

Abstract
Reducing excessive accumulation of amyloid-β (Aβ) in Alzheimer's disease (AD) is a key objective of most AD therapies, and inhibition of angiotensin-converting enzyme (ACE) may delay onset or progression of AD. The effects of an ACE-inhibitor (ACE-I) and an angiotensin II receptor blocker (ARB) on Aβ and tau pathology in a triple transgenic (3xTGAD) mouse model of AD were investigated. 9-10month 3xTGAD mice were treated with ARB, ACE-I or vehicle for 6 months. Mean arterial blood pressure (MABP) was measured periodically and mice were assessed behaviourally. Aβ, phospho-tau, amyloid precursor protein (APP) and ACE activity were analysed. MABP was significantly reduced at 2 weeks and 3 months in the ACE-I group and at 3 months in the ARB group, compared to vehicle. Neither drug altered performance of 3xTGAD mice in Morris Water Maze or T-maze, nor were Aβ, tau immunolabelling or APP levels altered. ACE-I significantly reduced ACE activity in kidney. Prolonged treatment with ACE-I or ARB does not affect Aβ or phospho-tau accumulation in brains of aged 3xTGAD mice.
Keywords: Angiotensin-converting enzyme inhibitor, angiotensin II receptor blocker, hypertension, triple transgenic mouse model, Alzheimer’s disease, amyloid-beta
Introduction
Alzheimer’s disease (AD) is now believed to affect almost 36 million people worldwide and its prevalence is estimated to rise to 115.4 million by 2050 [1]. One of the key pathological hallmarks of AD is the accumulation of amyloid-β peptide (Aβ) in the extracellular space, the vasculature and in neurons [2]. However, vascular risk factors, such as hypertension, hypercholesterolaemia and diabetes, have also long been implicated in the aetiology of AD (for review see [3]) and are likely not only to contribute to reduced cerebral perfusion, impaired regulation of cerebral perfusion and blood-brain barrier (BBB) dysfunction but may also increase Aβ production and impair its clearance.
In addition to increasing risk for cerebrovascular and coronary heart disease, hypertension is a risk factor for AD and vascular forms of cognitive decline [4]. Both epidemiological and neuropathological studies have demonstrated a link between mid-life hypertension and risk of AD, associated with increased accumulation of Aβ [5,6]. Hypertension is often treated by administration of angiotensin-converting enzyme inhibitors (ACE-Is) or angiotensin receptor blockers (ARBs) which act to inhibit the production and action of angiotensin II (AngII) respectively [7]. In addition to reducing the risk of atherothrombotic vascular diseases, these medications also appear to preserve cognitive function by reducing the incidence [8-10] and rate of progression of AD [9,11,12] and mild cognitive impairment [13-17]. This has led to the consideration of these drugs as potential treatments for AD [18-20]. However, there remains uncertainty as to whether or not all of these drugs are protective [17,21-23] and which sub-types are most likely to be helpful in AD [12,17,23].
The mechanisms of protection against AD is unclear; ACE-Is and ARBs are likely to benefit people by lowering blood pressure and improving cerebral blood flow [24]. There is also mounting evidence of interactions between blood pressure and Aβ pathology such that hypertension exacerbates Aβ accumulation [25-27]. Conversely, several in vitro, in vivo and ex vivo laboratory studies have demonstrated that ACE, as a mediator of Ang II formation, itself promotes the degradation of Aβ [24,28-36] and according to some animal studies ACE-Is administered to older animals for prolonged periods can exacerbate Aβ pathology [29,35]. Thus, it is possible that some anti-hypertensives improve vascular function and perhaps Aβ production in the short-term whilst others, ACE-Is in particular, may have deleterious effects when administered over prolonged periods, due to reduced degradation of Aβ and interference with Ang II activity (potentially reducing release of acetylcholine, altering G-protein signalling, and promoting inflammation [7,37-42]). The importance of the non-vascular actions of Ang II are supported by several in vivo studies in which ARBs improved cognitive performance and diminished Aβ pathology in animal models of AD [34,35,43,44], and from population based studies showing the superiority of ARBs over ACE-Is and other antihypertensive drugs in reducing the incidence of AD [8,9].
We recently examined whether treatment for 2 months with ACE-Is or ARBs accelerated the initiation of AD-type pathology in La Ferla 3xTGAD mice. We found no effect of either treatment on early AD pathology in mice aged 5-6 months, with no change in neuronal or total oligomeric Aβ [45]. We have now examined whether more prolonged (6 month) administration of an ACE-I (captopril) or an ARB (eprosartan) affects the progression of AD-related pathological changes and cognitive function in much older (16 month) 3xTGAD mice.
Materials and methods
Animals
Adult (9-10-month old), male triple transgenic (3xTGAD: PS1m146vKL; Thy1.2-APPswe; Thy1.2-tauP301L) mice [46] were used in this study. The animals were group housed under a 12-hour light-dark cycle with access to food and water ad libitum. Room temperature was maintained at approximately 21 ± 2°C. Prior to study commencement mice (n=60 in total) were randomly assigned to one of three groups: a control untreated group (n=22), and two representative drug treatment groups for ARBs (eprosartan, n=19) and ACE-Is (captopril, n=19). Researchers were blinded to drug treatment at all times. All animal procedures were performed according to the Animal Scientific Procedures Act of 1986 under license from the United Kingdom Home office.
Drug treatment
Drugs were administered in drinking water to avoid the stress of repeated injections over a 6-month period and to mimic the human route of administration. Eprosartan was administered at 0.8g/l, captopril at 2g/l. The control group received drinking water only. The doses were informed by previous work [45] and pilot studies. The dose of captopril was reduced from 5g/l, as used in our previous study [45], to 2g/l to reduce the hypotensive effects that we found to be mediated by the higher dose in younger animals [45]. Drugs were prepared as described previously [45]. All drinking solutions were freshly prepared twice each week, the containers wrapped in aluminium foil to avoid photochemical changes, and kept at room temperature to avoid precipitation from solution. Fluid consumption was monitored twice weekly throughout the study.
Blood pressure measurement
Mean arterial blood pressure (MABP) was monitored non-invasively with a tail sphygmomanometer at baseline, prior to the start of drug administration; at 2 weeks and 3 months after the commencement of drug administration; and 2 weeks prior to behavioural testing which was undertaken before termination of the experiment (i.e. following 6 months of treatment). Ten measurements of MABP were taken each day and the average for each mouse determined from the last 5 measurements. Mice were habituated to the procedure for 5 days before each measurement day to minimise any confounding effects on MABP that could result from any stress or anxiety. Visual inspection of the sphygmomanometer pulse trace was also carried out to ensure that movement artefacts had not occurred during the measurement; if these were detected the measurement was discarded. Any measurement that coincided with a heart rate of over 700 beats per minute (bpm) was discarded as this is indicative of excessive stress in the animal [47]. A preliminary analysis of the data was conducted to determine if there were any differences in baseline blood pressure (BP) using 1-way ANOVA with Dunns multiple comparisons post-hoc test. Thereafter a 1-way repeated measures ANOVA with Newman-Keuls multiple comparisons post-hoc test was used to assess the affect of each drug upon BP over time.
Behaviour
Morris water maze: After 6 months of drug administration, short and long-term memory was assessed using a validated 2-day water maze protocol [48], with minor modifications. Briefly, animals were trained to criterion (90% escaping under 60 seconds) on a series of visible platform trials on day 1 (D1) in a pool measuring 2m in diameter and 35cm deep, and monitored using Actimetrics Watermaze tracking software. The water temperature was 25 ± 1°C. Animals were given four visible platform trials (V1-V4) on D1 with trials staggered to ensure that each animal had the same inter-trial interval of 10 minutes. On day 2 (D2) animals were given three trials in which the platform was hidden beneath the surface of the water (T1-T3), followed by a probe trial 10 minutes (T4) and 24 hours (D3) after the final hidden platform trial. The platform was placed in two locations and animals were randomly assigned to one or other location, whereupon the platform remained in the chosen position throughout all trials. Animals were randomly delivered to the east and west quadrants, the target quadrant being designated as either northwest or southeast. The start points immediately adjacent to the platform were not used (i.e. north and west, or south and east, depending upon target quadrant). High contrast external visual cues were placed on the walls in the room. The data from each drug treatment group was individually assessed using repeated measures 1-way ANOVA, with D1V4 and D2T1 as the repeated measures, and Newman-Keuls multiple comparisons post-hoc test.
T-maze: At 6 months and 1 week after the commencement of drug administration, spatial-working memory was assessed using the T-maze spontaneous alternation test [49]. The T-maze comprised 3 arms constructed of black Plexiglas each 41cm long and 6cm wide, surrounded by transparent Plexiglas walls (15cm high). The start box (6cm x 7.5cm) was located at the base of the central arm. Vertical sliding doors gave access to/from the start box and each arm of the maze. 24 hours prior to each test day, mice were habituated to the maze during a 10 minute period of free exploration. During the test day, the mice underwent 1 test session made up of 5 trials. At the beginning of trial 1, mice were placed in the start box with the door closed for 30 seconds. The door was then opened and the mice were free to choose either the left or the right arm. Once a decision was made (all four limbs across threshold of arm) the mice were left in the goal arm for 30 seconds before being returned to the start box. This process was repeated over 5 consecutive trials separated by a 5 second inter-trial interval, during which mice remained in the start box. Any mouse failing to complete 5 consecutive trials within 10 minutes was discounted from that test session. Alternation rate (% alternation) of arm choice was measured over the 5 trials on three consecutive testing days. The mean of the three testing days are presented. Alternation rates were analysed by 1-way ANOVA with significance set at P<0.05. (Note there was no statistical significance so no post-hoc tests were conducted)
Tissue preparation
After 6 months and 2 weeks of treatment, the animals were anaesthetised with isoflurane and sacrificed by transcardial perfusion with cold saline. Brains were harvested and hemisected midsagitally. One hemisphere was snap frozen in liquid nitrogen for biochemical analyses, the other fixed in 4% paraformaldehyde for 48hours for histology and immunohistochemistry. From each mouse a kidney was also isolated and snap-frozen for subsequent analysis of peripheral ACE activity.
For western blots, fresh-frozen brain tissue was homogenized in 10 volumes (wt:vol) of tissue homogenization buffer (THB; 250mM Sucrose, 20mM Tris base, 1mM EDTA, 1mM EGTA). The homogenate was mixed 1:1 with 0.4% diethanolamine buffer (DEA; 200ul DEA, 1ml 5M NaCl, ddH2O to 50ml) and centrifuged at 135,000xg at 4°C for 1 hour. The supernatant was saved as the soluble fraction.
For the ACE activity assay and ACE ELISA, fresh-frozen brain and kidney were homogenised in a non-detergent buffer containing 50mM Tris (pH 7.4) with 2.3mm zirconia/silica beads (Thistle Scientific), in an automatic homogeniser (Stretton Scientific) for 2 x 15 seconds at 6000 rpm. The homogenates were then spun for 15 minutes at 13000 rpm in a centrifuge refrigerated to 4°C.
For immunohistochemistry the fixed brain tissue was sliced coronally at 3mm intervals and the slices embedded in paraffin wax. Sections 6μm in thickness were collected onto aminopropyltriethoxysilane-coated slides, dried on a hotplate and stored at room temperature until used.
ACE activity assay
ACE activity in mouse brain and kidney tissue was determined as described previously [45], by use of a fluorogenic substrate for angiotensin converting enzyme (Abz-FRK(Dnp)-P) (Biomol), prepared according to manufacturer's instructions. The protein concentration in the homogenates was measured using the total protein kit (Sigma-Aldrich). Samples containing 50μg of protein were diluted in HEPES buffer (595mg in 50ml dH2O, pH 6.5) and 50μl aliquots of each diluted sample were incubated in triplicate for 18 hours at 26°C with Abz-FRK(Dnp)-P. Further triplicate aliquots of each sample were incubated with 10μg of 100mM captopril (Biomol) to inhibit the reaction. The fluorescence of Abz-FRK(Dnp)-P was measured in a 96-well plate using a FLUOstar OPTIMA plate reader, and the difference in fluorescence between captopril-inhibited and non-inhibited samples used to calculate ACE activity (expressed in arbitrary units). Treatment groups were compared by 1-way ANOVA, with Dunn’s multiple comparison post-hoc test where appropriate, with significance set at P<0.05.
Mouse ACE protein ELISAs
Homogenates were prepared and, after total protein levels were measured as described above, stored at -80°C until used. A commercially available sandwich ELISA (R&D systems) was used on duplicate samples of mouse brain homogenate, 25μg total protein/ml diluted in 100μl of 1% PBS/BSA. Rat-anti mouse ACE (4μg/ml working concentration) was coated overnight on Costar EIA microplates, washed five times with PBS/0.05% tween 20 and blocked with 1% PBS/bovine serum albumin (BSA) (1%PBS/BSA) for 2 hours. After a further five washes, samples and recombinant human ACE standards (R&D Systems) were added to the plate for 2 hours with shaking. After 2 hours the plates were washed and incubated with biotinylated goat anti-mouse ACE antibody (400ng/ml working concentration) for 2 hours. After further washing of the plates, streptavidin HRP (1:200) was added for twenty minutes in the dark, followed by tetramethylbenzidine (TMB) which was left in the dark for 30 minutes. The reaction was stopped with 2N sulfuric acid and the emission read at 450nm. ACE concentrations were determined by interpolation from standard curves determined for each plate by simultaneous assay of recombinant human ACE diluted to a range of known concentrations. Data were analysed by 1-way ANOVA with Dunn’s multiple comparison post-hoc test, with significance set at p<0.05.
Immunohistochemistry
Glass microscope slides (3 x 1 inch, CellPath) were prepared by soaking in 10% Decon detergent (Fisher Scientific) overnight, then washed in running hot water for 1 hour and rinsed with distilled water before being left to dry in a 60°C oven overnight. Slides were then sequentially dipped for 15 second intervals in 2% aminopropyltriethoxysilane in ethanol, then ethanol and lastly distilled water. They were dried overnight at 37°C and stored in boxes at room temperature until use.
Sections were dewaxed in Clearene, dehydrated in 100% ethanol and immersed in 3% hydrogen peroxide in methanol for 30 minutes to block endogenous peroxidase activity. Non-specific binding was blocked by incubation with M.O.M mouse Ig blocking reagent (Vector M.O.M immunodetection kit, Vector Laboratories). Sections were washed in PBS and incubated for 5 minutes in a working solution of M.O.M diluent. Excess M.O.M diluent was tipped off the sections and the primary antibody added and left overnight. The sections were then washed with PBS and M.O.M biotinylated anti-mouse IgG reagent was added for 10 minutes. Sections were rinsed in PBS and incubated for 5 minutes with Vectastain Elite ABC reagent. Sections were again washed in PBS and then incubated with DAB (Vector Laboratories) for 7 minutes. After incubation with DAB, sections were washed in water and immersed in copper sulphate DAB enhancer for 4 minutes, counterstained with haematoxylin, dehydrated, cleared and mounted in Clearium.
The primary (murine) antibodies used were 4G8 (directed at Aβ17-24; Millipore, MAB1561) and AT8 (directed at phospho-tau; Autogen Bioclear, BR03). 4G8 labelling required a 20 minute pretreatment of sections with formic acid and was applied overnight at 1:16000. AT8 was applied overnight at 1:3000 without pre-treatment. Both were used at room temperature.
The immunohistochemically stained sections were examined under x10 and x20 objectives. Labelling of intraneuronal Aβ was scored in whole sections as 0 (absent), 1 (sparse) or 2 (abundant). Aβ plaques were scored as 0 (absent), 1 (few), 2 (moderate) and 3 (extensive). Vascular Aβ was scored as 0 (absent) or 1 (present), while phospho-tau positive neurons were scored 0 (none), 1 (few), 2 (moderate) and 3 (severe). Approximately 10% of sections were re-scored at a later date to check for consistency. Data were analysed using Kruskal-Wallis test for non-parametric data, with Dunn’s multiple comparison post-hoc test.
Quantification of full length APP
Soluble proteins were isolated from brain tissue as described and the levels of APP measured. 10μg of soluble protein was loaded onto a Tris-bis 4-12% polyacrylamide gel and the proteins separated by electrophoresis at 150V for 1.5 hours. Samples from each treatment group were randomly loaded onto one of 3 gels run concurrently, with a single control sample loaded onto all three gels as a reference sample. Proteins were transferred to a PVDF membrane and probed with MAB348 (Millipore) to detect full length APP, and with GAPDH antibody (Sigma-Aldrich) to assess equivalent loading. Bands were visualised using the appropriate Odyssey Infrared secondary antibodies (IRDye, LiCor) and scanned by Odyssey Infrared Imaging System (LiCor). The relative intensity of each band was normalised to the mean of the 3 reference sample intensities. Data were analysed by 1-way ANOVA and Newman-Keuls Multiple Comparison post-hoc test, with significance set at P<0.05.
Results
Mean arterial blood pressure (MABP)
Preliminary analysis of baseline mean arterial blood pressure (MABP) demonstrated that there were no significant differences in MABP between the three groups prior to any drug treatment. Thereafter 1-way ANOVA revealed significant reductions in MABP in the captopril group 2 weeks after treatment commenced, which persisted until the 6 month time point (p<0.05). In the eprosartan group, MABP was significantly reduced 3 months after treatment commenced (p<0.001); however, by 6 months MABP had returned to baseline. In the control group there was a significant increase in blood pressure by 6 months, most likely an effect of normal ageing (p<0.01) (Figure 1).
Figure 1.

Mean Arterial Blood Pressure (MABP): data represent mean ± sem of MABP measured in control, captopril and eprosartan treated animals at baseline and at 2 weeks, 3 and 6 months of treatment. Preliminary analysis with 1 -way ANOVA and Dunns multiple comparison post-hoc test demonstrated that there were no significant differences between the groups at baseline. Data within each treatment group were analysed using repeated measures 1-way ANOVA, with Newman-Keuls multiple comparisons post-hoc test. * indicates significantly different from baseline (p<0.05, **p<0.01, ***p<0.001)
Behaviour – Morris water maze
Aged 3xTGAD in all three treatment groups performed well in the visible trials, significantly reducing their escape latency by D1V4 compared with D1V1 (p<0.05) (Figure 2A and 2B). However, when the first hidden platform trial was conducted 24 h after the last visible platform trial, all 3 treatment groups demonstrated significant spatial memory deficits regardless of drug treatment (Figure 2A and 2C), A repeated measures 1-way ANOVA (with escape latencies on D1V4 and D2T1 as the repeated measures), showed escape latencies to be significantly longer in all three groups on D2T1 than on D1V4 (p<0.05, Figure 2A and 2C). Similarly, short-term memory was detrimentally affected in all three groups, as the escape latency did not significantly improve between D2T1 and D2T3, despite a relatively short inter-trial interval of 10 minutes (Figure 2A). In addition, the difference between each groups performance on the first trial of day 2 and the last trial of day 1 was generated (D2T1-D1V4; Figure 2C); the large number generated in all three groups is indicative of a poor performance in this test.
Figure 2.

Aged 3xTGAD mice have spatial memory deficits in the 2-day water maze protocol, irrespective of drug treatment. Performance in the visible platform trials (D1V1-D1V4), hidden platform trials (D2T1-D2T3) and the probe trial (D3) are shown in (A, B) and spatial memory in the water maze is assessed as absolute escape latency (A, C) or as the latency difference (D; D2T1-D1V4), where D2T1 was performed 24 hours after D1V4. Data from each treatment group were analysed by repeated measures 1-way ANOVA, with Newman-Keuls multiple comparison post-hoc test. * indicates significantly different from the escape latency on D1V4 in all three treatment groups, (*p<0.05, **p<0.01, ***p<0.001. None of the treatment groups achieved an escape latency greater than chance on any of the hidden trials, nor on the probe trial (D3).
T-maze
The arm choice over the 5 consecutive tasks was recorded for each animal, and alternation rate was calculated from the percentage of correct arm choices (alternation between left and right). Drug treatment did not affect performance in the spontaneous alternation task as 1-way ANOVA revealed no significant differences in performance between the three groups (Figure 3). Alternation reflects an animals’ motivation to explore its environment and typical alternation rates are around 75%. Neither the two treatment groups, nor the control group, achieved an alternation rate greater than 65% throughout the testing period (data not shown).
Figure 3.
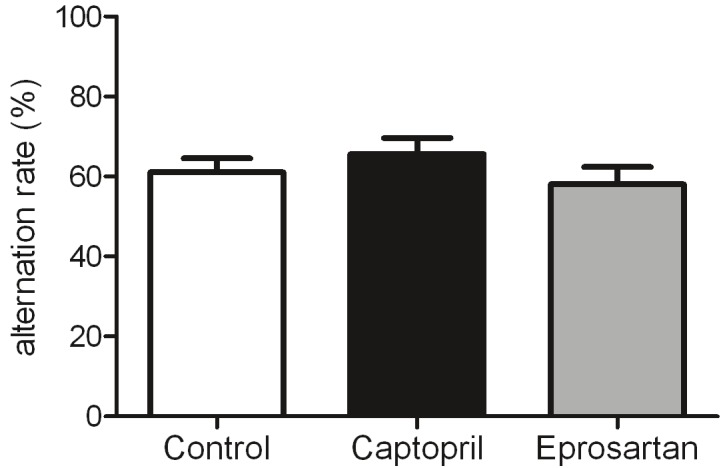
Drug treatment did not affect performance of aged 3xTGAD mice in the spotaneous alternation t-maze protocol. Data were analysed by 1-way ANOVA with significance set at p<0.05.
ACE activity
Measures of central and peripheral ACE activity were assessed in homogenates of brain and kidney respectively in all groups (Figure 4). In keeping with our previous work which involved young animals of the same strain [45], ACE activity tended to be higher in brain than kidney. A significant reduction in ACE activity was observed in the captopril group, in all other groups no significant effect of drug treatment was detected in either brain or kidney.
Figure 4.

Effects of ACE-inhibitor and ARB treatment upon ACE activity in the aged 3xTGAD mouse. ACE activity is expressed in arbitratry units and data presented represent mean ± SEM. 1-way ANOVA with Dunn's multiple comparison post-hoc test revealed no significant differences in ACE activity between the three treatment groups in brain tissue. In kidney tissue there was a significant reduction in ACE activity in the captopril treated group. ACE-activity tended to be lower in kidney tissue than in brain in all three treatment groups.
ACE ELISA
As the activity of ACE does not always mirror its level in brain and CSF in humans [50] we measured ACE concentration as well as activity, to assess the relationship between the two in these transgenic mice (Figure 5). ACE concentration differed significantly between the eprosartan, captopril and control groups (p=0.03); on post hoc analysis with Dunn’s test, only measurements in eprosartan- and captopril-treated mice differed significantly (p<0.05), the concentration of ACE being lower in mice given eprosartan.
Figure 5.
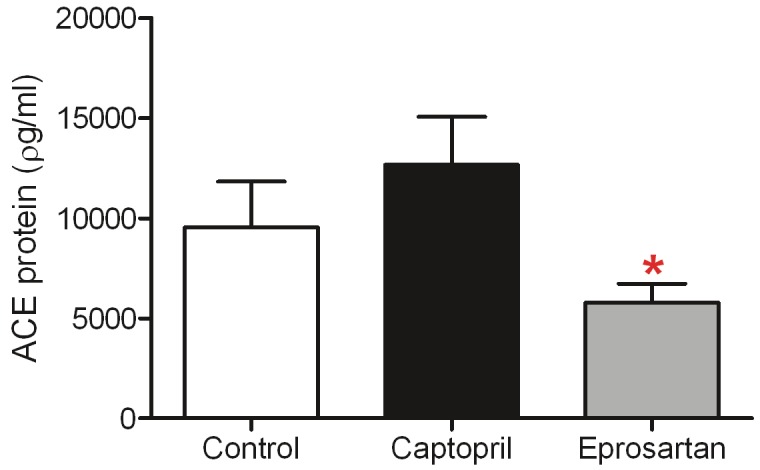
ACE-inhibitor and ARB treatment does not affect ACE protein levels in the aged 3xTGAD mouse. ACE protein level was assesed by ELISA and data represent mean pg/ml ± SEM. 1-way ANOVA with Dunn's post-test for multiple comparisons revealed no significant differences between control and the two drug-treatment groups, however ACE protein was significantly lower in the eprosartan group compared with captopril (p <0.05).
Immunohistochemistry
Both intraneuronal Aβ and Aβ plaques were detected in the hippocampus in all groups (Figure 6A and 6B). Grading of plaque load revealed modest variation between the groups: captopril-treated mice tended to have higher mean plaque scores but Kruskal-Wallis test revealed that the differences were not significant (Figure 6A). Little variation between groups was seen in the level of intraneuronal Aβ (Figure 6B) or the neuronal phospho-tau (Figure 6C). Accumulation of cerebrovascular Aβ was observed in all three groups; the captopril group had less than the other groups but the difference was not statistically significant (Figure 6D).
Figure 6.

Chronic ACE-inhibitor or ARB treatment does not affect the development of amyloid plaques (A, subiculum shown), intraneuronal amyloid (B, hippocampus shown), neuronal phospho-tau (C) or vessel amyloid (D) in aged 3xTGADmice. Images were analysed using a scoring system (see methods for details). Data were analysed by Kruskal-Wallis test with Dunn's multiple comparison post-hoc test where appropriate and significance set at p<0.05. Data presented are median values with representative images from each staining process illustrated at x20 magnification.
APP levels
The level of full-length APP did not differ significantly between the drug-treated mice and controls (Figure 7).
Figure 7.
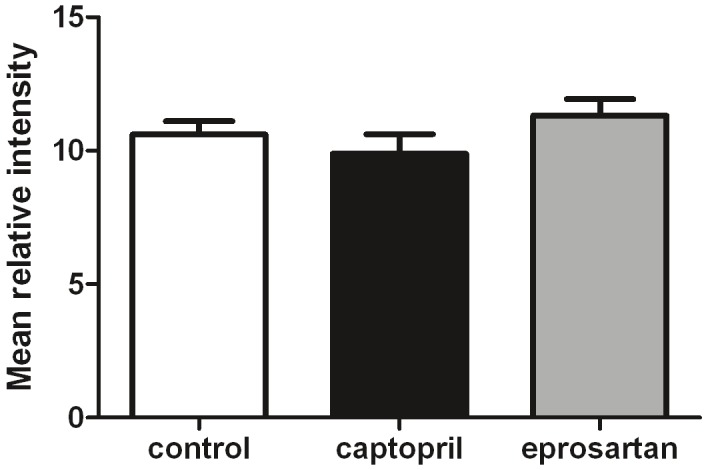
ACE inhibitor and ARB treatment does not affect expression of full length amyloid precursor protein (APP) in the aged 3xTGAD mouse. Full length APP was assessed by western blot in the soluble protein fraction extracted from fresh-frozen brain tissue. Samples from each treatment group were randomly loaded onto one of 3 gels run concurrently, with a single control sample loaded onto all three gels as a reference sample. The relative intensity of each band was normalised to the mean of the 3 reference sample intensities. Data were analysed by 1-way ANOVA with significance set at P<0.05.
Discussion
We previously demonstrated a lack of effect on neuronal or total oligomeric Aβ or extracellular Aβ pathology following short-term (2-month) treatment of younger 3xTGAD mice (aged 5-6 months) with either the ACE-I captopril or the ARBs eprosartan and valsartan [45]. In the present study we have examined whether a longer period of treatment (for 6 months) extending into older age affects the clinical or pathological development of Aβ and tau pathology in 3xTGAD mice. Since our previous study showed no differences between eprosartan and valsartan, and preliminary findings from the Observational Study on Cognitive function and systolic blood pressure Reduction (OSCAR) trial [51] showed eprosartan to improve cognitive performance whilst lowering blood pressure [52], we elected to concentrate on eprosartan for the longer-treatment study.
Present findings in relation to previous research
Consistent with expectations for this transgenic model [46], intraneuronal Aβ, extracellular Aβ plaques, vessel-associated Aβ and neuronal phospho-tau were detected in all 3 groups of mice at 16 months of age, enabling assessment of the effect of anti-hypertensives on the development of these pathologies. In keeping with our data from younger animals [45], we found little evidence that 6 months of treatment with either captopril or eprosartan influenced the level of intraneuronal Aβ, extracellular Aβ, vascular Aβ or neuronal phospho-tau. Although captopril treated mice tended to have higher mean plaque scores and lower cerebrovascular Aβ (in keeping with the suggestion that ACE can degrade Aβ), neither of these differences were statistically significant.
Drug dose and blood pressure
In our previous study in a younger cohort of 3xTGAD mice, captopril at a dose of 5g/l produced an unexpected but significant progressive and sustained reduction in MABP. To try to reduce the potential influence of the hypotensive actions of captopril in the older mice and to make our study more comparable to that of Hemming et al. [53] we reduced the captopril dose to 2g/l in the present study. Even the lower dose, the same as that used by Hemming and colleagues [53], produced a reduction in MABP at 2 weeks that was sustained at 3 months. However, by 6 months MABP had reverted to baseline level. We cannot compare our observations with those of Hemming and colleagues as they did not record MABP in their mice.
In eprosartan-treated mice a similar reduction in MABP was observed at 3 months but which was no longer present at 6 months. Whilst we used doses that would be expected to have minimal, if any impact on MABP, the fact that reductions were observed (as in our previous study) suggests that the 3xTGAD may be particularly susceptible to changes in MABP. The subsequent recovery in MABP over the treatment period may reflect changes to physiological processes that regulate blood pressure; this has been reported in transgenic models of AD [54]. It is unclear whether a similar phenomenon occurs in AD patients, although reductions in blood pressure have been observed both before and after the development of AD [55,56]. The changes may also be a function of the advancing age of the animals since control untreated mice demonstrated a significant rise in blood pressure by 6 months.
It is possible that any positive cognitive effects of these drugs could have been masked by effects of hypotension the drugs produced. Non-hypotensive doses of the ARB telmisartan improved cognitive function and white matter changes in hypoperfused mice but these positive outcomes were lost at a hypotensive dose of the drug [57]. Another ARB, losartan, was associated with reduced Aβ plaques in a transgenic mouse at a dose considerably below the blood pressure-lowering threshold [34]. Likewise olmesartan in low-doses, but not in a highdose, which induced hypotension improved cognitive function in another experimental model [44]. These results suggest that there may be a fine balance between a dose of ARBs that benefits cognitive function and a dose that produces hypotension.
Effects upon cognitive function
Several groups have demonstrated an improvement in cognitive function in rodent and mouse models of AD following ACE-I or ARB (including olmesartan) treatment, sometimes with and sometimes without changes in Aβ pathology [34,35,43,44,58-60]. However, in the present study, no improvement was observed in cognitive function in either of the two drug-treatment groups, with the data demonstrating no effects upon long-term memory or spatia-lworking memory as tested in a 2 day Morris Water Maze paradigm. Although this task was previously shown to be sufficiently sensitive to detect impairment in longer-term memory in 3xTGAD as compared to wild-type mice, other watermaze paradigms may be more sensitive to detect subtle alterations in serial spatial memory [61,62]
A T-maze task for assessment of spontaneous alternation behaviour was previously used to demonstrate restoration of cognitive function in Aβ25-35-injected mice by the centrally-acting ACE-inhibitor, perindopril [58]. Captopril is also reported to be centrally-active and was found to slow cognitive decline in hypertensive patients [12] and to have protective effect along with perindopril in slowing cognitive decline in AD [12]. Typical alternation rates for the spontaneous alternation test are around 75% [63]. However, the rates were lower than expected in all three groups in the present study: 61% (untreated control), 66% (captopril) and 58% (eprosartan). These lower than normal levels of alternation across all groups may indicate a reduction in exploratory drive [64] – perhaps a consequence of age or genotype.
ACE activity and ACE protein level
In keeping with our previous work [45], ACE activity was broadly comparable in brain and kidney in this study, although activity in the kidney tended to be slightly lower. In the captopril treated group activity in the kidney was significantly lower than that in brain, possibly because captopril is more potent in the periphery than the brain or that captopril is not as centrally active as is suggested in other studies. In comparison to our previous study in younger animals from the same transgenic line, ACE activity in both brain and kidney was somewhat lower, which may be an effect of age. Age-related reduction in serum ACE activity was previously reported in Fischer 344 rats [65]. In contrast, ACE activity is significantly elevated in human post-mortem brain tissue [50,66]. There was no significant difference in ACE protein level between the three animal groups; however a modest reduction in ACE concentration in the eprosartan group which coincided with a modest increase in the captopril-treated group resulted in a significant difference between these two groups. It is not clear why these opposing effects occurred. The different mechanisms of actions of the drugs may induce different feedback responses.
Limitations of the study
Despite our use of a lower dose of captopril to that used in our previous study [45] and its selection based on use by other groups [53], the older 3xTGAD mice still experienced reductions in blood pressure. Similarly, the dose of eprosartan used, which was well tolerated in younger mice with no hypotensive effects, also produced a reduction in blood pressure at 3 months of treatment. These effects reduce our ability to isolate any potential non-vascular benefits of these drugs from any blood pressure-related changes. A limitation of our earlier study [45] was a lack of assessment of cognitive function. Here we have included 2 behavioural tests, but the study design may have benefited from having a further untreated group of younger 3xTGAD mice, to clarify if these mice have a normal capacity to learn the procedures or more sensitive measures of spatial learning and memory in mice.
Implications and conclusions
Our results suggest that prolonged treatment with captopril or eprosartan does not affect accumulation of Aβ or neurofibrillary pathology in the brains of aged 3xTGAD mice. This is in keeping with some studies but not others in which various ACE-Is and ARBs have been shown to benefit cognitive function and Aβ pathology in animal models of AD. Similarly in humans there have been conflicting findings. ARBs appear to outperform ACE-Is in terms of having risk-reducing effects in AD [8,9]. When ACE-Is are sub-divided according to their blood brain barrier-penetrating or chemical properties there appear to be differential effects on risk and progression in AD [23] and in the risk of incident mild cognitive impairment [17]. Although we observed a significant reduction in ACE activity in the periphery of the captopril-treated group, we found a surprising lack of inhibition of central ACE activity which is difficult to explain. The differences in outcome between current and previous studies most likely relate to the different mouse strains used as well as factors relating to the mechanisms of action of individual drugs; there is ongoing debate as to whether the different ACE-Is [67] or ARBs [68] are truly interchangeable from a functional perspective. Overall the current findings suggest that ACE-Is (and ARBs) do not have a major effect on the development and progression of Aβ pathology in this mouse model of AD, and that any potential benefit of these drugs for the treatment of AD may be critically influenced by the dose administered and the extent to which it reduces blood pressure.
Acknowledgements
This work was funded by a Network grant from the Alzheimer’s Research UK. Linda Ferrington is funded by a Lloyds TSB/Royal Society of Edinburgh Personal Research Fellowship. Laura E Palmer is funded by a PhD studentship from BRACE
References
- 1.Prince M, Bryce R, Ferri C. Alzheimer's Disease International; 2010. World Alzheimer Report 2010. [Google Scholar]
- 2.Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Duron E, Hanon O. Hypertension, cognitive decline and dementia. Arch Cardiovasc Dis. 2008;101:181–189. doi: 10.1016/s1875-2136(08)71801-1. [DOI] [PubMed] [Google Scholar]
- 5.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider P, Buerger K, Teipel S, Uspenskaya O, Hartmann O, Hansson O, Minthon L, Rujescu D, Moeller HJ, Zetterberg H, Blennow K, Ernst A, Bergmann A, Hampel H. Antihypertensive therapy is associated with reduced rate of conversion to Alzheimer's disease in midregional proatrial natriuretic Peptide stratified subjects with mild cognitive impairment. Biol Psychiatry. 2011;70:145–151. doi: 10.1016/j.biopsych.2011.01.036. [DOI] [PubMed] [Google Scholar]
- 7.Kehoe PG, Miners S, Love S. Angiotensins in Alzheimer's disease - friend or foe? Trends Neurosci. 2009;32:619–628. doi: 10.1016/j.tins.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Davies NM, Kehoe PG, Ben-Shlomo Y, Martin RM. Associations of Anti-Hypertensive Treatments with Alzheimer's Disease, Vascular Dementia, and Other Dementias. J Alzheimers Disease. 2011;26:699–708. doi: 10.3233/JAD-2011-110347. [DOI] [PubMed] [Google Scholar]
- 9.Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, Wolozin B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. doi: 10.1136/bmj.b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohrui T, Matsui T, Yamaya M, Arai H, Ebihara S, Maruyama M, Sasaki H. Angiotensin-converting enzyme inhibitors and incidence of Alzheimer's disease in Japan. J Am Geriatr Soc. 2004;52:649–650. doi: 10.1111/j.1532-5415.2004.52178_7.x. [DOI] [PubMed] [Google Scholar]
- 11.Hajjar IM, Keown M, Lewis P, Almor A. Angiotensin converting enzyme inhibitors and cognitive and functional decline in patients with Alzheimer's disease: an observational study. Am J Alzheimers Dis Other Demen. 2008;23:77–83. doi: 10.1177/1533317507309803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohrui T, Tomita N, Sato-Nakagawa T, Matsui T, Maruyama M, Niwa K, Arai H, Sasaki H. Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology. 2004;63:1324–1325. doi: 10.1212/01.wnl.0000140705.23869.e9. [DOI] [PubMed] [Google Scholar]
- 13.Hajjar I, Catoe H, Sixta S, Boland R, Johnson D, Hirth V, Wieland D, Eleazer P. Cross-sectional and longitudinal association between antihypertensive medications and cognitive impairment in an elderly population. J Gerontol A Biol Sci Med Sci. 2005;60:67–73. doi: 10.1093/gerona/60.1.67. [DOI] [PubMed] [Google Scholar]
- 14.He M, Ohrui T, Maruyama M, Tomita N, Nakayama K, Higuchi M, Furukawa K, Arai H. ACE activity in CSF of patients with mild cognitive impairment and Alzheimer disease. Neurology. 2006;67:1309–1310. doi: 10.1212/01.wnl.0000238102.04582.ec. [DOI] [PubMed] [Google Scholar]
- 15.Rozzini L, Chilovi BV, Bertoletti E, Conti M, Del Rio I, Trabucchi M, Padovani A. Angiotensin converting enzyme (ACE) inhibitors modulate the rate of progression of amnestic mild cognitive impairment. Int J Geriatr Psychiatry. 2006;21:550–555. doi: 10.1002/gps.1523. [DOI] [PubMed] [Google Scholar]
- 16.Rozzini L, Vicini Chilovi B, Trabucchi M, Padovani A. Antihypertensive medications influence the rate of conversion from mild cognitive impairment to Alzheimer disease. Arch Neurol. 2008;65:993–994. doi: 10.1001/archneur.65.7.993. author reply 994-995. [DOI] [PubMed] [Google Scholar]
- 17.Solfrizzi V, Scafato E, Frisardi V, Seripa D, Logroscino G, Kehoe PG, Imbimbo BP, Baldereschi M, Crepaldi G, Di Carlo A, Galluzzo L, Gandin C, Inzitari D, Maggi S, Pilotto A, Panza F. for the Italian Longitudinal Study on Aging Working Group. Angiotensin-Converting Enzyme Inhibitors, Incident Mild Cognitive Impairment, and Progression to Dementia. The Italian Longitudinal Study on Aging. AGE (Journal of the American Aging Association) 2011 doi: 10.1007/s11357-011-9360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fournier A, Oprisiu-Fournier R, Serot JM, Godefroy O, Achard JM, Faure S, Mazouz H, Temmar M, Albu A, Bordet R, Hanon O, Gueyffier F, Wang J, Black S, Sato N. Prevention of dementia by antihypertensive drugs: how AT1-receptor-blockers and dihydropyridines better prevent dementia in hypertensive patients than thiazides and ACE-inhibitors. Expert Rev Neurother. 2009;9:1413–1431. doi: 10.1586/ern.09.89. [DOI] [PubMed] [Google Scholar]
- 19.Kehoe PG. Angiotensins and Alzheimer's disease: a bench to bedside overview. Alzheimers Res Ther. 2009;1:3. doi: 10.1186/alzrt3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou K, Michikawa M. Angiotensin-converting enzyme as a potential target for treatment of Alzheimer's disease: inhibition or activation? Rev Neurosci. 2008;19:203–212. doi: 10.1515/revneuro.2008.19.4-5.203. [DOI] [PubMed] [Google Scholar]
- 21.Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, Tschanz JT, Mayer LS, Welsh-Bohmer KA, Breitner JC. Antihypertensive Medication Use and Incident Alzheimer Disease: The Cache County Study. Arch Neurol. 2006;63:7. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- 22.Rosenberg PB, Mielke MM, Tschanz J, Cook L, Corcoran C, Hayden KM, Norton M, Rabins PV, Green RC, Welsh-Bohmer KA, Breitner JC, Munger R, Lyketsos CG. Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. Am J Geriatr Psychiatry. 2008;16:883–892. doi: 10.1097/JGP.0b013e318181276a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sink KM, Leng X, Williamson J, Kritchevsky SB, Yaffe K, Kuller L, Yasar S, Atkinson H, Robbins M, Psaty B, Goff DC., Jr Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension: results from the cardiovascular health study. Arch Intern Med. 2009;169:1195–1202. doi: 10.1001/archinternmed.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kehoe PG, Passmore AP. The Renin-Angiotensin System and Antihypertensive Drugs in Alzheimer’s Disease: Current Standing of the Angiotensin Hypothesis? Journal of Alzheimers Disease. 2012 doi: 10.3233/JAD-2012-111376. [DOI] [PubMed] [Google Scholar]
- 25.Diaz-Ruiz C, Wang J, Ksiezak-Reding H, Ho L, Qian X, Humala N, Thomas S, Martinez-Martin P, Pasinetti GM. Role of Hypertension in Aggravating Aβ Neuropathology of AD Type and Tau-Mediated Motor Impairment. Cardiovascular Psychiatry and Neurology. 2009;2009:Article ID 107286. doi: 10.1155/2009/107286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gentile MT, Poulet R, Di Pardo A, Cifelli G, Maffei A, Vecchione C, Passarelli F, Landolfi A, Carullo P, Lembo G. Beta-amyloid deposition in brain is enhanced in mouse models of arterial hypertension. Neurobiol Aging. 2009;30:222–228. doi: 10.1016/j.neurobiolaging.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman LB, Schmeidler J, Lesser GT, Beeri MS, Purohit DP, Grossman HT, Haroutunian V. Less Alzheimer disease neuropathology in medicated hypertensive than nonhypertensive persons. Neurology. 2009;72:1720–1726. doi: 10.1212/01.wnl.0000345881.82856.d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemming ML, Selkoe DJ. Amyloid beta -protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem. 2005;280:37644–37650. doi: 10.1074/jbc.M508460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou DR, Wang Y, Zhou L, Chen K, Tian Y, Song Z, Bao J, Yang QD. Altered angiotensin-converting enzyme and its effects on the brain in a rat model of Alzheimer disease. Chin Med J (Engl) 2008;121:2320–2323. [PubMed] [Google Scholar]
- 30.Hu J, Igarashi A, Kamata M, Nakagawa H. Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta ); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–47868. doi: 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- 31.Oba R, Igarashi A, Kamata M, Nagata K, Takano S, Nakagawa H. The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide. Eur J Neurosci. 2005;21:733–740. doi: 10.1111/j.1460-9568.2005.03912.x. [DOI] [PubMed] [Google Scholar]
- 32.Sun X, Becker M, Pankow K, Krause E, Ringling M, Beyermann M, Maul B, Walther T, Siems W-E. Catabolic attacks of membrane-bound angiotensin-converting enzyme on the N-terminal part of species-specific amyloid-[beta] peptides. European Journal of Pharmacology. 2008;588:18–25. doi: 10.1016/j.ejphar.2008.03.058. [DOI] [PubMed] [Google Scholar]
- 33.Toropygin IY, Kugaevskaya EV, Mirgorodskaya OA, Elisseeva YE, Kozmin YP, Popov IA, Nikolaev EN, Makarov AA, Kozin SA. The N-domain of angiotensin-converting enzyme specifically hydrolyzes the Arg-5-His-6 bond of Alzheimer's Abeta-(1-16) peptide and its isoAsp-7 analogue with different efficiency as evidenced by quantitative matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2008;22:231–239. doi: 10.1002/rcm.3357. [DOI] [PubMed] [Google Scholar]
- 34.Danielyan L, Klein R, Hanson LR, Buadze M, Schwab M, Gleiter CH, Frey WH. Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res. 2010;13:195–201. doi: 10.1089/rej.2009.0944. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117:3393–3402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zou K, Yamaguchi H, Akatsu H, Sakamoto T, Ko M, Mizoguchi K, Gong JS, Yu W, Yamamoto T, Kosaka K, Yanagisawa K, Michikawa M. Angiotensin-converting enzyme converts amyloid beta-protein 1-42 (Abeta(1-42)) to Abeta(1-40), and its inhibition enhances brain Abeta deposition. J Neurosci. 2007;27:8628–8635. doi: 10.1523/JNEUROSCI.1549-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gard PR. Angiotensin as a target for the treatment of Alzheimer's disease, anxiety and depression. Expert Opin Ther Targets. 2004;8:7–14. doi: 10.1517/14728222.8.1.7. [DOI] [PubMed] [Google Scholar]
- 38.Zhu D, Shi J, Zhang Y, Wang B, Liu W, Chen Z, Tong Q. Central angiotensin II stimulation promotes beta amyloid production in Sprague Dawley rats. PLoS One. 2011;6:e16037. doi: 10.1371/journal.pone.0016037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnes JM, Barnes NM, Costall B, Horovitz ZP, Ironside JW, Naylor RJ, Williams TJ. Angiotensin II inhibits acetylcholine release from human temporal cortex: implications for cognition. Brain Res. 1990;507:341–343. doi: 10.1016/0006-8993(90)90294-l. [DOI] [PubMed] [Google Scholar]
- 40.Barnes JM, Barnes NM, Costall B, Horovitz ZP, Naylor RJ. Angiotensin II inhibits the release of [3H] acetylcholine from rat entorhinal cortex in vitro. Brain Research. 1989;491:136–143. doi: 10.1016/0006-8993(89)90095-4. [DOI] [PubMed] [Google Scholar]
- 41.AbdAlla S, Lother H, el Missiry A, Langer A, Sergeev P, el Faramawy Y, Quitterer U. Angiotensin II AT2 receptor oligomers mediate G-protein dysfunction in an animal model of Alzheimer disease. J Biol Chem. 2009;284:6554–6565. doi: 10.1074/jbc.M807746200. [DOI] [PubMed] [Google Scholar]
- 42.AbdAlla S, Lother H, el Missiry A, Sergeev P, Langer A, el Faramawy Y, Quitterer U. Dominant negative AT2 receptor oligomers induce G-protein arrest and symptoms of neurodegeneration. J Biol Chem. 2009;284:6566–6574. doi: 10.1074/jbc.M808277200. [DOI] [PubMed] [Google Scholar]
- 43.Mogi M, Tsukuda K, Li JM, Iwanami J, Min LJ, Sakata A, Fujita T, Iwai M, Horiuchi M. Inhibition of cognitive decline in mice fed a high-salt and cholesterol diet by the angiotensin receptor blocker, olmesartan. Neuropharmacology. 2007;53:899–905. doi: 10.1016/j.neuropharm.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 44.Takeda S, Sato N, Takeuchi D, Kurinami H, Shinohara M, Niisato K, Kano M, Ogihara T, Rakugi H, Morishita R. Angiotensin receptor blocker prevented beta-amyloid-induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension. 2009;54:1345–1352. doi: 10.1161/HYPERTENSIONAHA.109.138586. [DOI] [PubMed] [Google Scholar]
- 45.Ferrington L, Miners JS, Palmer LE, Bond SM, Povey JE, Kelly PAT, Love S, Horsburgh KJ, Kehoe PG. Angiotensin II-inhibiting drugs have no effect on intraneuronal Aβ or oligomeric Aβ levels in a triple transgenic mouse model of Alzheimer's disease. Am J Transl Res. 2011;3:12. [PMC free article] [PubMed] [Google Scholar]
- 46.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 47.Lorenz JN. A practical guide to evaluating cardiovascular, renal, and pulmonary function in mice. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1565–R1582. doi: 10.1152/ajpregu.00759.2001. [DOI] [PubMed] [Google Scholar]
- 48.Gulinello M, Gertner M, Mendoza G, Schoenfeld BP, Oddo S, LaFerla F, Choi CH, McBride SM, Faber DS. Validation of a 2-day water maze protocol in mice. Behav Brain Res. 2009;196:220–227. doi: 10.1016/j.bbr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scullion GA, Kendall DA, Marsden CA, Sunter D, Pardon MC. Chronic treatment with the alpha(2)-adrenoceptor antagonist fluparoxan prevents age-related deficits in spatial working memory in APPxPS1 transgenic mice without altering beta-amyloid plaque load or astrocytosis. Neuropharmacology. 2011;60:223–234. doi: 10.1016/j.neuropharm.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 50.Miners S, Ashby E, Baig S, Harrison R, Tayler H, Speedy E, Prince JA, Love S, Kehoe PG. Angiotensin-converting enzyme levels and activity in Alzheimer's disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res. 2009;1:163–177. [PMC free article] [PubMed] [Google Scholar]
- 51.Pathak A, Hanon O, Negre-Pages L, Sevenier F. Rationale, design and methods of the OSCAR study: observational study on cognitive function and systolic blood pressure reduction in hypertensive patients. Fundam Clin Pharmacol. 2007;21:199–205. doi: 10.1111/j.1472-8206.2006.00465.x. [DOI] [PubMed] [Google Scholar]
- 52.Shlyakhto E. Observational Study on Cognitive function And systolic blood pressure Reduction (OSCAR): preliminary analysis of 6-month data from > 10,000 patients and review of the literature. Curr Med Res Opin. 2007;23(Suppl 5):S13–18. doi: 10.1185/030079907X260719. [DOI] [PubMed] [Google Scholar]
- 53.Hemming ML, Selkoe DJ, Farris W. Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid beta-protein metabolism in mouse models of Alzheimer disease. Neurobiol Dis. 2007;26:273–281. doi: 10.1016/j.nbd.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Claassen JA, Zhang R. Cerebral autoregulation in Alzheimer's disease. J Cereb Blood Flow Metab. 2011;31:1572–1577. doi: 10.1038/jcbfm.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanon O, Latour F, Seux ML, Lenoir H, Forette F, Rigaud AS. Evolution of blood pressure in patients with Alzheimer's disease: a one year survey of a French Cohort (REAL. FR) J Nutr Health Aging. 2005;9:106–111. [PubMed] [Google Scholar]
- 56.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, Persson G, Oden A, Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 57.Washida K, Ihara M, Nishio K, Fujita Y, Maki T, Yamada M, Takahashi J, Wu X, Kihara T, Ito H, Tomimoto H, Takahashi R. Nonhypotensive dose of telmisartan attenuates cognitive impairment partially due to peroxisome proliferator-activated receptor-gamma activation in mice with chronic cerebral hypoperfusion. Stroke. 2010;41:1798–1806. doi: 10.1161/STROKEAHA.110.583948. [DOI] [PubMed] [Google Scholar]
- 58.Yamada K, Uchida S, Takahashi S, Takayama M, Nagata Y, Suzuki N, Shirakura S, Kanda T. Effect of a centrally active angiotensin-converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer's disease. Brain Res. 2010;1352:176–186. doi: 10.1016/j.brainres.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 59.Mogi M, Li JM, Tsukuda K, Iwanami J, Min LJ, Sakata A, Fujita T, Iwai M, Horiuchi M. Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun. 2008;375:446–449. doi: 10.1016/j.bbrc.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 60.Takeda S, Sato N, Rakugi H, Morishita R, editors. In Improvement of cognitive decline and cerebrovascular dysfunction in a mouse model of Alzheimer's disease by angiotensin receptor blocker, Olmesartan, Alzheimer's Association International Conference on Alzheimer's Disease (ICAD), McCormick Place, Chicago, 2008. Alzheimer's & Dementia; 2008; McCormick Place, Chicago. T479 pp. [Google Scholar]
- 61.Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- 62.Daumas S, Sandin J, Chen KS, Kobayashi D, Tulloch J, Martin SJ, Games D, Morris RG. Faster forgetting contributes to impaired spatial memory in the PDAPP mouse: deficit in memory retrieval associated with increased sensitivity to interference? Learn Mem. 2008;15:625–632. doi: 10.1101/lm.990208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dember WN, Richman CL. Spontaneous Alternation Behaviour. New York: Springer-Verlag Publishing; 1989. [Google Scholar]
- 64.Bontempi B, Whelan KT, Risbrough VB, Lloyd GK, Menzaghi F. Cognitive enhancing properties and tolerability of cholinergic agents in mice: a comparative study of nicotine, donepezil, and SIB-1553A, a subtype-selective ligand for nicotinic acetylcholine receptors. Neuropsychopharmacology. 2003;28:1235–1246. doi: 10.1038/sj.npp.1300150. [DOI] [PubMed] [Google Scholar]
- 65.Mooradian AD, Lieberman J. Age-related decrease in serum angiotensin converting enzyme activity: the role of thyroidal status and food intake. J Gerontol. 1990;45:B24–27. doi: 10.1093/geronj/45.1.b24. [DOI] [PubMed] [Google Scholar]
- 66.Savaskan E, Hock C, Olivieri G, Bruttel S, Rosenberg C, Hulette C, Muller-Spahn F. Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer's dementia. Neurobiol Aging. 2001;22:541–546. doi: 10.1016/s0197-4580(00)00259-1. [DOI] [PubMed] [Google Scholar]
- 67.Furberg CD, Pitt B. Are all angiotensin-converting enzyme inhibitors interchangeable? J Am Coll Cardiol. 2001;37:1456–1460. doi: 10.1016/s0735-1097(01)01161-5. [DOI] [PubMed] [Google Scholar]
- 68.Hudson M, Humphries K, Tu JV, Behlouli H, Sheppard R, Pilote L. Angiotensin II receptor blockers for the treatment of heart failure: a class effect? Pharmacotherapy. 2007;27:526–534. doi: 10.1592/phco.27.4.526. [DOI] [PubMed] [Google Scholar]