PegIFN/RBV treatment and remained at low for the following 11 weeks of the study period (MFI at 0 h: 407±37; at 24h: 279±25; at week 12: 181±24, p=0.039). The same kinetics was observed when the in vitro inducibility of pSTAT1 was normalized either to in vivo pSTAT1 levels (Fig. 4B) or to total STAT1 levels at each individual treatment time point (Suppl. Fig. 2). These results demonstrate that maximal pSTAT1 induction was reached very early during PegIFN/RBV therapy (between 6h and 48h) and that NK cells remained refractory to further stimulation. To ensure that these observations were not a result of sampling at nadir time points, i.e. just prior to the weekly PegIFN injection, we studied two patients after the first injection and after the week 12 injection of pegIFN. As shown in figure 4C, in vivo pSTAT1 levels increased in NK cells within 6h after the first PegIFN injection. However, no increase was observed in the 6h following the week 12 PegIFN injection. Thus, prolonged exposure to IFN-α appears to render NK cell refractory over the course of treatment.
Figure 4. Refractoriness of pSTAT 1 inducibility in NK cells during PegIFN/RBV therapy.

(A) pSTAT1 levels in CD3-CD56+ NK cells after in vitro stimulation with IFN-α (pSTAT1 inducibility).
(B) In vitro pSTAT1 inducibility in CD3-CD56+ NK cells at the indicated PegIFN/RBV time points relative to in vivo pSTAT1 expression at the same time points. Inducibility was calculated as the fold change in pSTAT1 MFI after in vitro treatment with IFN at each time point. Mean ± SEM are shown for 21 patients. *P 0.05, ** P≤0.01 by repeated measures ANOVA.
(C) In vivo pSTAT1 levels in NK cells prior to and 6h following PegIFN injection at start (left panel) and 12 weeks (right panel) of PegIFN/RBV therapy. h, hour; wk, week.
Patients with a weak first phase virological response do not reach maximal pSTAT1 induction in vivo
To evaluate a potential association of NK cell responsiveness with treatment efficacy, we determined the decline in HCV RNA in the peripheral blood during the first 48h of treatment. This is defined as the first phase virological response and predicts treatment outcome (13). Because chronic infection with HCV genotypes 1 and 4 requires a longer course of treatment than chronic infection with HCV genotypes 2 and 3 (15), we limited this analysis to patients infected with HCV genotypes 1 and 4 (Table 2). As shown in figure 5A, patients with a strong first phase virological response (defined as greater than 2 log reduction in HCV RNA titer in the first 48h), displayed a significantly greater increase of in vivo pSTAT1 levels in NK cells during the first 6h (Fig. 5A, B) and 24h (Fig. 5C) of therapy than patients with a weak first phase virological response (less than 2 log reduction). This was independent of the IL-28 genotype (another determinant of treatment outcome)(16) because neither in vivo pSTAT1 levels nor in vitro pSTAT1 inducibility in NK cells correlated to the IL28B SNP at rs12979860, an independent factor of treatment responsiveness.
Table 2.
Epidemiological and clinical data of patients with and without a strong first phase virological response.
| Patients with a greater than 2log10 48-h decrease in HCV titer (n=4) | Patients with a less than 2log10 48-h decrease in HCV titer (n=10) | |
|---|---|---|
| Decrease in HCV titer 0h-48h, median log10 (IQRa) | 2.59 (2.2– 2.9) | 1.15 (0.7–1.6) |
| Early virological response, n (%) | 4 (100%) | 9 (100%) c |
| Gender (male/female) | 3/1 | 6/4 |
| Age at start of treatment, median (IQR) years | 57.5 (54.3–60.8) | 53.0 (47.3–55.5) |
| Ethnicity (asian/african american/ caucasian/hispanic) | 1/1/2/0 | 1/3/5/1 |
| Body mass index, median (IQR) | 30.8 (22.9– 34.5) | 29 (25.6–35.9) |
| IL-28B rs12979860 SNPb (CC/CT/TT) | 3/0/0 | 4/4/2 |
| HCV genotype (1/4) | 4/0 | 7/1 |
| Serum HCV RNA titer at start of treatment, mean (±SEM) log10 IU/mL | 6.6 (±0.15) | 6.6 (±0.31) |
| ALT level at start of treatment, median (IQR) U/mL | 61 (36– 98) | 83 (62–179) |
IQR: interquartile range
SNP: single nucleotide polymorphism
one patient did not yet reach the week 12 time, thus was not evaluated for SVR.
Figure 5. Correlation of pSTAT1 expression and first phase virological response.

(A) Fold change in in vivo pSTAT1 expression by CD3-CD56+ NK cells during the first 6h of PegIFN/RBV therapy in individual patients with (solid lines) and without (broken lines) a greater than 2 log10 first phase decline in HCV RNA titer.
(B–C) Fold change in in vivo pSTAT1 expression by CD3-CD56+ NK cells during the first 6h (B) and 24h (C) of PegIFN/RBV therapy in individual patients with and without a greater than 2 log10 first phase decline in HCV RNA titer.
(D) Fold change in pSTAT1 inducibility (pSTAT1 MFI after in vitro treatment with IFN normalized to in vivo levels prior to (0h) or 6h after initiation of PegIFN/RBV therapy) in individual patients with (solid lines) and without (broken lines) a greater than 2 log10 first phase decline in HCV RNA titer.
(E–F) Fold change in pSTAT1 inducibility in NK cells (pSTAT1 MFI after in vitro treatment with IFN normalized to in vivo levels prior to (0h) or 6h (panel E) or 24h (panel F) after initiation of PegIFN/RBV therapy) in individual patients with and without a greater than 2 log10 first phase decline in HCV RNA titer.
There are two possible explanations for the lower IFN-responsiveness of NK cells in patients with a weak first phase virological response. One possibility is that their level of NK cell responsiveness to IFN is genetically predetermined. The other possibility is that their NK cells are suboptimally stimulated in vivo. To differentiate between both possibilities we subjected PBMC of patients with and without a strong first phase virological response to further in vitro stimulation with IFN-α. Interestingly, NK cells from patients with a < 2 log10 first phase HCV RNA decline exhibited greater in vitro inducibility of pSTAT1 than NK cells from patients with a greater first phase response (Fig. 5D–F). These results suggest that NK cells of patients with a weak first phase virological response are suboptimally stimulated in vivo.
Discussion
This study shows that IFN-α-induced modulation of STAT1 phosphorylation underlies the in vivo polarization of NK cells towards increased cytotoxicity and decreased IFN-γ production. This result is consistent with the observation that LCMV-induced IFN-α secretion in mice has been shown to increase STAT1 expression in NK cells resulting in preferential STAT1 over STAT4 phosphorylation (6, 9, 11). It also extends the findings by Miyagi et al. on preferential STAT1 phosphorylation in HCV-infected patients (11) because we show that IFN-α exposure in vivo results in increased pSTAT1 levels and that it correlates to increased TRAIL production and degranulation and decreased IFN-γ production (Fig. 3). The clinical relevance of IFN-α signaling in NK cells is suggested by our observation that NK cell responsiveness and refractoriness correlate with the first phase virological response to IFN-α-based therapy (Fig. 5).
This analysis of NK cells is relevant for current research on biomarkers predicting interferon responsiveness and treatment outcome. Advantages of using NK cells as biomarkers of IFN responsiveness are that they are readily accessible from the peripheral blood and that both in vivo and in vitro NK cell responsiveness can easily be assessed in a short, standardized flow cytometry-based assay by checking pSTAT1 levels. How does our system compare to other biomarkers of IFN-responsiveness? A well-established biomarker for interferon responsiveness is the intrahepatic expression of interferon-stimulated genes (ISGs). Typically, ISGs are most highly expressed pretreatment in HCV-infected patients who respond poorly to IFN-α-based therapy (17). As potential explanation it has been proposed that high baseline activation of the endogenous interferon system does not allow a further increase of ISGs during interferon-based therapy, possibly because the ISG response has already reached maximal levels and/or inhibitory autocrine feedback mechanisms have been induced (18). In contrast to these ISG data, we did not find any evidence that pretreatment pSTAT1 levels or in vitro inducibility differed among HCV-infected patients (data not shown). Thus, pSTAT1 induction is an independent measure for IFN responsiveness and may complement ISG analysis.
How does the NK cell response correlate to the treatment response? Because all patients in our study achieved an early virological response to PegIFN/RBV therapy at week 12 we were not able to assess NK cell responses in the context of the ultimate treatment outcome. On the other hand, we believe that late time points of PegIFN/RBV therapy are less relevant for our study because NK cells exhibited their greatest response within the first days of therapy in parallel to the first phase virological response (Fig. 1–4). Our data clearly indicate that near-maximal NK cell activation can be reached within hours of the first injection of PegIFN because the response to additional in vitro stimulation with IFN-α was significantly reduced at later treatment time points (Fig. 4). Here, we made the interesting observation that NK cells from patients with a weak first phase decline in HCV titer, who displayed lower levels of in vivo pSTAT1 induction than patients with a strong first phase decline in HCV titer, nevertheless retained responsiveness to in vitro stimulation with IFN-α. Thus, both patient groups differed in their in vivo responsiveness to IFN-based therapy, but not in their overall response to IFN-α (Fig. 5A-C). These results suggest that NK cell responsiveness depends to a certain extent on the environment. One explanation is that in vivo levels and pharmacokinetics of IFN differ among patients. Another possible explanation is that certain factors, such as suppressive cytokines, interfere with the responsiveness of NK cells to PegIFN therapy in vivo, and that these are overcome once NK cells are stimulated with high doses of IFN-α in vitro. However, removal of inhibitory factors can be excluded, because the in vitro NK cell stimulation was performed in whole blood. A third possibility is that genetic determinants, such as IL28B SNP at rs12979860 (16) and KIR/HLA compound genotype (19) cannot completely be ruled out due to the small size of the analyzed patient cohort (Tables 1, 2). However, if rs12979860 SNPs play a role, it would be an indirect rather than direct effect on NK cells because NK cells retain their responsiveness to in vitro stimulation with IFN-α (Fig. 5D–F) and because they do not respond directly to type III IFN including IL28B (20). Thus our study opens the interesting possibility that in vivo responsiveness to IFN-α-based therapy may be improved.
Another relevant result of this study was the observed refractoriness of NK cells to in vitro IFN-α stimulation, which occurred in all patients within the first week of IFN-α-based therapy and was maintained for the entire study (Fig. 4A, B). NK cells were not only refractory to in vitro IFN-α stimulation but exhibited refractoriness in vivo as shown in the patients who consented to a blood draw prior to and 6h after the week 12 PegIFN injection and did not exhibit an increase in vivo pSTAT1 levels during this period (Fig. 4C). This refractoriness to STAT1 phosphorylation striking because STAT1 levels continued to increase while pSTAT1 levels declined in NK cells. There are at least three possible explanations:
First, the half-life time of STAT1 is longer than that of pSTAT1, because STAT1 has been shown to persist for many days in response to IFNs whereas pSTAT1 levels decrease by SHP1, SHP2 and SOCS1-dependent negative regulation and tyrosine phosphatase-mediated dephosphorylation. Second, the accumulated unphosphorylated STAT1 itself is able to induce the expression of a subset of ISGs such as 2’5’ OAS, Mx1 and STAT1 creating a pSTAT1-independent positive feed-back loop (21). Third, STAT1 can also be induced independently from signaling via the IFN-alpha/beta receptor in NK cells as suggested by the observation that NK cells from STAT1-deficient mice show a greater level of impairment in cytotoxicity and ability to reject transplanted tumors than NK cells from mice that lack IFN receptors (22).
Collectively, the results suggest that continued exposure to high levels of IFN-α may reign in the NK cell response to prevent collateral damage. Similar mechanisms may be operative in acute HCV infection, which is known to induce high levels of type I IFN-induced genes without evidence of significant liver injury throughout the incubation phase of 1–2 months (23). Thus IFN-α-induced NK cell refractoriness may contribute to the often observed, but in its mechanisms not yet understood, clinically asymptomatic nature of acute HCV infection.
Supplementary Material
Acknowledgments
Financial Support: This study was supported by the NIDDK, NIH intramural research program.
This study was supported by the NIDDK, NIH intramural research program. We thank Dr. Xiongce Zhao, NIDDK, for statistical analysis.
Abbreviations
- ALT
alanine aminotransferase
- EVR
early virological response
- HCV
hepatitis C virus
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate buffer saline
- pegIFN
pegylated interferon-alfa
- RBV
ribavirin
References
- 1.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, et al. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Billiau A, Matthys P. Interferon-gamma: a historical perspective. Cytokine Growth Factor Rev. 2009;20:97–113. doi: 10.1016/j.cytogfr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115:2167–2176. doi: 10.1182/blood-2009-08-238469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci U S A. 2011;108:728–732. doi: 10.1073/pnas.1012356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahlenstiel G, Martin MP, Gao X, Carrington M, Rehermann B. Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J Clin Invest. 2008;118:1017–1026. doi: 10.1172/JCI32400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlenstiel G, Titerence RH, Koh C, Edlich B, Feld JJ, Rotman Y, Ghany MG, et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology. 2010;138:325–335. e321–322. doi: 10.1053/j.gastro.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, De Filippi F, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology. 2009;137:1151–1160. 1160 e1151–1157. doi: 10.1053/j.gastro.2009.05.047. [DOI] [PubMed] [Google Scholar]
- 8.Stegmann KA, Bjorkstrom NK, Veber H, Ciesek S, Riese P, Wiegand J, Hadem J, et al. Interferon-alpha-induced TRAIL on natural killer cells is associated with control of hepatitis C virus infection. Gastroenterology. 2010;138:1885–1897. doi: 10.1053/j.gastro.2010.01.051. [DOI] [PubMed] [Google Scholar]
- 9.Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. 2007;204:2383–2396. doi: 10.1084/jem.20070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, Biron CA. Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat Immunol. 2000;1:70–76. doi: 10.1038/76940. [DOI] [PubMed] [Google Scholar]
- 11.Miyagi T, Takehara T, Nishio K, Shimizu S, Kohga K, Li W, Tatsumi T, et al. Altered interferon-alpha-signaling in natural killer cells from patients with chronic hepatitis C virus infection. J Hepatol. 2010;53:424–430. doi: 10.1016/j.jhep.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 12.Davis GL, Wong JB, McHutchison JG, Manns MP, Harvey J, Albrecht J. Early virologic response to treatment with peginterferon alfa-2b plus ribavirin in patients with chronic hepatitis C. Hepatology. 2003;38:645–652. doi: 10.1053/jhep.2003.50364. [DOI] [PubMed] [Google Scholar]
- 13.Parruti G, Polilli E, Sozio F, Cento V, Pieri A, Di Masi F, Mercurio F, et al. Rapid prediction of sustained virological response in patients chronically infected with HCV by evaluation of RNA decay 48h after the start of treatment with pegylated interferon and ribavirin. Antiviral Res. 2010;88:124–127. doi: 10.1016/j.antiviral.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Ahlenstiel G, Edlich B, Hogdal LJ, Rotman Y, Noureddin M, Feld JJ, Holz LE, et al. Early Changes in Natural Killer Cell Function Indicate Virologic Response to Interferon Therapy for Hepatitis C. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335–1374. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 17.He XS, Ji X, Hale MB, Cheung R, Ahmed A, Guo Y, Nolan GP, et al. Global transcriptional response to interferon is a determinant of HCV treatment outcome and is modified by race. Hepatology. 2006;44:352–359. doi: 10.1002/hep.21267. [DOI] [PubMed] [Google Scholar]
- 18.Feld JJ, Nanda S, Huang Y, Chen W, Cam M, Pusek SN, Schweigler LM, et al. Hepatic gene expression during treatment with peginterferon and ribavirin: Identifying molecular pathways for treatment response. Hepatology. 2007;46:1548–1563. doi: 10.1002/hep.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knapp S, Warshow U, Hegazy D, Brackenbury L, Guha IN, Fowell A, Little AM, et al. Consistent beneficial effects of killer cell immunoglobulin-like receptor 2DL3 and group 1 human leukocyte antigen-C following exposure to hepatitis C virus. Hepatology. 2010;51:1168–1175. doi: 10.1002/hep.23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witte K, Gruetz G, Volk HD, Looman AC, Asadullah K, Sterry W, Sabat R, et al. Despite IFN-lambda receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun. 2009;10:702–714. doi: 10.1038/gene.2009.72. [DOI] [PubMed] [Google Scholar]
- 21.Cheon H, Yang J, Stark GR. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J Interferon Cytokine Res. 2011;31:33–40. doi: 10.1089/jir.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee CK, Rao DT, Gertner R, Gimeno R, Frey AB, Levy DE. Distinct requirements for IFNs and STAT1 in NK cell function. J Immunol. 2000;165:3571–3577. doi: 10.4049/jimmunol.165.7.3571. [DOI] [PubMed] [Google Scholar]
- 23.Shin EC, Seifert U, Kato T, Rice CM, Feinstone SM, Kloetzel PM, Rehermann B. Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J Clin Invest. 2006;116:3006–3014. doi: 10.1172/JCI29832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.