

Abstract
The survival kinase Akt and the tumor suppressor LKB1 elicit opposite effects on cell proliferation and tumorigenesis. The present study demonstrates that Akt acts as an upstream kinase of LKB1 to promote the phosphorylation at Ser334 and facilitate its binding to 14-3-3 proteins, resulting in a decreased interaction with STE20-related adaptor protein α (STRADα) and an enhanced nuclear accumulation of LKB1. The S334A mutant of LKB1 exhibits impaired binding with 14-3-3 proteins and is localized predominantly in the cytoplasm, whereas the phosphorylation-mimic mutant, S334D, is sequestrated in the nuclei and unable to elicit the tumor suppressor function. On the other hand, S334A exerts more potent activity than wild type LKB1 in inhibiting the breast cancer cell proliferation and tumor growth in mice. These findings suggest that Akt blocks the anti-growth signal of LKB1 by triggering a phosphorylation-dependent nuclear sequestration of LKB1 through 14-3-3 proteins.
Keywords: Akt kinase, 14-3-3, LKB1, nuclear/cytoplasmic shuttling, tumorigenesis
Introduction
LKB1 is a serine/threonine protein kinase mutated in patients with Peutz-Jeghers syndrome, an autosomal dominant genetic condition that predisposes to a wide spectrum of benign and malignant tumors [1,2]. LKB1 functions as a tumor suppressor by controlling cell division [3], regulating cell polarity [4], and coupling cell growth to energy metabolism [5]. Restoring LKB1 expression in a number of tumor cell lines suppresses their growth by inducing G1 cell-cycle arrest [6]. LKB1 is a major upstream kinase of AMP-activated protein kinase (AMPK), a master regulator of energy homeostasis and cell growth [7]. In addition, it activates at least 12 other members of AMPK-related protein kinase family through phosphorylation of a conserved threonine in the “T-loop” of the kinase domain [8].
LKB1 signaling is regulated by posttranslational modification and sub-cellular translocation [9-11]. It is phosphorylated on multiple residues, and some of these phosphorylations (such as Thr336 and Ser428) have been shown to modulate the catalytic activity or sub-cellular localizations of this kinase [9,12,13]. LKB1 forms a heterotrimeric complex with the pseudokinase STE20-related adaptor protein α (STRADα) and the scaffolding protein mouse protein 25 (MO25), which facilitate the activation and localization of LKB1 in the cytoplasm [14-16]. Various mutants of LKB1 discovered in human cancer patients accumulate predominantly to the nucleus and are unable to suppress cell growth [11,17], suggesting that the cytosol pool of LKB1 plays a key role in mediating its tumor suppressive activities. On the other hand, the precise upstream signaling events that control the posttranslational modification and subcellular translocation of LKB1 remain poorly characterized.
The present study demonstrated that the survival kinase Akt acts as an immediate upstream regulator of LKB1 to mediate the phosphorylation at Ser334, leading to an enhanced interaction with 14-3-3 proteins and augmented nuclear retention of this kinase. Importantly, phosphorylation at Ser334 abolishes the tumor suppressive activity of LKB1 in both human breast cancer cell cultures and mammary carcinoma implanted in nude mice.
Materials and methods
Antibodies, reagents and expression vectors
Antibodies for Akt, LKB1, β-tubulin and CREB (48H2) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies for mouse anti-Flag M2 and β-actin were from Sigma (Saint Louis, MO, USA). Antibodies for pan-14-3-3 and 14-3-3ζ, and the specific siRNA for 14-3-3ζ were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Akt inhibitor (Akti-1/2) was kindly provided by Dr Peter R. Shepherd [18]. Activated His-Akt1 was purchased from Calbiochem (San Diego, CA, USA). The mammalian expression vector for murine LKB1 (pcDNA3-LKB1) was a gift from Dr David Carling [7]. The vectors for prokaryotic expression of 14-3-3 protein isoforms and His-tagged STRADα were obtained from Addgene (Cambridge, MA, USA). Prokaryotic expression vector encoding GST-tagged LKB1 (GST-LKB1) was generated by subcloning the mouse LKB1 cDNA into pGEX-4T-1 vector (GE Healthcare). Site-directed mutagenesis was performed to construct vectors for expressing LKB1 mutants in mammalian cells (pcDNA3-LKB1 S334A and pcDNA-LKB1 S334D), using the primers listed in Supplementary Table 1. The Recombinant proteins were produced and purified as described [19].
In vitro phosphorylation assay
In vitro phosphorylation was performed by co-incubating ten microgram of recombinant proteins with 250 nanogram of activated Akt1 in 25 μl kinase assay buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM EGTA, 1 mM PMSF, 0.01% Brij-35, 1 mM DTT, 1 mM Na3VO4, 1 mM NaF, 1×Protease Inhibitor cocktail, 2.5 μCi of [γ-32P] ATP and 100 μM unlabelled ATP) for one hour at 30°C. Reactions were stopped by addition of SDS-PAGE loading buffer and boiling for five minutes. Samples were then separated by 8% SDS-PAGE and radioactive signaling detected by autoradiography.
Cell culture and establishment of stable cell lines
MDA-MB-231 and HEK293 cells were obtained from the American Type Culture Collection and maintained at 37°C in DMEM (Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum. Cells were transfected with mammalian expression vectors encoding Flag-tagged LKB1 or two types of mutants (S334A and S334D), followed by drug-resistance selection with G418 (1 mg/ml) [19]. Positive single clones were selected and then pooled together for subsequent characterization or tumor implantation. The expression of Flag-tagged LKB1 or its mutants was confirmed by Western blotting analysis as described [20].
Co-immunoprecipitation and GST pull-down assay
For co-immunoprecipitation, cells were solubilized in RIPA lysis buffer and the lysates incubated with anti-Flag M2 affinity agarose beads (Sigma) at 4°C overnight on a shaking platform. After washing extensively with ice-cold PBS, the immune complexes were eluted with 0.1 M glycine HCl (pH 3.0) and neutralized for SDS-PAGE and Western blotting analysis.
GST pull-down was performed by incubating the cell lysates with proteins bound to Glutathione Sepharose 4B beads for 2 hours at 4°C on a rotating platform. The beads were recovered by centrifugation (500 g, 15 seconds, 4°C), washed three times with phosphate buffer, resuspended in SDS-PAGE loading buffer and then subjected to Western blotting analysis.
Subcellular fractionation
Cells were resuspended in a hypotonic buffer [10 mM Tris-HCl (pH 7.5), 25 mM KCl, 2 mM magnesium acetate, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride plus protease inhibitor cocktails] and incubated on ice for 10 minutes. Cell membranes were disrupted by 20 passes through a 25-gauge needle, and the nuclei integrity was monitored under a microscope. After centrifugation for five minutes at 1,000 g, the supernatant was harvested as the “cytoplasmic fraction”. The nuclear pellet was washed with the hypotonic buffer and lysed with RIPA buffer. The lysates were centrifuged at 12,000 g for 5 minutes and the supernatant collected as the “nuclear fraction”. Protein concentrations were determined by BCA method (Pierce Biotechnology Inc. Rockford, USA). Equal amount of proteins from cytoplasmic and nuclear fractions were separated by SDS-PAGE, transferred to PVDF membrane, and probed with specific primary antibodies to determine the expression of the target proteins.
Immunocytochemistry
Twenty-four hours prior to transfection, HEK293 cells were seeded on coverslips. The cells were transiently transfected with various vectors and fixed with methanol. Stably-transfected MDA-MB-231 cells were cultured on coverslips and then fixed with 4% paraformaldehyde. After blocking with 5% FBS in 1 × TBST (0.1% Triton X-100, 0.15 M NaCl, 0.05 M Tris-HCl, pH7.4) for one hour, the cells were incubated with goat anti-LKB1 antibody at 4°C for 16 hours. After three washes, the slides were incubated with an Alex Fluor 594 chicken anti-goat secondary antibody (Life Technologies, Grand Island, NY, USA) at a dilution of 1:500 for 1 hour at room temperature in dark. The slides were then rinsed, mounted with ProLong® Gold antifade reagent with DAPI (Life Technologies) and observed under a fluorescence microscope (Carl Zeiss), and images were captured using AxioVision plus software.
RNA interference and transfection
Cells were seeded at a density of 5 × 105 cells/well in 6-well plates and grown overnight. Specific 14-3-3ζ siRNA and control scramble siRNA were transfected using jetPEITM reagent (Polyplus-transfection SA, New York, NY, USA) according to manufacturer’s instructions. 48 hours after transfection, cells were harvested and subjected to cellular fractionation and Western blotting analysis. DeliverXTM Peptide Transfection kit (Panomics, Santa Clara, CA, USA) was used for the peptide transfection.
Cell counting and 3H-thymidine incorporation assay
Cells were seeded at a density of 1 × 103 per well in 96-well plates. After starving in DMEM with 0.5% FBS for 24 hours, cells were subsequently stimulated with 10% FBS. At different time points, cells were harvested by trypsin digestion and stained with trypan blue. The numbers of viable cells were manually counted under a microscope using a hemocytometer. For [3H]thymidine incorporation experiment, equal number of cells (2 × 104 per well) was seeded in a 24-well plate. After starvation in DMEM containing 0.5% FBS for 24 hours, the cells were subsequently stimulated with DMEM containing 10% FBS. Cells were labeled with 1 μCi/ml of [3H-methyl] thymidine during the last six hours to determine the rate of DNA synthesis [21].
Cell cycle analysis
Cells fixed with 70% ethanol were resuspended in the staining buffer [100 mM Tris pH 7.4, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2, 0.1 mg/ml RNase, 0.1% NP-40 and 50 μg/ml propidium iodide] and incubated for 30 minutes. The analysis was carried out with EPICS® Elite ESP Flow Cytometer and EXPO software (Beckman Coulter, Miami, FL). Data were acquired from 1 x 104 cells per sample. Gating of G0/G1-, S-, and G2/M-populations was performed using modfit 3.1 software (Verity Software House, Topsham, ME, USA).
Inoculation of breast cancer cells into nude mice
A total number of 5×106 stably-transfected MDA-MB-231 cells were injected into the right thoracic mammary fat pad of 4-week old female nude mice under anesthetic condition to investigate the effects of LKB1 and its two mutants on breast cancer development. Tumors were measured using digital vernier calipers, with tumor volume calculated using the formula [sagittal dimension (mm) × cross dimension (mm)] 2/2 and expressed in mm3. All animals were sacrificed at 8 weeks after the initial implantation. Tumor tissues were collected, fixed in 10% neutral-buffered formalin and subjected to further analysis. All the experimental protocols were approved by the Animal Ethics Committee at the University of Hong Kong.
Data analysis
All experiments were performed with four to six samples per group and results derived from at least three independent experiments. The density of protein bands were quantitatively analyzed by ImageJ software for calculating the expression ratios. Representative Western blotting images were shown. Values are expressed as mean ± SEM. All the statistical calculations were analyzed with the Statistical Package for the Social Sciences version 11.5 software package (SPSS). Comparison between groups was done using Student’s unpaired t test. In all statistical comparisons, P < 0.05 was used to indicate a significant difference.
Results
LKB1 is phosphorylated at Ser334 by Akt
Tandem mass spectrometric analysis was performed for Flag-tagged LKB1 purified from a stably-transfected HEK293 cells using 4800 MALDI-TOF/TOFTM instrument. This analysis revealed that phosphorylation occurred at six amino acid residues of LKB1, including Ser31, Thr32, Ser69, Thr71, Ser334 and Thr336. Among them, phosphorylations at Ser31 and Thr336 have been reported previously [12]. Analysis of LKB1 protein sequence using the Scansite 2.0 program (http://scansite.mit.edu) indicated that there was an Akt consensus phosphorylation motif containing the highly conserved Ser334 and Thr336 residues (RXRXXS/ T) (Figure 1A). Therefore, an in vitro phosphorylation assay was performed to test whether Akt indeed acted as an upstream kinase of LKB1. LKB1 protein was purified from E.coli. Incubation of this recombinant LKB1 with activated Akt1 led to a distinct 32P-radiolabelled band (Figure 1B, top panel). In-gel trypsin digestion was subsequently performed for the corresponding Coomassie Brilliant Blue-stained protein bands (Figure 1B, bottom panel). Mass spectrometric analysis identified a unique peptide with a molecular weight of 2038.04 Da existing only in LKB1 protein samples incubated with Akt1. De novo sequencing using QSTAR® XL Hybrid quadrupole-TOF mass spectrometer confirmed that this peptide was derived from amino acid 332-347 of LKB1, containing only one phosphorylated residue at Ser334 (Supplementary Figure 1). The direct interaction between LKB1 and Akt was further verified by GST-pull down assay (Figure 1C, top panel). Coimmunoprecipitation analysis also demonstrated that endogenous Akt interacted with LKB1 in HEK293 cells (Figure 1C, bottom panel).
Figure 1.
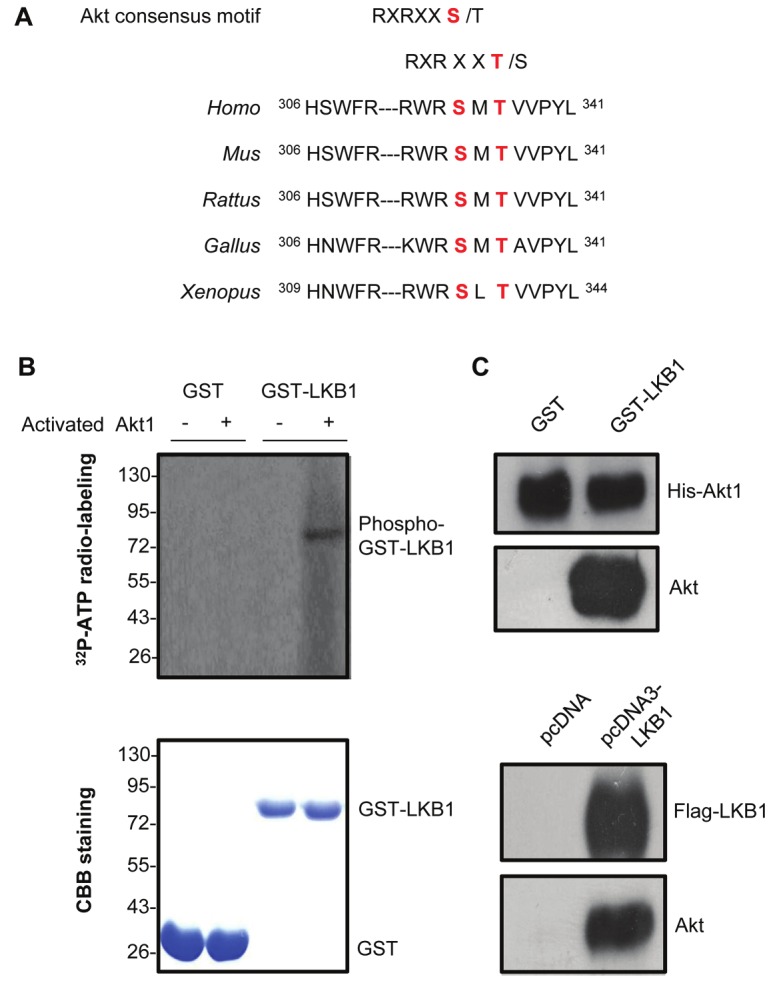
LKB1 is a downstream target of Akt. (A). Two conserved Akt consensus motifs containing Ser334 and Thr336 residues were found within the amino acid sequences of LKB1 from different species. (B). Top panel: In vitro phosphorylation assay was performed by incubating recombinant active Akt1 with GST-LKB1 or GST. 32P-labeled phosphorylated LKB1 was detected by autoradiography. Bottom panel: The total proteins were visualized by Coomassie Brilliant Blue (CBB) staining. (C). Top panel: GST-LKB1 or GST proteins bound to Glutathione Sepharose 4B beads were used for incubating with equal amount of His-tagged Akt1, and the associated Akt was detected by Western blot using its specific antibody. Bottom panel: Coimmunoprecipitation was performed by adding anti-Flag M2 agarose beads into the lysates of HEK293 cells transfected with pcDNA or pcDNA3-LKB1 vectors. Immune complexes were resolved by SDS-PAGE and the endogenous Akt associated with LKB1 detected by anti-Akt antibody.
14-3-3 proteins interact with LKB1 via Akt-mediated phosphorylation of Ser334
Using the Scansite program, a conserved 14-3-3 binding site (RXXSX) was found to locate within the same Akt consensus motif of LKB1, containing the Ser334 residue (Figure 2A). 14-3-3 proteins are a family of conserved regulatory molecules that bind to a multitude of intracellular signaling proteins and regulate the functions of their interacting partners [22,23]. In vitro pulldown analysis demonstrated that among the seven 14-3-3 proteins, robust interactions could be detected between LKB1 and 14-3-3 ζ, γ and η (Figure 2B). The interaction of LKB1 with endogenous 14-3-3 proteins in HEK293 cells was further confirmed by coimmunoprecipitation using the pan 14-3-3 antibody (Figure 2C).
Figure 2.
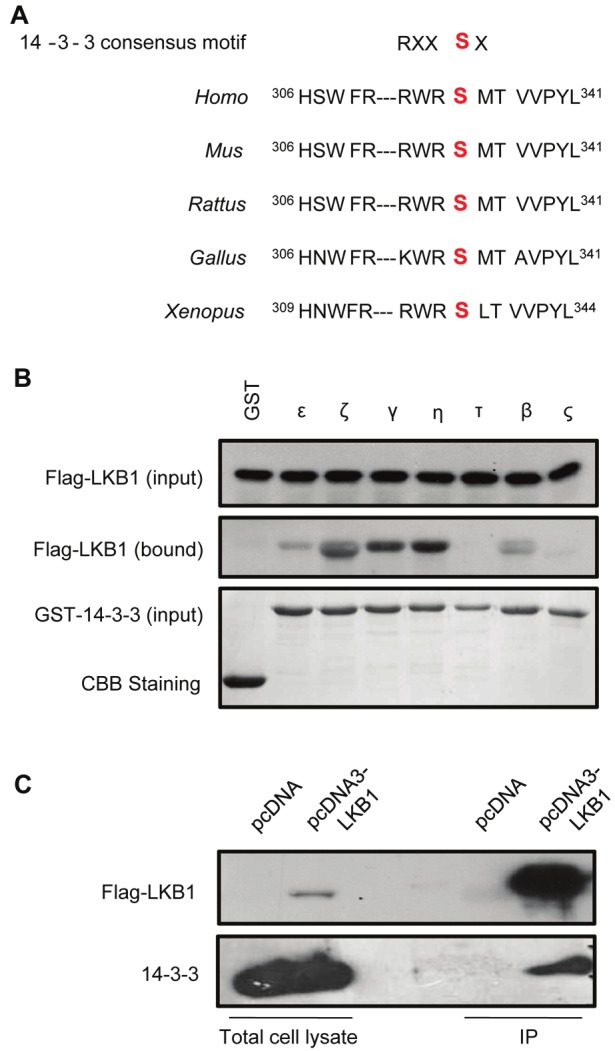
LKB1 interacts with 14-3-3 proteins. (A). A conserved 14-3-3 binding site was found in LKB1 sequences containing the Ser334 residue. (B). Equal amounts of the seven 14-3-3 isoforms were cross-linked to the Glutathione Sepharose 4B beads and incubated with cell lysates from HEK293 cells overexpressing Flag-tagged wild type LKB1. The proteins bound to 14-3-3 isoforms were detected by Western blotting analysis using an anti-Flag antibody. (C). The lysates of HEK293 cells transfected with pcDNA or pcDNA3-LKB1 vectors were incubated with anti-Flag M2 agarose beads. Total cell lysates or immunoprecipitated complexes (IP) were resolved by SDS-PAGE and analyzed by Western blotting using anti-Flag or anti-pan-14-3-3 antibody.
To investigate whether Akt-induced phosphorylation was involved in the binding of LKB1 to 14-3-3 proteins, HEK293 cells were pre-treated with Akt inhibitor (Akti-1/2) and then subjected to both GST-pull down and co-immunoprecipitation analysis. The results demonstrated that the interactions between LKB1 and 14-3-3 proteins were significantly suppressed by treatment with the Akt inhibitor (Akti-1/2) (Figure 3A and 3B). Furthermore, mutation of Ser334 to non-phosphorylatable alanine residue (S334A) abolished the interactions between LKB1 and 14-3-3 proteins. By contrast, S334D, a LKB1 mutant mimicking Akt-mediated phosphorylation, exhibited much stronger interactions with 14-3-3 proteins when compared to wild type LKB1 (Figure 3C and 3D).
Figure 3.

The interaction between 14-3-3 and LKB1 is dependent on the phosphorylation of LKB1 at Ser334. (A). GST-14-3-3ζ bound to Glutathione Sepharose 4B beads was incubated with the cell lysates of HEK293 cells treated with DMSO (vehicle control) or 10 μM Akti-1/2. The endogenous LKB1 associated with GST-14-3-3ζ was detected by Western blotting using an anti-LKB1 antibody. (B). HEK293 cells overexpressing Flag-tagged wild type LKB1 were treated with or without 10 μM Akti-1/2, followed by co-immunoprecipitation using anti-Flag M2 agarose beads. 14-3-3 proteins associated with Flag-tagged LKB1 were detected by Western blotting using an anti-pan-14-3-3 antibody. (C). HEK293 cells overexpressing wild type LKB1 (WT), S334A, or S334D were incubated with GST-14-3-3ζ-bound Glutathione Sepharose 4B beads, and the immunoprecipitated proteins were detected by Western blotting using an anti-Flag antibody. (D). HEK293 cells overexpressing wild type LKB1 (WT), S334A or S334D were subjected to co-immunoprecipitation using anti-Flag M2 agarose beads, and endogenous 14-3-3 proteins associated with Flag-LKB1 were detected by Western blotting using an anti-pan-14-3-3 antibody. The relative ratios of the protein bands in Western blotting results were calculated and presented as the means ± SEM from at least three independent experiments. * P <0.05 vs corresponding controls.
Phosphorylation at Ser334 promotes the nuclear accumulation of LKB1
Sub-cellular fractionation and immunofluorescence staining analysis demonstrated that wild type LKB1 was present in both nucleus and cytoplasm, whereas treatment with Akt inhibitor Akti-1/2 promoted the translocation of LKB1 from nuclei to cytosol (Figure 4A and 4B). S334A mutation enhanced the cytosolic localization of LKB1 (Figure 4C and 4D). By contrast, the S334D mutant was located predominantly in the nucleus and barely detectable in the cytoplasm.
Figure 4.

Phosphorylation at Ser334 modulates the sub-cellular localization of LKB1 in HEK293 cells. (A). Cells stably overexpressing Flag-tagged wild type LKB1 were treated with vehicle control (DMSO) or 10 μM Akti-1/2 for 24 hours. The distribution of LKB1 in nuclear (Nuc) and cytosolic (Cyt) fractions was analyzed by Western blotting using an anti-Flag antibody. CREB and β-tubulin were used as the nuclear and cytosolic marker, respectively. (B). Immunofluorescence staining was performed for LKB1 (red) in cells treated with vehicle control or Akti-1/2 as in panel A. The images were merged with those of DAPI-stained nuclei (blue). Magnification, 400×. (C). Sub-cellular distribution of wild type LKB1 and its two mutants (S334A and S334D) in HEK293 cells was determined by cell fractionation and Western blotting analysis as in panel A. (D). Immunofluorescence staining for LKB1 and its two mutants expressed in HEK293 cells was performed and results presented as in panel B. The relative ratios of the protein bands in Western blotting results were calculated and presented as the means ± SEM from at least three independent experiments. * P <0.05 vs corresponding controls.
In HEK293 cells, 14-3-3ζ was the most abundant isoform of 14-3-3 proteins (data not shown). Transfection with specific siRNA reduced the amount of 14-3-3ζ by ~50% (Figure 5A). The reduction of 14-3-3ζ significantly promoted the translocation of LKB1 from nucleus to cytoplasm (Figure 5B). Moreover, treatment with difopein, the dimeric 14-3-3 peptide inhibitor [24], profoundly reduced the amount of 14-3-3 proteins associated with LKB1 (Figure 5C). Notably, the decreased interaction between LKB1 and 14-3-3 proteins was accompanied by an enhanced translocation of LKB1 from nucleus to cytoplasm (Figure 5D). Downregulation or inhibition of 14-3-3 proteins elicited similar effects on promoting the nuclear-cytoplasmic shuttling of S334D mutant (Supplementary Figure 2).
Figure 5.
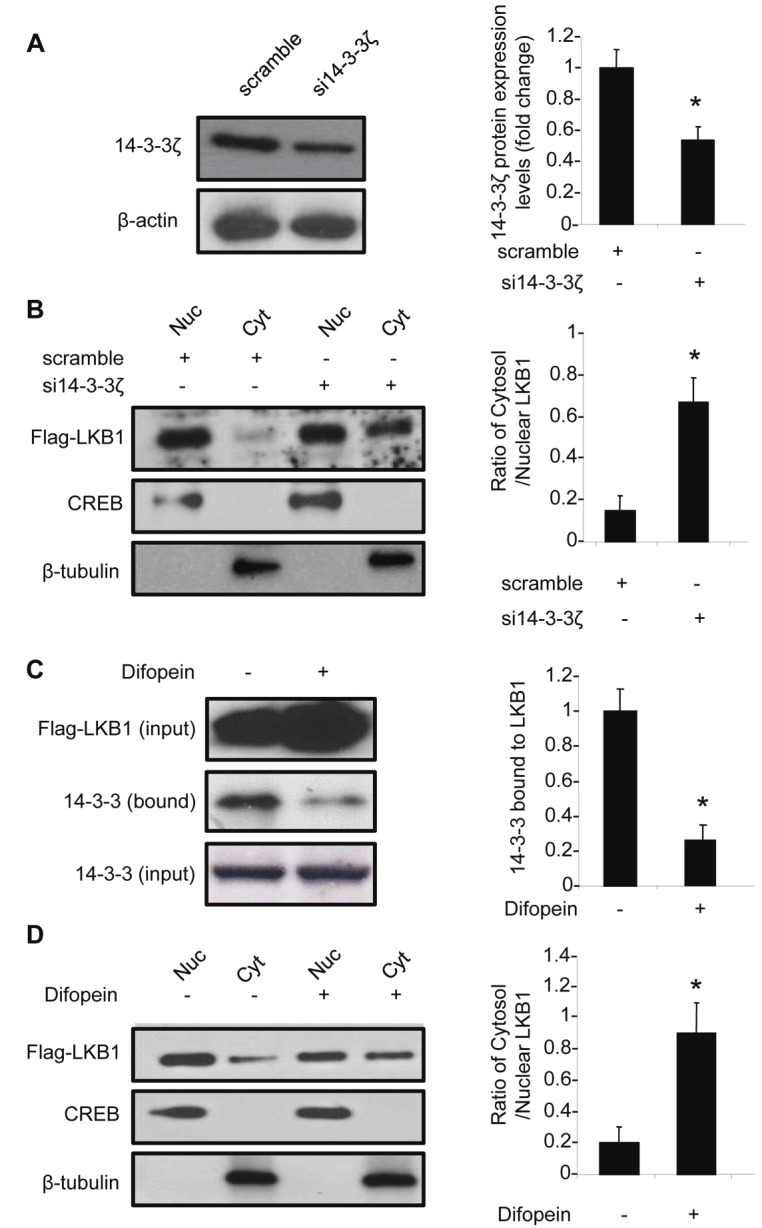
14-3-3 proteins are required for Akt-mediated nuclear accumulation of LKB1. (A). HEK293 cells were transfected with scramble control or 14-3-3ζ-specific siRNA (si14-3-3ζ). After 48 hours, the level of 14-3-3ζ was detected by Western blotting using an anti-14-3-3ζ antibody. (B). The effects of 14-3-3ζ knocking down on sub-cellular distribution of LKB1 were evaluated in the nuclear (Nuc) and cytosolic (Cyt) fractions of HEK293 cells stably overexpressing Flag-tagged wild type LKB1. Western blotting was performed using an anti-Flag antibody. (C). HEK293 cells stably overexpressing Flag-tagged wild type LKB1 were transfected with or without difopein, and subjected to co-immunoprecipitation using anti-Flag M2 agarose beads. 14-3-3 proteins associated with LKB1 were detected by Western blotting using an anti -pan-14-3-3 antibody. (D). The nuclear and cytosolic fractions of Flag-tagged wild type LKB1 in cells treated with or without difopein were detected using an anti-Flag antibody. The relative ratios of the protein bands in Western blotting results were calculated and presented as the means ± SEM from at least three independent experiments. * P <0.05 vs corresponding controls.
The association of STRADα, a cofactor of LKB1 that facilitates its cytoplasmic localization [16], was significantly enhanced by S334A, but attenuated by S334D mutagenesis (Figure 6A). Inhibition of Akt further elevated the amount of STRADα associated with LKB1 (Figure 6B). Taken together, these results demonstrate that Akt-mediated phosphorylation and the interaction with 14-3-3 proteins act synergistically to facilitate the nuclear retention of LKB1.
Figure 6.

Phosphorylation of LKB1 at Ser334 reduces the complex formation with STRADα. (A). HEK293 cells stably overexpressing wild type LKB1 (WT), S334A or S334D were transfected with plasmid encoding His-tagged STRADα or an empty control vector (pcDNA). After 48 hours, cells were harvested and subjected to co-immunoprecipitation using anti-Flag M2 agarose beads. STRADα bound to LKB1 was detected by Western blotting using the specific antibody. (B). HEK293 cells stably overexpressing Flag-tagged wild type LKB1 were transfected with plasmid encoding His-tagged STRADα. After starvation in DMEM containing 0.5 % FBS for 24 hours, cells were treated with or without 10 μM Akti-1/2 for another 24 hours. The association of STRADα with LKB1 was determined by co-immunoprecipitation followed by Western blotting analysis as in panel A. The relative ratios of the protein bands in Western blotting results were calculated and presented as the means ± SEM from at least three independent experiments. * P <0.05 vs corresponding controls.
Phosphorylation of LKB1 at Ser334 modulates its tumor suppressor activity
The anti-proliferative effects of wild type LKB1, S334A and S334D mutants were evaluated in MDA-MB-231 human breast cancer cells. To this end, stable-expression clones of MDA-MB-231 cells were established by transfection with plasmids encoding wild type LKB1 or each of the two mutants (S334A and S334D). The protein expression levels of the three versions of Flag-tagged LKB1 were comparable as demonstrated by Western blotting analysis (Supplementary Figure 3A). Their intracellular distributions were similar to those observed in HEK293 cells, with S334A mutant mainly localized in the cytoplasm and S334D mutant accumulated predominantly within the nucleus (Supplementary Figure 3B). Cell number counting results showed that wild type LKB1 and S334A dramatically reduced the cell proliferation rate of MDA-MB-231 cells, whereas S334D lost the anti-growth activity (Figure 7A). Consistently, 3H-thymidine incorporation experiment revealed that S334D virtually had no effects on DNA synthesis (Figure 7B). Note that the suppressive effects of LKB1 on DNA synthesis were significantly augmented by S334A mutation. Flow cytometric analysis showed that ~62% of cells overexpressing wild type LKB1 and ~73% of cells overexpressing S334A were arrested in G1 phase after serum stimulation (Figure 7C). By contrast, much less percentage of cells was distributed in G1 phase for the control group and those overexpressing S334D (~48% and ~45%, respectively).
Figure 7.
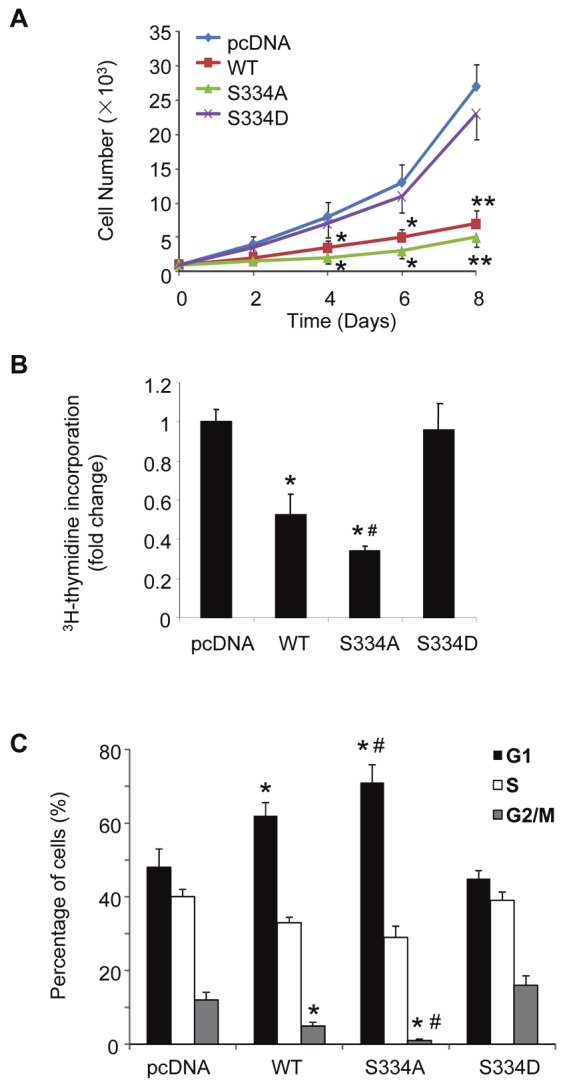
Phosphorylation at Ser334 inhibits the antiproliferative activity of LKB1 in human MDA-MB-231 breast cancer cells. Cells stably overexpressing wild type LKB1 (WT), S334A or S334D were established as described in Figure 4. (A). The proliferation rates of the stably-transfected MDA-MB-231 cells were analyzed by manually counting the cell numbers at different time points. (B). DNA synthesis rates were determined by 3H-thymidine incorporation assay and expressed as fold changes over pcDNA-transfected control cells. (C). The stably-transfected MDA-MB-231 cells were starved with DMEM containing 0.5% FBS for 48 hours and stimulated with DMEM containing 10% FBS for another 24 hours. The cell cycle distribution was analyzed by flow cytometry. The results are presented as the means ± SEM. * P < 0.05 and ** P < 0.01 vs pcDNA groups; # P < 0.05 vs WT groups (unpaired t test, n = 6).
The stably-transfected MDA-MB-231 cells were subsequently implanted into the mammary fat pads of female nude mice (106 cells per animal) and the tumor development monitored regularly after initial implantation. Tumors originated from control cells and those overexpressing S334D were palpable as early as 6 days after initial inoculation, whereas the onset time of tumors derived from cells overexpressing wild type LKB1 and S334A was delayed to around 14 and 18 days, respectively (Figure 8A). Tumor growth was significantly suppressed by overexpression of wild type LKB1 (Figure 8B). The growth of MDA-MB-231 tumors was almost abolished by S334A mutation. The tumor weights of cells overexpressing wild type LKB1 and S334A were significantly reduced by ~69 % and ~88 %, respectively, compared to the control group (Figure 8C). On the contrary, the tumors derived from cells overexpressing LKB1 S334D showed similar tumor growth rate and slightly higher tumor weight compared with those of the control group (Figure 8B and 8C).
Figure 8.

Phosphorylation of LKB1 at Ser334 attenuates its tumor-suppressive activity in nude mice transplanted with human MDA-MB-231 breast cancer cells. Same numbers (5 × 105) of stably transfected MDA-MB-231 cells were implanted into the mammary fat pads of female nude mice. (A). Tumor onset day was recorded as the first day of the palpable tumor formation. (B). Tumor growth was monitored weekly by measuring the tumor sizes and calculated as tumor volume as described in Methods. (C). At eight weeks after implantation, mice were sacrificed for tissue collection. Representative tumor development images were shown in left panel and the wet weights of tumors measured and recorded in the right panel. (D). Representative microscopic images of PCNA-stained tumor sections from the four groups of mice. Positive-stained cells were manually counted and calculated as percentage against total cells. Scale bar = 200 μm. The results are exhibited as the means ± SEM. * P < 0.05 and ** P < 0.01 vs pcDNA groups; # P < 0.05 vs WT groups (unpaired t test, n = 6).
Immunohistochemical staining of proliferating cell nuclear antigen (PCNA) was performed in tumor sections derived from the above mice. The analysis revealed that over 70% and 80% of cells showed positive staining in control and S334D groups, respectively (Figure 8D). On the other hand, there were only ~20 % of PCNA positive cells in wild type LKB1 group and less than 5 % in S334A group. These results demonstrated that phosphorylation occurred at Ser334 essentially blocked the tumor suppressive activity of LKB1 in mice models.
Discussion
Akt kinases are the key regulators of diverse cellular processes, including cell survival and growth, protein synthesis, angiogenesis and glucose metabolism [25,26]. Akt signaling is frequently altered in cancer [27,28]. Hyperactivation of Akt has been documented in a wide range of sporadic human cancers as well as in dominantly inherited cancer syndromes [29-33]. The present study demonstrates that the tumor suppressor LKB1 is a downstream target of Akt. The suppressive effects of LKB1 on cell proliferation and tumorigenesis are abolished by Akt-mediated phosphorylation at Ser334.
LKB1 is generally considered to be constitutively active [9]. Nuclear-cytoplasmic shuttling of LKB1 represents an important mechanism contributing to its signaling specificity and target accessibility in mammalian cells [16]. For example, activation of AMPK, a key downstream target of LKB1, occurs mainly in the cytoplasm [5]. STRADα enhances LKB1 activity primarily by promoting its translocation from nuclei to cytoplasm [16]. The cytoplasmic portion of LKB1 is responsible for its cell cycle arrest and tumor suppressive activities [14]. Previous studies have demonstrated that phosphorylation of LKB1 at Ser307 and Ser428 may increases its cytoplasmic localization [13,34]. However, the upstream signaling pathways that control the nuclear-cytoplasmic shuttling of LKB1 remain unclear. Evidence in the present study demonstrated that Akt regulates the sub-cellular localization of LKB1 by promoting a phosphorylation-dependent interaction with 14-3-3 proteins. LKB1 mutant S334A, which cannot be phosphorylated by Akt, localizes predominantly to cytoplasm and exhibits much more potent inhibition on human breast cancer cell proliferation and tumorigenesis. By contrast, the LKB1 mutant S334D, which mimics Akt-mediated phosphorylation, is accumulated mainly in the nucleus and displays impaired anti-tumor activity.
The 14-3-3 proteins play important roles in regulating cell fate and tumor development by integrating diversed signaling pathways [23]. They interact with a plethora of intracellular signaling molecules in a serine/theronine phosphorylation-dependent manner [22]. One of the important regulatory functions of 14-3-3 proteins is to control the sub-cellular distributions of their binding partners [35]. For example, 14-3-3 proteins increase the nuclear localization of telomerase and p53 [36,37]. Results of the present study suggest that phosphorylation-dependent interactions with 14-3-3 proteins promote the nuclear sequestration of LKB1 and prevent the association of this kinase with STRADα. Note that LKB1 mutants with defective binding to STRADα also loss their cell-cycle arrest activity [14]. Likewise, the S334D mutant that exhibits augmented interaction with 14-3-3 proteins but impaired association with STRADα is mainly localized within nucleus and unable to elicit the tumor suppressor activity.
The present study uncovers a molecular pathway whereby the sub-cellular localization of LKB1 is regulated by the survival kinase Akt, through both phosphorylation modification and protein-protein interactions (Figure 9). Spatial regulation represents a key mechanism whereby activated Akt exerts its functions to promote cell survival and tumorigenesis [38]. Notably, Akt-induced sub-cellular redistribution of its substrates is often mediated by 14-3-3 proteins [39]. For instance, Akt inactivates FKHRL1 and p27Kip1 by inducing a phosphorylation-dependent association with 14-3-3 proteins [40,41]. Unlike these two examples showing increased cytoplasmic localization, Akt-induced association of LKB1 with 14-3-3 proteins leads to augmented nuclear retention of LKB1, indicating a complex role of Akt in regulating the nuclear-cytoplasmic transport of its downstream targets. In fact, aberrant Akt signaling is an important contributor to the development of LKB1-deficient tumors [25]. On the other hand, ectopic expression of LKB1 negatively regulates the Akt signaling pathway [42]. These findings collectively suggest the existence of a negative feedback regulation between LKB1 and Akt, two kinases with opposite effects on cell survival, proliferation and tumorigenesis.
Figure 9.

Schematic illustration of the mechanisms by which Akt promotes the nuclear accumulation and prevents cytosolic translocation of LKB1. Left panel: Activated Akt phosphorylates LKB1 at Ser334 and induces phosphorylation-dependent association with 14-3-3 proteins, which block STRADα binding and lead to the nuclear retention of LKB1. Right panel: When Akt is inactive, LKB1 binds to STRADα, which promotes the cytoplasmic translocation of LKB1 and enhances the tumor suppressor activity of this kinase.
Acknowledgements
This work is supported by Collaborative Research Fund (HKU4/10C) and General Research Fund (HKU781311M) from the Research Grant Council of Hong Kong; the matching fund for the National Basic Research Program of China (2010CB9455000) and Seeding Fund for Basic Research from the University of Hong Kong.
Supporting Information
References
- 1.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. A serine/ threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 2.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 3.Williams T, Brenman JE. LKB1 and AMPK in cell polarity and division. Trends Cell Biol. 2008;18:193–198. doi: 10.1016/j.tcb.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Spicer J, Ashworth A. LKB1 kinase: master and commander of metabolism and polarity. Curr Biol. 2004;14:R383–385. doi: 10.1016/j.cub.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 5.Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev. 2009;89:777–798. doi: 10.1152/physrev.00026.2008. [DOI] [PubMed] [Google Scholar]
- 6.Tiainen M, Ylikorkala A, Makela TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA. 1999;96:9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 8.Alexander A, Walker CL. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett. 2011;585:952–957. doi: 10.1016/j.febslet.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 10.Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1) Hum Mol Genet. 2002;11:1497–1504. doi: 10.1093/hmg/11.13.1497. [DOI] [PubMed] [Google Scholar]
- 12.Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, Alessi DR. Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz-Jeghers cancer syndrome. Biochem J. 2002;362:481–490. doi: 10.1042/0264-6021:3620481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–962. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–3072. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boudeau J, Scott JW, Resta N, Deak M, Kieloch A, Komander D, Hardie DG, Prescott AR, van Aalten DM, Alessi DR. Analysis of the LKB1-STRAD-MO25 complex. J Cell Sci. 2004;117:6365–6375. doi: 10.1242/jcs.01571. [DOI] [PubMed] [Google Scholar]
- 16.Dorfman J, Macara IG. STRADalpha regulates LKB1 localization by blocking access to importin-alpha, and by association with Crm1 and exportin-7. Mol Biol Cell. 2008;19:1614–1626. doi: 10.1091/mbc.E07-05-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boudeau J, Kieloch A, Alessi DR, Stella A, Guanti G, Resta N. Functional analysis of LKB1/STK11 mutants and two aberrant isoforms found in Peutz-Jeghers Syndrome patients. Hum Mutat. 2003;21:172. doi: 10.1002/humu.9112. [DOI] [PubMed] [Google Scholar]
- 18.Lam JB, Chow KH, Xu A, Lam KS, Liu J, Wong NS, Moon RT, Shepherd PR, Cooper GJ, Wang Y. Adiponectin haploinsufficiency promotes mammary tumor development in MMTV-PyVT mice by modulation of phosphatase and tensin homolog activities. PLoS One. 2009;4:e4968. doi: 10.1371/journal.pone.0004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Law IK, Liu L, Xu A, Lam KS, Vanhoutte PM, Che CM, Leung PT, Wang Y. Identification and characterization of proteins interacting with SIRT1 and SIRT3: implications in the anti-aging and metabolic effects of sirtuins. Proteomics. 2009;9:2444–2456. doi: 10.1002/pmic.200800738. [DOI] [PubMed] [Google Scholar]
- 20.Zu Y, Liu L, Lee MY, Xu C, Liang Y, Man RY, Vanhoutte PM, Wang Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res. 2010;106:1384–1393. doi: 10.1161/CIRCRESAHA.109.215483. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Lam JB, Lam KS, Liu J, Lam MC, Hoo RL, Wu D, Cooper GJ, Xu A. Adiponectin modulates the glycogen synthase kinase-3beta/beta-catenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res. 2006;66:11462–11470. doi: 10.1158/0008-5472.CAN-06-1969. [DOI] [PubMed] [Google Scholar]
- 22.Hermeking H. The 14-3-3 cancer connection. Nat Rev Cancer. 2003;3:931–943. doi: 10.1038/nrc1230. [DOI] [PubMed] [Google Scholar]
- 23.Morrison DK. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009;19:16–23. doi: 10.1016/j.tcb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang B, Yang H, Liu YC, Jelinek T, Zhang L, Ruoslahti E, Fu H. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry. 1999;38:12499–12504. doi: 10.1021/bi991353h. [DOI] [PubMed] [Google Scholar]
- 25.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 26.Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 27.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 28.Hsieh AC, Truitt ML, Ruggero D. Oncogenic AKTivation of translation as a therapeutic target. Br J Cancer. 2011;105:329–336. doi: 10.1038/bjc.2011.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han S, Polizzano C, Nielsen GP, Hornicek FJ, Rosenberg AE, Ramesh V. Aberrant hyperactivation of akt and Mammalian target of rapamycin complex 1 signaling in sporadic chordomas. Clin Cancer Res. 2009;15:1940–1946. doi: 10.1158/1078-0432.CCR-08-2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, Woodman SE, Calderone TC, Ju Z, Lazar AJ, Prieto VG, Aldape K, Mills GB, Gershenwald JE. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clin Cancer Res. 2009;15:7538–7546. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 32.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/ threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA. 1992;89:9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bedogni B, Welford SM, Cassarino DS, Nickoloff BJ, Giaccia AJ, Powell MB. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell. 2005;8:443–454. doi: 10.1016/j.ccr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Xie Z, Dong Y, Zhang J, Scholz R, Neumann D, Zou MH. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol Cell Biol. 2009;29:3582–3596. doi: 10.1128/MCB.01417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol. 2002;156:817–828. doi: 10.1083/jcb.200112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seimiya H, Sawada H, Muramatsu Y, Shimizu M, Ohko K, Yamane K, Tsuruo T. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 2000;19:2652–2661. doi: 10.1093/emboj/19.11.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol. 2003;23:7096–7107. doi: 10.1128/MCB.23.20.7096-7107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang R, Brattain MG. AKT can be activated in the nucleus. Cell Signal. 2006;18:1722–1731. doi: 10.1016/j.cellsig.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 39.Liu MY, Cai S, Espejo A, Bedford MT, Walker CL. 14-3-3 interacts with the tumor suppressor tuberin at Akt phosphorylation site(s) Cancer Res. 2002;62:6475–6480. [PubMed] [Google Scholar]
- 40.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 41.Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277:28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- 42.Jimenez AI, Fernandez P, Dominguez O, Dopazo A, Sanchez-Cespedes M. Growth and molecular profile of lung cancer cells expressing ectopic LKB1: down-regulation of the phosphatidylinositol 3'-phosphate kinase/PTEN pathway. Cancer Res. 2003;63:1382–1388. [PubMed] [Google Scholar]