

Abstract
The prevalence of cardiovascular disease (CVD) increases with advancing age. While the long-term exposure to cardiovascular risk factors plays a major role in the etiopathogenesis of CVD, intrinsic cardiac aging enhances the susceptibility to developing heart pathologies in late life. The progressive decline of cardiomyocyte mitochondrial function is considered to be a major mechanism underlying heart senescence. Damaged mitochondria not only produce less ATP, but also generate increased amounts of reactive oxygen species (ROS) and display a greater propensity to trigger apoptosis. Given the post-mitotic nature of cardiomyocytes, the efficient removal of dysfunctional mitochondria is critical for the maintenance of cell homeostasis, as damaged organelles cannot be diluted by cell proliferation. The only known mechanism whereby mitochondria are turned over is through macroautophagy (MA). The efficiency of this process declines with advancing age which may play a critical role in heart senescence as well as in age-related CVD. This review illustrates the putative mechanisms whereby alterations in the autophagic removal of damaged mitochondria intervene in the process of cardiac aging as well as in the pathogenesis of specific heart diseases especially prevalent in late life (e.g., left ventricular hypertrophy, ischemic heart disease, heart failure, and diabetic cardiomyopathy). Interventions proposed to counter cardiac aging through improvements in MA (e.g., calorie restriction and calorie restriction mimetics) are also presented.
Keywords: heart senescence, mitophagy, oxidative stress, resveratrol, calorie restriction
1. Introduction
The prevalence of cardiovascular disease (CVD) increases dramatically with advancing age. Over 80% of cases of coronary artery disease (CAD) and more than 75% of those of congestive heart failure (CHF) are observed in geriatric patients.1 The incidence of CVD, including CAD, CHF and stroke, increases from 4–10/1,000 person-years in adults aged 45–54 years to 65–75/1,000 person-years in those aged 85+ years.1 CVD is a major cause of chronic disability in the elderly.2 Notably, in older persons, subclinical CVD is associated with a decline in physical and cognitive function equivalent to over 5 years of aging.3 The disproportionate prevalence of CVD at advanced age is largely attributable to the long-term exposure to cardiovascular risk factors such as hypertension, dyslipidemia, diabetes mellitus, physical inactivity, etc. In addition, intrinsic cardiac aging, defined as the development of structural and functional alterations during aging, may render the heart more vulnerable to various stressors, ultimately favoring the development of CVD.4
One major challenge in the investigation of cardiac senescence is to discern the effects of age per se from those produced by conditions such as hypertension, body composition changes or diabetes mellitus, which are highly prevalent in late life. However, age-associated alterations in cardiac structure/function also develop in experimental animals, such as the mouse, which are typically exempt from hypertension, diabetes and atherosclerosis.5 In addition, longitudinal studies in human cohorts with very low cardiovascular risk profile (e.g., the Baltimore Longitudinal Study on Aging) have shown that advanced age is associated with abnormalities in cardiac performance and structure, such as a decline in early diastolic left ventricular filling and increases in wall thickness, respectively.6,7 These observations indicate that the heart undergoes anatomical and functional changes over the course of aging, whose interaction with CVD-specific mechanisms may eventually result in an excess risk for CVD in late life. Furthermore, the aging heart is characterized by an impaired responsiveness to stress as well as by a reduced efficiency of endogenous protective mechanisms (e.g., ischemic preconditioning and postconditioning), resulting in increased vulnerability to injury.8
Although the intimate mechanisms involved in cardiac senescence are not fully understood, the progressive accrual of macromolecular oxidative damage over the lifetime is invoked as a major factor.9 Reactive oxygen species (ROS) are constantly generated within cells by several enzymatic reactions, including those catalyzed by cyclooxygenases, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and xanthine oxidase. However, the bulk of ROS production occurs as a byproduct of mitochondrial oxidative phosphorylation (OXPHOS). Experimental evidence indicates that mitochondrial function decreases over the course of aging, resulting in increased ROS generation, enhanced free radical-inflicted damage and further mitochondrial decay.10 In this scenario, the removal of dysfunctional mitochondria through autophagy is crucial for the maintenance of cell viability.11 The efficiency of this process declines with advancing age which may be critically involved in heart senescence as well as in age-related CVD.12,13
In the next sections, mechanisms linking mitochondrial dysfunction and abnormal ROS production to defective mitochondrial autophagy in cardiac senescence will be reviewed. Subsequently, the involvement of mitochondrial dysfunction and altered autophagy in heart diseases common in advanced age (i.e., left ventricular hypertrophy, ischemic heart disease, heart failure, diabetic cardiomyopathy) will be discussed. Finally, interventions aimed at delaying cardiac aging by targeting autophagy will be presented.
2. Mechanisms and consequences of cardiac mitochondrial dysfunction in advanced age
2.1. The dual nature of mitochondria-generated oxidants
Mitochondria are essential for cardiomyocyte function and viability. Indeed, the myocardium is a highly energy demanding tissue, with mitochondria supplying over 90% of ATP. Free radicals are constantly generated during mitochondrial respiration. In physiologic conditions, 0.2–2% of oxygen is converted into superoxide anion (O2˙−) mainly at complex I and III of the electron transport chain (ETC).14 To counteract the burden of ROS production, the mitochondrion is equipped with a multileveled defense network comprising detoxifying enzymes and non-enzymatic antioxidants.15 Within the mitochondrial matrix, manganese-containing superoxide dismutase (MnSOD, SOD2) converts O2˙− into hydrogen peroxide (H2O2), which is further detoxified into O2 and H2O by glutathione peroxidase (Gpx-I) and peroxiredoxine (Prx-III). Alternatively, O2˙− can be released in the intermembrane space where it is converted to H2O2 by copper-zinc-containing SOD (CuZnSOD, SOD1). In addition, O2˙− leaked in the intermembrane space can be scavenged by cytochrome c.16
Once merely considered unwanted byproducts of OXPHOS, mitochondria-derived oxidants are now viewed as essential signaling molecules necessary for the induction of endogenous defense mechanisms that culminate in increased stress resistance (mitochondrial hormesis or mitohormesis).17 In contrast, excessive ROS generation and/or defective oxidant scavenging have been implicated in the aging process as well as in the pathogenesis of several chronic degenerative diseases, including CVD.18 The mechanisms responsible for abnormal mitochondrial oxidant generation during aging and the impact of oxidative stress on heart physiology are presented in the next subsections.
2.2. Mechanisms of mitochondrial free radical generation in the aging heart
Damaged cardiac mitochondria can release up to 10-fold more H2O2 than intact organelles.19 Furthermore, in the presence of non-protein-bound redox cycling metals (e.g., iron and copper), H2O2 can be converted into the highly reactive hydroxyl radical (•OH), through Fenton's and Haber-Weiss' reactions. In such circumstances, the mitochondrion is exposed to a high burden of oxidative stress, resulting in the primary damage to its own constituents. It is worth mentioning that the mitochondrial iron content increases with aging in rodent post-mitotic tissues, including the myocardium, which may exacerbate the extent of oxidative damage in late life.20
The mitochondrial DNA (mtDNA) is especially prone to oxidative damage due to its proximity to the ETC, the lack of protective histones and a less efficient repair system compared with nuclear DNA (nDNA).21 As a result, the level of oxidatively-modified bases in mtDNA is several-fold higher than that in nDNA.21 Moreover, due to the compactness of the mitochondrial genome (i.e., lack of introns), each mutation is likely to affect gene integrity and, hence, protein function.22,23 It follows that mtDNA mutations can lead to the synthesis of defective ETC components, resulting in impairment of OXPHOS, decreased ATP production and further ROS generation.22 The vicious cycle originating from ROS-inflicted mtDNA damage represents the main tenet of the mitochondrial free radical theory of aging and is believed to play a central role in the aging process as well as in the pathogenesis of age-associated degenerative diseases, including CVD.22
Sahin and colleagues24 have recently provided experimental evidence linking the mitochondrial free radical and the telomere shortening theories of aging. These authors used mice with impaired telomere maintenance caused by the targeted deletion of telomerase reverse transcriptase (Tert−/−). Tert−/− rodents develop severe telomere dysfunction when backcrossed for four or more generations (G4) and display pathologies not only in highly proliferative tissues, but also in post-mitotic organs, such as the heart. Specifically, G4 Tert−/− mice develop dilated cardiomyopathy during aging, with left ventricular wall thinning and reduced contractile performance. Mitochondria isolated from the heart and liver of G4 Tert−/− mice exhibit reduced mtDNA content, decline in complex I and IV activity, impaired respiration and increased ROS generation. These abnormalities are linked to p53-mediated repression of peroxisome proliferator-activated receptor-γ coactivator-1α and 1β (PGC-1α and PGC-1β) and their downstream targets nuclear respiratory factor-1 (NRF-1) and mitochondrial transcription factor A (TFAM). Hence, the age-related telomerase dysfunction might represent a primary instigator of mitochondrial decay, which in turn would lead to decreased bioenergetic efficiency and increased ROS production through sustained p53 activation and further repression of PGC signaling.25
2.3. Consequences of abnormal mitochondrial free radical generation on heart physiology: evidence from rodent models
Elevated levels of oxidative damage to mitochondrial proteins, lipids and nucleic acids have been detected in the myocardium of old rodents.26–29 The frequency of mtDNA point mutations and deletions is ~3-fold higher in the aged mouse heart compared with young adult controls.5 Similarly, the frequency of the common 4977 bp deletion of mtDNA increases during aging in the human heart, and is 5 to 15-fold higher in persons over 40 years of age than in younger individuals.30,31 However, the proof of principle that the accumulation of mtDNA damage and subsequent mitochondrial dysfunction may be causative to mammalian aging has been provided by the characterization of mice expressing a proofreading-deficient mtDNA polymerase γ (PolG).32,33 These mutants accumulate a high load of mtDNA mutations and deletions, and are characterized by the early appearance of many aging-like phenotypes, including cardiac enlargement. Heart mitochondria of PolG mice exhibit abnormal ETC with depressed activity of complex I and IV, reduced ATP production, and accumulation of enlarged, irregularly shaped mitochondria.32 Furthermore, levels of protein carbonyls are increased in cardiac mitochondria from mtDNA mutator mice compared with wild-type rodents.34 PolG mice die prematurely with dilated cardiomyopathy. Severe cardiomyopathy has also been observed in mice expressing a heart-specific proofreading-deficient mtDNA polymerase.35 Remarkably, the PolG heart phenotype, the cardiac mtDNA mutation load and the extent of mitochondrial protein oxidation are partially rescued by the overexpression of catalase targeted to the mitochondrial matrix (mCAT).34
Further experimental support to the involvement of the mitochondrial vicious cycle in mammalian aging and heart senescence has been provided by the observation that mCAT overexpression extends mean and maximum lifespan and delays the development of cardiac pathology in mice.36,37 The extent of mitochondrial oxidative damage, including mtDNA deletions, and the rate of H2O2 generation, are significantly attenuated in the heart of old mCAT mice compared with age-matched wild-type littermates.36,37
Collectively, studies in mouse models have made a strong argument in favor of the mitochondrial vicious cycle as a contributing factor to cardiac senescence. However, definite evidence of the involvement of mitochondrial decay in the aging process requires the reciprocal experiment, that is the generation of experimental rodents genetically engineered to suffer a reduced rate of mtDNA mutations during aging. If these animals lived longer and maintained a youthful heart performance in late life, the contribution of mitochondrial damage to heart senescence would be conclusively established.
3. The autophagic machinery and the relevance of mitochondrial quality control to cardiomyocyte homeostasis
Regardless of the mechanism(s) primarily responsible for mitochondrial decay during aging, mitochondrial quality control is essential for the preservation of cardiomyocyte homeostasis. This task is accomplished through the complex coordination of several processes (reviewed in38). An intramitochondrial proteolytic system selectively removes damaged proteins. A second line of defense is provided by the dynamic nature of the mitochondrial population. For instance, the functionality of damaged mitochondria can be restored by their fusion with neighboring, intact organelles. Finally, severely damaged mitochondria are eliminated through autophagy.
3.1. Types of autophagy
Autophagy is a self-eating process through which cells degrade their own components, recycling amino acids and other building blocks that can be eventually reutilized.39 Such degradation is performed by lysosomal acidic hydrolases. Depending on the pathway cellular components are delivered to lysosomes, three types of autophagy can be distinguished: macroautophagy (MA), microautophagy and chaperone-mediated autophagy (CMA). MA involves the degradation of long-lived proteins and whole cellular organelles through a multistep process (Figure 1).39 MA begins with the formation of a double-layered isolation membrane (phagophore) around the molecules and/or organelles to be degraded. The phagophore engulfs cytosolic components and seals around the content, forming an autophagosome. Eventually, the autophagosome fuses with a lysosome, evolving into an autophagolysosome (or autolysosome), wherein lysosomal hydrolases digest the cargo.39 Microautophagy involves the direct sequestration of cytosolic components through invaginations or arm-like projections of the lysosomal membrane.40 Microautophagy may serve for the turnover of long-lived proteins; however, the significance and regulation of this type of autophagy remain poorly understood.40 Finally, CMA is a highly selective process devoted to the degradation of soluble cytosolic proteins.41
Figure 1. Schematic representation of the macroautophagy machinery.

Macroautophagy begins with the formation of a double-layered isolation membrane (phagophore) around the molecules and/or organelles to be degraded. The phagophore grows in size and completely engulfs the cargo, forming an autophagosome. The autophagosome subsequently fuses with a lysosome evolving into an autolysosome wherein the cargo is digested.
Among the three types of autophagy described, MA is the best characterized in mammalian cells. Starvation is the strongest stimulus of MA.39,42,43 During nutrient deprivation, MA breaks down cellular components generating amino acids, fatty acids and carbohydrates, which can be harnessed for energy production as well as for the synthesis of essential cellular molecules. MA is also involved in specific cytosolic rearrangements during embryogenesis and postnatal development.44 Furthermore, MA is induced during viral or bacterial infections,45 hypoxia,46 and under various stress conditions, including radiation exposure and increased ROS generation.47,48 In these circumstances, MA is essential for the maintenance of cell homeostasis by promoting the removal of damaged components.11 Indeed, impairments in MA induce premature aging and shorten the lifespan in several organisms.49–51 Conversely, up-regulation of MA is proposed to be a major mechanism underlying the lifespan-extending properties of calorie restriction (CR).49,52,53
The execution of MA involves the coordination of a complex molecular machinery which is briefly described in the next subsection.
3.2. The autophagic machinery and the molecular regulation of macroautophagy
To date, over 35 AuTophaGy-related (Atg) proteins have been identified in yeasts and mammals; however, the precise role each Atg protein plays during autophagy is not fully established.54 As illustrated in Figure 1, the process of MA can be divided into discrete steps, namely, induction and nucleation, expansion, fusion, and degradation. The induction phase is mediated by the ULK1-Atg13-FIP200 kinase complex.55 The regulation of the nucleation stage, which consists in the recruitment of Atg proteins to the phagophore assembly site, is not yet completely understood. However, the vacuolar protein sorting-34 (Vps34), a class III phosphatidylinositol-3-kinase (PI3K), is required for this step.56 Vps34 associates with Beclin1, the mammalian homologue of yeast Atg6, and subsequently recruits Atg14 and Vps15 (p150) to the pre-autophagosomal structure.56 The elongation and expansion of the phagophore membrane require two ubiquitin-like conjugation systems involving Atg12 (conjugated to Atg5) and Atg8/light chain-3 (LC3, conjugated to phosphatidyl ethanolamine), along with other Atg proteins such as Atg9 and Atg16.55 The fusion of the autophagosome with a lysosome relies on the canonical cellular fusion machinery consisting of the Rab-SNARE (Soluble Nethylmaleimide-sensitive factor Attachment protein REceptor) system and requires the presence of lysosomal membrane-associated protein-2 (LAMP-2) and the UV radiation resistance-associated gene (UVRAG).57,58 Finally, the digestion of the cargo is carried out by lysosomal hydrolases, followed by the transportation of degraded components into the cytoplasm by lysosomal efflux transporters such as Atg22.58
With regard to the regulation of MA, the mammalian target of rapamycin (mTOR) is considered to be a major check-point, linking the cellular nutritional state with the level of ongoing autophagy (Figure 2).59 Under nutrient rich conditions, mTOR is active and inhibits the ULK1-Atg13-FIP200 complex required for the induction of MA.60 Energy deprivation leads to mTOR inactivation and stimulation of AMP-activated protein kinase (AMPK), which both induce MA.59,61 AMPK functions as an energy-sensing kinase and is activated by increases in the cellular AMP:ATP ratio. Under such circumstances, AMPK promotes autophagy by directly activating ULK1 as well as by relieving the mTOR-mediated inhibition of MA.59
Figure 2. Schematic overview of the molecular regulation of macroautophagy in the context of mitochondrial homeostasis.
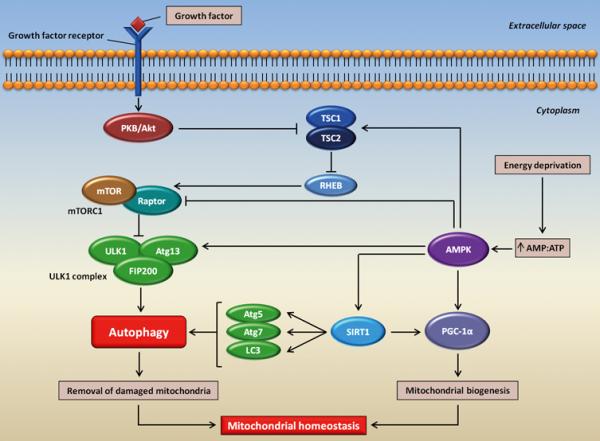
In the presence of nutrients and growth factors, the PKB/Akt (protein kinase B) pathway is activated, which blocks the TSC1/TSC2 (tuberous sclerosis complex 1/2), thus relieving its inhibitory effect on RHEB (Ras homolog enriched in brain). The latter, in turn, activates mTORC1 [mammalian target of rapamycin (mTOR) complex 1]. mTORC1 inhibits macroautophagy by blocking the ULK1-Atg13-FIP100 complex through Atg13 hyperphosphorylation. In cases of energy depletion, the cellular AMP:ATP ratio increases and AMPK (AMP-activated protein kinase) becomes activated, which in turn stimulates macroautophagy through multiple mechanisms. For instance, AMPK can phosphorylate and activate TSC1/TSC2, thereby relieving the mTORC1-mediated inhibition of macroautophagy. In addition, AMPK can phosphorylate Ulk1 at specific serine residues leading to the initiation of macroautophagy. AMPK can also inhibit the mTORC1 complex by phosphorylating Raptor (mTORC1 containing rapamycin-associated TOR protein), the binding partner required for mTORC1 activity. Once activated, macroautophagy provides to several tasks, including the clearance of damaged mitochondria. In addition AMPK induces mitochondrial biogenesis by activating PGC-1α (peroxisome proliferator-activated receptor-γ coactivator-1α) either directly or through SIRT1(sirtuin-1). SIRT1 can also deacetylate and activate several autophagy-related proteins, such as Atg5, Atg7 and LC3. The autophagic removal of damaged mitochondria coupled with mitochondrial biogenesis is essential for the maintenance of mitochondrial homeostasis.
Although MA might seem to be a random, bulk digestion process, evidence is accumulating that intracellular components can be selectively targeted for degradation.62 For instance, MA can be specifically directed towards the removal of peroxisomes (pexophagy), endoplasmic reticulum (reticulophagy), and ribosomes (ribophagy).62 Likewise, mitochondria can be selectively targeted for degradation via mitophagy.63 The molecular machinery and the regulation of this cellular pathway are outlined in the next subsection.
3.4. Mitophagy: a specialized form of macroautophagy
Mitophagy is a highly selective process that can promote the elimination of dysfunctional or unnecessary mitochondria.63 The loss of mitochondrial membrane potential (Δψm) represents a major trigger of mitophagy.63 Indeed, laser-induced photo-damage of selected mitochondria inside living hepatocytes results in the rapid dissipation of Δψm, followed by the quick removal of depolarized mitochondria through mitophagy.64 In addition, oxidative damage can lead to the formation of asymmetric daughter mitochondria characterized by different Δψm, with autophagy specifically targeting mitochondria with lower Δψm.65 Apart from the degradation of damaged mitochondria under stress conditions, mitophagy is essential for the mitochondrial turnover in the basal state as well as during cell differentiation, such as the maturation of reticulocytes into mature red blood cells.66 The occurrence of selective mitophagy in cardiomyocytes has not yet been conclusively demonstrated.
Investigations into the molecular regulation of mitophagy have unveiled several mitophagy-specific proteins.67 Parkin and Pink1 are believed to play important roles in the selective degradation of damaged mitochondria, at least under certain circumstances.68 Parkin is a cytosolic E3-ubiquitin ligase which is selectively recruited to dysfunctional mitochondria, and assists in their removal by mitophagy.68 Pink1 is imported into healthy mitochondria through a Δψm-dependent process and is degraded by the presenilins-associated rhomboid-like (PARL) protease.69 The dissipation of Δψm results in the accumulation of Pink1 on the mitochondrial surface, leading to the recruitment of Parkin, which ubiquitinates outer membrane proteins including the voltage-dependent anion channel (VDAC).70 It is proposed that ubiquitin-tagged mitochondria are directly targeted to autophagic vacuoles through the interaction of ubiquitinated proteins with the autophagosomal marker LC3.70 In addition, Parkin can ubiquitinate B cell leukemia-2 (Bcl-2), therefore derepressing Beclin1.71
Recent evidence also suggests that the opening of the mitochondrial permeability transition pore (mPTP) may be required for the selective removal of damaged mitochondria.72,73 Opening of the mPTP causes a sudden increase of the inner membrane permeability to solutes with molecular weight up to 1,500 Da.74 This results in mitochondrial depolarization, activation of the mitochondrial ATPase (i.e., ATP synthase operating in reverse), and swelling and rupture of the outer membrane.74 The loss of Δψm subsequent to permeability transition targets individual mitochondria for degradation.72 Notably, in cultured cardiomyocytes, starvation-induced MA is preceded by mitochondrial depolarization.72 The loss of Δψm and the activation of MA are prevented by cyclosporin A, an inhibitor of the mPTP component cyclophilin D (CypD).72 Furthermore, starvation fails to induce MA in CypD-deficient murine cardiomyocytes, whereas in cardiac cells from mice overexpressing CypD autophagy is enhanced even under fed conditions.72 The NAD-dependent deacetylase sirtuin-3 (SIRT3) appears to be critically involved in the control of mPTP by modulating CypD.75 Indeed, in transgenic mice, the loss of SIRT3 activity leads to increased activation of the mPTP in cardiac mitochondria in response to Ca2+ increases, hemodynamic stress and aging.75
Similar to the mPTP, the apoptotic proteins Bnip3 (Bcl-2 and adenovirus E1B 19 kDa-interacting protein-3) and Nix (Nip3-like protein X) are thought to trigger selective mitophagy through mitochondrial depolarization.76 Moreover, Bnip3 may induce mitophagy by competitively disrupting the inhibitory interaction between Bcl-2 and Beclin1.76 Finally, Nix associates with mitochondrial membranes and directly interacts with LC3.77
Although the molecular regulation of mitophagy has not yet been completely elucidated, the mTOR/AMPK pathway is proposed to be a major check-point.78 AMPK, in addition to stimulating the mitochondrial removal through autophagy, enhances the activity of sirtuin-1 (SIRT1) and its downstream target PGC-1α, resulting in the stimulation of mitochondrial biogenesis (Figure 2).79Hence, through the activity of AMPK, mitophagy and mitochondrial biogenesis are coordinately regulated, maintaining a healthy and functional pool of mitochondria in the cell.
4. Impaired macroautophagy: why and how dysfunctional mitochondria accumulate within old cardiomyocytes
Metabolically, the heart is highly active and its mitochondria are challenged with a substantial burden of oxidative stress. Moreover, cardiomyocytes are terminally differentiated, post-mitotic cells with a lifespan of several decades. Hence, the maintenance of a healthy pool of mitochondria and the efficient removal of damaged and potentially harmful organelles are vital for the preservation of cardiomyocyte homeostasis.
Recent evidence indicates that cardiomyocyte MA becomes impaired during aging.12,13 This may result in the accumulation within cardiomyocytes of dysfunctional mitochondria that are bioenergetically inefficient and prone to ROS leakage.80 Indeed, the ultrastructural analysis of myocardium from aged rodents has revealed the presence of enlarged mitochondria, characterized by swelling, loss of cristae and matrix derangement.81 Biochemically, these senescent mitochondria exhibit reduced ATP production and increased ROS generation.82 It is hypothesized that giant mitochondria may progressively displace functional organelles over the course of aging.83 This phenomenon is attributed to a replicative advantage of damaged mitochondria secondary to their partially deleted genome.84 Alternatively, giant mitochondria may benefit from a survival advantage, being less likely to be autophagocytosed by virtue of their dimensions. Along these lines, the so-called survival of the slowest (SOS) theory, postulates that damaged mitochondria would suffer from less ROS damage on their own membranes due to a reduced respiratory function, and would consequently be less targeted for autophagy compared with intact mitochondria.85 Thus, the SOS hypothesis is in apparent contradiction with the widely-accepted mitochondrial free radical theory of aging. However, it needs to be considered that O2˙−, which constitutes the main free radical directly generated by the ETC, is not reactive enough to cause significant membrane lipid damage, unless it becomes protonated into the more reactive perhydroxyl radical (HO2˙).86 This conversion may occur at a slower rate in mitochondria with defective ETC, because of the reduced proton gradient, which might translate into a milder mitochondrial membrane oxidative damage.
It is worth noting that both the mitochondrial free radical and the SOS theory of aging present a major pitfall: within cells, mitochondria frequently fuse with one another forming large syncytia. As a result, a constant mixing of the total cellular mtDNA pool occurs, disrupting the link between genotype (damaged mtDNA) and phenotype (defective OXPHOS), which is the actual basis of the two theories. Nevertheless, the hypothesis linking mtDNA damage, ETC dysfunction and abnormal ROS generation may still hold true inasmuch as damaged mitochondria are not efficiently broken down because of defective MA in aged cardiomyocytes. Indeed, advanced age is associated with the accumulation within post-mitotic cells of a non-degradable, polymeric, toxic, yellow-brown pigment, called lipofuscin or age pigment.87 Lipofuscigenesis results from peroxide-induced Fenton reactions elicited by intralysosomal materials producing highly reactive hydroxyl radicals (Figure 3). ROS-derived modifications to proteins and lipids cause crosslinking inside lysosomes/autolysosomes, generating lipofuscin.87 Peroxides involved in these Fenton reactions can diffuse into lysosomes from cytosolic damaged mitochondria or may originate from autophagocytosed, yet undegraded mitochondria.88 The accumulation of such intracellular garbage eventually overburdens the autophagosomal-lysosomal degradative capacity, by acting as a sink for lysosomal hydrolases.89 It follows that attempts to digest lipofuscin, accumulated within a growing number of lysosomes, eventually result in the incapacity of MA to keep up with the cell's needs.89 This series of events is thought to trigger a vicious cycle in which the autophagic failure and the accumulation of damaged mitochondria perpetuate each other, resulting in further oxidative stress and enhanced lipofuscinogenesis.89 The collapse of the catabolic machinery will eventually become incompatible with the maintenance of cell homeostasis and survival. This assumption represents the basis of the garbage catastrophe theory of aging, also known as the mitochondrial-lysosomal axis theory of aging.80,89
Figure 3. Lipofuscinogenesis inside a lysosome.
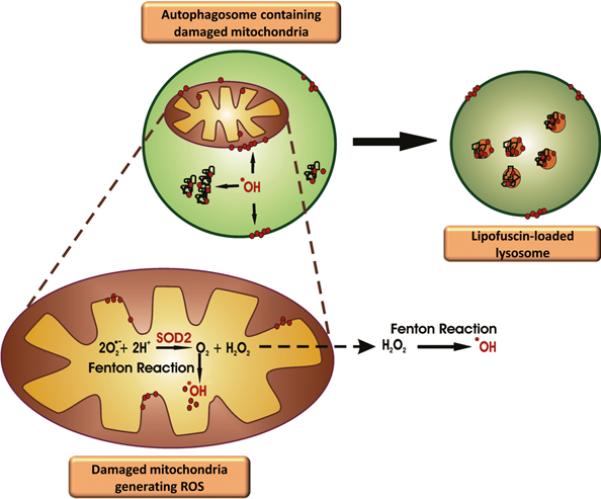
Damaged mitochondria engulfed within an autolysosome generate hydroxyl radicals (˙OH) via Fenton reactions. Hydrogen peroxide (H2O2) participating in these reactions is generated by the action of SOD2 on superoxide anion (O2˙−) produced by the electron transport chain. ˙OH causes crosslinking between intralysosomal proteins, lipids and/or proteins, forming an indigestible polymer, called lipofuscin.
In addition to removing cellular waste, MA also provides to the elimination of dysfunctional mitochondria whose persistence could lead to the induction of apoptosis.90,91 Indeed, mitochondria are a major check-point for the integration of apoptotic stimuli. Notably, cardiomyocyte removal through apoptosis increases with advancing age, which, combined with insufficient replenishment by cardiac stem cells, may contribute to the age-related heart remodeling.9 However, whether the increased severity of apoptosis suffered by the aged myocardium is directly attributable to autophagic failure is yet to be established.
Although recent evidence supports the involvement of impaired MA in the accumulation of abnormal mitochondria within old cardiomyocytes,12,13 several research questions remains to be addressed. First of all, the impact of dysfunctional mitochondrial-lysosomal axis on cardiac aging needs to be clearly established. For instance, whether the manipulation of MA rescues the premature heart senescence phenotype in animal models characterized by high loads mtDNA mutation and severe mitochondrial dysfunction is yet to be determined. However, the inhibition of MA via cardiac-specific Atg5 deficiency has recently been shown to induce the early appearance of anatomical and functional features of heart senescence.13 This was accompanied by biochemical and morphological abnormalities, including decreased mitochondrial respiratory function and accumulation of collapsed mitochondria, respectively.13 Finally, whether the optimization of MA preserves cardiomyocyte mitochondrial function and delays cardiac aging in humans is currently unknown.
5. Mitochondrial dysfunction and impaired regulation of autophagy increase the vulnerability to injury of the aged heart
As previously discussed, evidence from animal models suggests that mitochondrial dysfunction plays a pivotal role in cardiac aging (reviewed in18). According to the mitochondrial-lysosomal theory of aging, the progressive impairment of MA over the course of aging may represent a primary mechanism responsible for the accumulation of damaged mitochondria within aged cardiomyocytes, resulting in decreased ATP availability, enhanced oxidative stress, and eventually cell dismissal.80 Hence, the efficient removal of defective mitochondria through MA is essential for maintaining cardiomyocyte homeostasis and viability in the basal state.92 Mitochondrial dysfunction is also implicated in the pathogenesis of a host of heart diseases highly prevalent in old age, including CAD, heart failure (HF), left ventricular hypertrophy (LVH) and diabetic cardiomyopathy (DC).93 It is worth noting that an altered regulation of MA has been shown to contribute to the pathogenesis of these conditions, further supporting the relevance of the mitochondrial-lysosomal axis to cardiac physiology.
In the following sections, the role played by cardiomyocyte mitochondrial dysfunction and abnormal regulation of MA in specific heart diseases especially common in advanced age will be discussed. This overview will highlight the prospect of targeting MA as a novel means for achieving therapeutic gain in age-related CVD.
5.2. Left ventricular hypertrophy
Mitochondrial dysfunction is implicated in the pathogenesis of LVH as well as in the transition from compensated LVH to HF.94 In cultured cardiomyocytes, hypertrophy induced by angiotensin II, endothelin 1, norepinephrine, tumor necrosis factor α (TNF-α) or mechanical stress is associated with increased levels of oxidative stress and ROS-mediated activation of several intracellular signaling pathways, including mitogen activated protein kinases (MAPKs) and nuclear factor κB (NF-κB).95In vivo, the development of LVH in animal models of pressure overload is blunted by the administration of antioxidants.95 Strong support to the involvement of mitochondria-derived ROS in cardiac hypertrophy has been provided by the observation that mCAT mice are resistant to LVH.96 In contrast, mice overexpressing wild-type peroxisomal catalase (pCAT) accumulate high levels of mitochondrial protein carbonyls and mtDNA deletions, while displaying an altered MA regulation.96 These animals are prone to developing LVH in response to angiotenin II-induced mitochondrial dysfunction. Furthermore, mitochondrial misalignment and aggregation are observed in adult mice with heart-specific deficiency of Atg5 in response to pressure overload induced by thoracic transverse aortic constriction. These animals develop LVH, contractile dysfunction and heart dilation.97 Moreover, a reduced autophagic flux with concomitant appearance of clustered mitochondria has been observed in double transgenic rats harboring human renin and angiotensinogen genes.98 These rodents develop angiotensin II-induced cardiac hypertrophy and die prematurely from HF. Four weeks of 40% CR increased the activation of cardiomyocyte MA and mitigated heart macroscopic and ultrastructural remodeling, while reducing mortality.98 These findings suggest that an abnormal regulation of MA may contribute to the development of LVH, possibly through the accumulation of dysfunctional mitochondria and subsequent increased ROS generation.
Interestingly, chronically enhanced MA activity may be involved in the transition from stable LVH to HF.99 Indeed, in Beclin1+/− rodents subjected to aortic banding, the blunted autophagic response is concomitant with reduced left ventricular remodeling.99 Conversely, overexpression of Beclin1 results in severe pathological remodeling.99
Collectively, these findings suggest that MA may be either beneficial or deleterious in the setting of pressure overload, depending on the circumstances under which it is induced, the extent to which it is stimulated and the signaling pathways responsible for MA activation, assuming that both an insufficient and an excessive autophagic response are maladaptive.100 Understanding the optimal level of autophagic activation is mandatory to design treatments harnessing this cellular process to counter the development of LVH and its transition to HF.
5.3. Ischemia/reperfusion
Oxidative stress plays a central role in the pathogenesis of myocardial damage during ischemia/reperfusion (I/R). In this setting, mitochondria contribute to cardiac dysfunction and cardiomyocyte injury via both a loss of metabolic function and an increased generation of oxidants (reviewed in93). Mitochondrial ultrastructural and functional abnormalities in cardiomyocytes develop early during ischemia and progress over its course.93 The extent of mitochondrial damage is commensurate to the duration and severity of the ischemic insult. Following oxygen deprivation, OXPHOS ceases, leading to a decline in ATP availability, which becomes inadequate to meet the energetic demand of contractile elements. The energetic crisis also leads to disruption of the transmembrane ionic homeostasis, resulting in cellular calcium overload eventually leading to mPTP opening. This event can induce cardiomyocyte dropout. Simultaneously, an abnormal ROS production ensues, mainly at complex I and III of the ETC, resulting in extensive oxidative damage, whose severity is proportional to the duration of ischemia.101 Oxidative stress is further exacerbated by the concomitant impairment of endogenous antioxidant defenses.102
MA is activated in response to myocardial ischemia and promotes cardiomyocyte survival, likely via maintaining energy production during acute nutrient deprivation.103 The degradation of proteins and organelles by MA generates amino acids and fatty acids, which can be used to maintain mitochondrial ATP production and promote survival of cardiac cells.104 A further means whereby MA may protect the ischemic myocardium is through the removal of damaged mitochondria. In mouse embryonic fibroblasts, this adaptation is mediated by up-regulation of Bnip3 promoted by the hypoxia-inducible factor-1α (HIF-1α), a transcription factor activated by low oxygen concentrations.46
The recovery of mitochondrial function during the reperfusion phase is largely dependent upon the duration of the ischemic insult. Following prolonged oxygen deprivation, extensive damage to ETC complexes ensues, which, in conjunction with the concomitant disruption of antioxidant defenses, leads to further ROS generation and additional mitochondrial and cardiomyocyte injury during reperfusion.102 The combination of oxidative stress and calcium overload can eventually trigger the opening of the mPTP resulting in cardiomyocyte dismissal.
The involvement of mitochondrial dysfunction in myocardial damage during reperfusion would suggest that the activation of MA might be beneficial. Indeed, MA is up-regulated during reperfusion; however, this adaptation may not necessarily be protective. In a study by Hamacher-Brady et al,90 inhibition of MA with wortmannin, 3-methyladenine, RNAi knockdown of Beclin1 or overexpression of dominant negative Atg5, sensitized cultured cardiomyocytes to apoptosis after simulated I/R. Conversely, enhancement of MA through rapamycin treatment or Beclin1 overexpression reduced the extent of cardiomyocyte apoptosis in this experimental model. In contrast, Matsui et al103 showed that the up-regulation of MA during reperfusion is maladaptive and is accompanied by enhanced cardiomyocyte apoptosis and increased infarct size. These authors demonstrated that AMPK is rapidly inactivated during reperfusion, whilst Beclin1 is up-regulated.103 Both the induction of MA and cardiac injury are significantly attenuated in mice with heterozygous disruption of the Beclin1 gene. Several mechanisms can be invoked to explain the detrimental effects of cardiac MA during reperfusion. For instance, excessive activation of MA could culminate in autophagic cell death. In addition, Beclin1 contains a conserved pro-apoptotic BH3 domain.105 Thus, overexpression of Beclin1 during reperfusion might result in further deterioration of mitochondrial function and excessive elimination of cardiomyocytes via apoptosis. This eventuality is also promoted by calpain-mediated truncation of Atg5, followed by its translocation to mitochondria, where it interacts with B cell leukemia-X long (Bcl-XL) to trigger permeabilization of the outer mitochondrial membrane.106
Further investigations are needed to obtain a clearer understanding of the role of MA in the context of I/R. This knowledge is necessary for the design of novel therapeutic strategies exploiting the homeostatic nature of MA, while avoiding the deleterious consequences of abnormal autophagic activation.
5.4. Heart failure
Various stressors, including humoral factors (e.g., catecholamines and renin-angiotenin-aldosterone), pressure overload and toxins, may be responsible for mitochondrial dysfunction and enhanced ROS generation in the failing heart regardless of the etiology.107 HF mitochondria generate larger amounts of O2˙− in the presence of NADH compared with normal mitochondria.108 This enhanced ROS production has been associated with increased levels of lipid peroxidation to mitochondrial membranes, decreased mtDNA copy number, reduced abundance of mitochondrial RNA transcripts, and impaired OXPHOS capacity.108 In addition, oxidative stress directly impacts cardiomyocyte structure and function by activating signaling pathways involved in myocardial remodeling and failure.108,109 This suggests the existence of a pathogenetic link between enhanced ROS production, mitochondrial dysfunction and the development of HF.
Recent evidence also suggests that an altered regulation of cellular quality control mechanisms, including MA, may contribute to the pathogenesis of HF. For instance, cardiomyocytes isolated from Atg5-deficient mice show an increased sensitivity to β-adrenergic stimulation compared with wild-type cells.97 Indeed, 7-day isoproterenol treatment resulted in left ventricular dilation and cardiac dysfunction in Atg5-deficient mice, but not in wild-type controls.97 This suggests that up-regulation of MA protects cardiomyocytes against the detrimental effects of excessive β-adrenergic stimulation. In contrast, in transgenic mice expressing the human diphtheria toxin receptor in the heart, HF induced by intramuscular injections of diphtheria toxin was accompanied by the appearance of degenerated cardiomyocytes showing morphological features of autophagic cell death.110
Therefore, the question remains as to whether MA is protective or maladaptive in the context of HF. As observed with regard to pressure overload, the level of MA may be critical in determining whether this process will be protective or detrimental. Indeed, the up-regulation of MA might represent an attempt to cope with increased levels of mitochondrial dysfunction and cellular damage, with autophagic cell death resulting from the failure of the autophagy-mediated survival machinery. However, the possibility exists that an excessive up-regulation of MA may be the primary cause of cardiomyocyte dismissal in the failing myocardium, therefore contributing to the deterioration of cardiac performance. Further studies are needed to decipher the role of MA in mediating either cardioprotection or cardiomyocyte death in the failing heart, in order to properly manipulate this cellular pathway in patients affected by HF.
5.5. Diabetic cardiomyopathy
DC is a major cause of HF in diabetic patients and develops independent of underlying CAD.111 DC is characterized by reduced cardiomyocyte contractility, mitochondrial dysfunction and increased levels of apoptosis. Decreased mitochondrial respiration and reduced expression of OXPHOS components have been observed in the heart of obese type 2 diabetic mice.112 These alterations are thought to contribute to cardiac dysfunction by diminishing high-energy phosphate reserves, thereby impairing myocardial contractility. In addition to the reduced OXPHOS capacity, an increased ROS generation has been documented in cardiac mitochondria from diabetic mice.113 Notably, overexpression of ROS-detoxifying systems (metallothionein, catalase and MnSOD) reverses mitochondrial dysfunction and cardiomyopathy induced by diabetes, suggesting a central role for mitochondria-derived oxidants in the pathogenesis of DC.111
The inefficient autophagic removal of damaged mitochondria may promote the accumulation of dysfunctional organelles in the diabetic heart. Indeed, down-regulation of cardiac MA, secondary to reduced AMPK activity, has been documented in diabetic mice.114 Ultrastructurally, hearts from these rodents display several morphological aberrations, including aggregation of chaotically distributed mitochondria.114 Inhibition of AMPK by a cardiac-specific dominant negative AMPK gene further reduced MA, exacerbated ultrastructural aberrations, worsened cardiac dysfunction and increased mortality in diabetic mice.114 In contrast, treatment with metformin significantly enhanced MA and ameliorated cardiomyocyte ultrastructural abnormalities, while preserving cardiac function. Such benefits were not observed in rodents expressing a dominant negative AMPK, indicating that cardioprotection by metformin is accomplished through AMPK-mediated up-regulation of MA.114
Collectively, these findings suggest that depression of MA may promote the accumulation of dysfunctional mitochondria in the diabetic myocardium, thereby contributing to the development of DC. Therefore, interventions that up-regulate MA appear as promising means to prevent and/or treat this relevant complication of diabetes.
6. Macroautophagy as a therapeutic target against cardiac aging
The critical role postulated for mitochondria-driven oxidative damage in cardiac aging and CVD would suggest that the administration of antioxidants might mitigate the burden of cardiomyocyte injury. However, the efficacy of antioxidant supplementation is still a matter of debate. Indeed, most clinical trials failed to show any positive effect of antioxidants on cardiovascular outcomes.115 Chronic administration of β-carotene, vitamin A or vitamin E may even increase cardiovascular mortality.115 Exploiting the ability of cells to repair or replace oxidatively-damaged molecules and organelles represents an appealing alternative against heart senescence and associated pathologies.116 In this context, interventions aimed at improving mitochondrial turnover through the fine-tuning of MA (e.g., CR, resveratrol administration and sirtuin pathway activation) might be especially relevant to delay cardiac aging and manage age-related CVD.67
CR, defined as a reduction in food intake without malnutrition, is a robust anti-aging intervention and the most powerful physiological inducer of MA.117 The modulation of the autophagic response represents a primary mechanism underlying the lifespan-extending properties of CR.49,52,53 Indeed, the inhibition of autophagy prevents the anti-aging effects of CR in lower organisms.118 Whether CR also stimulates mitochondrial biogenesis is controversial.119,120 CR can induce MA through different pathways: the insulin-like growth factor-1 (IGF-1)/insulin signaling pathway,52 the sirtuin pathway,53 the AMPK pathway,121 and the mTOR pathway.59 These pathways are intimately interconnected and all play important roles in mediating different aspects of the response. With regard to cardiac aging, Wohlgemuth et al122 showed that lifelong 40% CR increased the protein expression of Atg7, Atg9 and lipidated LC3 (LC3-II) in the heart of old rats. More recently, Shinmura et al123 demonstrated that a similar dietary regimen enhanced the autophagic flux in the heart of aged rats through mTOR suppression. These adaptations were associated with reduced lipofuscin accumulation in the myocardium, down-regulation of cardiomyocyte apoptosis, decreases in fiber cross-sectional area, and preservation of left ventricular diastolic function.123 Similar echocardiographic findings have been reported in late-middle aged humans on long-term CR.124 However, whether these effects were linked with changes in MA activity was not investigated. Furthermore, it is presently unclear if the cardioprotective effects of CR are primarily mediated by improvements in autophagy.
Despite the host of health benefits brought about by CR, it is likely that most people will not be able to sustain drastic food restrictions for the long term. Furthermore, persons practicing chronic severe CR may experience several adverse events, including undesired changes in physical appearance, loss of strength and stamina, menstrual irregularities, infertility, loss of libido, osteoporosis, cold sensitivity, slower wound healing, and psychological conditions such as food obsession, depression, and irritability.9 Moreover, CR may not be advisable in non-obese older persons, given the fact that low body mass index is associated with increased risk of disability and mortality in advanced age.125 Thus, considerable effort has been directed toward the discovery of drugs that could mimic the effects of CR, without requiring food restriction and its detrimental consequences.126
The first CR-mimetic identified was 2-deoxy-D-glucose (2DG), an analog of glucose, shown to extend both mean and maximum lifespan in Caenorhabditis elegans.127 However, a recent study demonstrated that chronic 2DG administration to rats, although reproducing a CR-like phenotype, caused cardiotoxicity and increased mortality.128 Promising CR-mimetics with autophagy-inducing properties are those that intersect with the critical signaling pathways identified above and include biguanides, such as metformin that targets the AMPK and insulin signaling pathways,129 resveratrol that affects sirtuin activity,53 and rapamycin that interacts with mTOR signaling.130
Resveratrol has been shown to recapitulate the transcriptional profile and some of the physiological changes that develop under CR.131,132 Indeed, both CR and resveratrol supplementation inhibit gene expression profiles associated with cardiac aging in mice.131,132 In addition, resveratrol improved survival and reduced the prevalence of cardiac pathology in mice fed a high-calorie diet.131 Studies in rodents have also shown that resveratrol inhibits cardiomyocyte apoptosis, protects the myocardium against I/R injury, prevents LVH, improves endothelial function, inhibits platelet aggregation, and reduces inflammation (reviewed in133). However, while low doses of resveratrol induce MA, eliciting a preconditioning-like effect, and generate a survival signal in H9c2 heart myoblasts, higher doses of this compound inhibit MA.134 Similar effects were observed in the heart of rats supplemented with resveratrol.134 SIRT1, which is directly or indirectly activated by resveratrol, exerts a similar hormetic action on cardiomyocyte physiology.135 Low (2.5-fold) to moderate (7.5-fold) transgenic overexpression of SIRT1 in the mouse heart attenuates the age-dependent hypertrophy and reduces the severity of apoptosis/fibrosis as well as cardiac dysfunction.135 In contrast, high levels (12.5-fold) of SIRT1 expression increase the extent of cardiomyocyte apoptosis and the degree of hypertrophy, while decreasing cardiac function, thereby inducing the development of cardiomyopathy.135
In conclusion, interventions aimed at modulating MA may represent a novel strategy to prevent cardiac deterioration with age. However, a deeper understanding of the role of MA in heart physiology is necessary to determine the “therapeutic amount of stress” and the “hormetic window” that elicit an autophagic response within the adaptive range.
7. Future directions
The critical role of mitochondrial autophagy in cardiomyocyte physiology suggests that the development of therapeutic interventions that exploit the homeostatic properties of MA, without stimulating its maladaptive effects, would be of great value to preserve cardiac function into old age and manage age-related CVD. To accomplish this challenging task, several critical issues need to be addressed. First, the actual contribution of dysfunctional MA to the pathogenesis of cardiac senescence and CVD remains to be clearly established. In addition, MA is not just a random degradation process; rather, it is a highly regulated and potentially selective machinery in the service of the cell's needs. Therefore, it is mandatory to identify specific pathways or substrates of MA (dysfunctional mitochondria?) whose derangements are primarily involved in heart senescence. Moreover, the identification of signaling pathways linking mitochondrial dynamics and selective mitophagy is necessary for the development of therapeutics that maximize the removal of damaged organelles, while sparing functional mitochondria. In this context, it also needs to be established whether the effects produced by the specific induction of cardiac mitophagy are comparable to those elicited by general MA. Furthermore, it should be considered that most MA mediators have multiple functions, meaning that their manipulation may produce unrelated, and perhaps undesirable effects. Therefore, a deeper understanding of the various actions performed by autophagy-related factors is warranted. Another major caveat is the lack of suitable assays for measuring the ongoing autophagic flux in humans. This limitation makes extremely challenging the identification of the optimal window of autophagic activation to exploit the cardioprotective effects of MA without disrupting cardiac homeostatic mechanisms. Finally, most data on the effects of pharmacological or behavioral modulation of MA on cardiac aging and physiology derive from model organisms in which the role of autophagy may differ from what impacts human health, partly because of difficulties in modeling complex human diseases and degenerative processes in experimental settings.
Answering these critical research questions will likely provide cardiologists and geriatricians with novel therapeutic means to postpone the degenerative fate of cardiomyocytes and relieve the burden associated with CVD at old age.
8. Conclusions
The optimal regulation of mitochondrial autophagy is critical for the maintenance of cell homeostasis. This is especially true for cardiomyocytes, due to their post-mitotic nature and the high reliance on mitochondrial oxidative metabolism for energy supply. Over their lifespan, cardiac cells suffer from a high burden of mitochondria-derived oxidative damage, which cannot be diluted through cell proliferation. This implies that the maintenance of a healthy pool of mitochondria and the removal of damaged organelles are vital for the preservation of cardiomyocyte function and viability. Autophagy serves this essential homeostatic function. The relevance of the mitochondrial-lysosomal axis to cardiomyocyte homeostasis is witnessed by the observation that mitochondrial dysfunction, abnormal oxidant generation and the accumulation of intracellular waste material are observed during cardiac aging. This suggests that the optimization of the housekeeping function of autophagy may be harnessed as a therapeutic means against heart senescence. Indeed, preclinical studies indicate that interventions that up-regulate MA (e.g., CR, resveratrol administration, SIRT1 overexpression) preserve cardiac function during aging. An abnormal regulation of MA is also involved in the pathogenesis of a wide spectrum of heart diseases, whose prevalence increases with advancing age. In this scenario, up-regulation of MA is usually part of a cardioprotective response. Nevertheless, an excessive activation of autophagy may be maladaptive and contribute to disease progression.
In summary, the relevance of mitochondrial autophagy to cardiac physiology suggests the possibility that therapeutic interventions targeting this cellular pathway may represent effective means to counter heart senescence and age-related CVD. Untangling the complexity of autophagic regulation and managing the dual nature of autophagy are major tasks the field of geriatric cardiology is called to pursue.
Acknowledgements
The authors recognize that not all of the excellent scientific work in this area could be included or cited due to the vast literature on the subject and space limitations.
Sources of funding This research was supported by a grant to C.L. (NIA RO1-AG21042, NIDDK RO1-DK090115-01A1), the University of Florida's Institute on Aging and Claude D. Pepper Older Americans Independence Center (NIA 1P30AG028740) and by the Centro Studi Achille e Linda Lorenzon (R.C., R.B., E.M.). DD is supported by a fellowship from the American Heart Association (10PRE4310091).
Non-standard Abbreviations and Acronyms
- 2DG
2-deoxy-d-glucose
- AMPK
AMP-activated protein kinase
- Atg protein
AuTophaGy-related protein
- Bcl-2
B cell leukemia-2
- Bcl-XL
B cell leukemia-X long
- Bnip3
Bcl-2 and adenovirus E1B 19 kDa-interacting protein-3
- CMA
chaperone-mediated autophagy
- CR
calorie restriction
- CuZnSOD
copper-zinc-containing superoxide dismutase
- CVD
cardiovascular disease
- CypD
cyclophilin D
- DC
diabetic cardiomyopathy
- ETC
electron transport chain
- Gpx
glutathione peroxidase
- HF
heart failure
- HIF-1α
hypoxia-inducible factor-1α
- I/R
ischemia/reperfusion
- IGF-1
insulin-like growth factor-1
- LAMP-2
lysosomal membrane-associated protein-2
- LC3
light chain-3
- LVH
left ventricular hypertrophy
- MA
macroautophagy
- MAPKs
mitogen activated protein kinases
- mCAT
catalase targeted to the mitochondrial matrix
- MnSOD
manganese-containing superoxide dismutase
- mPTP
mitochondrial permeability transition pore
- mtDNA
mitochondrial DNA
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- nDNA
nuclear DNA
- NF-κB
nuclear factor κB
- Nix
Nip3-like protein X
- NRF-1
nuclear respiratory factor-1
- OXPHOS
oxidative phosphorylation
- PARL
presenilins-associated rhomboid-like
- pCAT
peroxisomal catalase
- PGC
peroxisome proliferator-activated receptor-γ coactivator
- PI3K
phosphatidylinositol-3-kinase
- PKB/Akt
protein kinase B
- PolG
mtDNA polymerase γ
- Prx
peroxiredoxine
- Raptor
mTORC1 containing rapamycin-associated TOR protein
- RHEB
Ras homolog enriched in brain
- ROS
reactive oxygen species
- SNARE
Soluble N-ethylmaleimide-sensitive factor Attachment protein Receptor
- SOS
survival of the slowest
- Tert
telomerase reverse transcriptase
- TFAM
mitochondrial transcription factor A
- TNF-α
tumor necrosis factor α
- TSC1/TSC2
tuberous sclerosis complex 1/2
- UVRAG
UV radiation resistance-associated gene
- VDAC
voltage-dependent anion channel
- Vps
vacuolar protein sorting
Footnotes
Disclosures: none
Subject codes: [137] Cell biology/structural biology; [138] Cell signalling/signal transduction; [140] Energy metabolism; [107] Biochemistry and metabolism; [108] Other myocardial biology; [91] Oxidant stress
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, De SG, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O'Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–6. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 2.Odding E, Valkenburg HA, Stam HJ, Hofman A. Determinants of locomotor disability in people aged 55 years and over: the Rotterdam Study. Eur J Epidemiol. 2001;17:1033–41. doi: 10.1023/a:1020006909285. [DOI] [PubMed] [Google Scholar]
- 3.Newman AB, Arnold AM, Naydeck BL, Fried LP, Burke GL, Enright P, Gottdiener J, Hirsch C, O'Leary D, Tracy R. “Successful aging”: effect of subclinical cardiovascular disease. Arch Intern Med. 2003;163:2315–22. doi: 10.1001/archinte.163.19.2315. [DOI] [PubMed] [Google Scholar]
- 4.Lakatta EG. Heart aging: a fly in the ointment? Circ Res. 2001;88:984–6. doi: 10.1161/hh1001.091963. [DOI] [PubMed] [Google Scholar]
- 5.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–20. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swinne CJ, Shapiro EP, Lima SD, Fleg JL. Age-associated changes in left ventricular diastolic performance during isometric exercise in normal subjects. Am J Cardiol. 1992;69:823–6. doi: 10.1016/0002-9149(92)90518-4. [DOI] [PubMed] [Google Scholar]
- 7.Gerstenblith G, Frederiksen J, Yin FC, Fortuin NJ, Lakatta EG, Weisfeldt ML. Echocardiographic assessment of a normal adult aging population. Circulation. 1977;56:273–8. doi: 10.1161/01.cir.56.2.273. [DOI] [PubMed] [Google Scholar]
- 8.Jahangir A, Sagar S, Terzic A. Aging and cardioprotection. J Appl Physiol. 2007;103:2120–8. doi: 10.1152/japplphysiol.00647.2007. [DOI] [PubMed] [Google Scholar]
- 9.Marzetti E, Wohlgemuth SE, Anton SD, Bernabei R, Carter CS, Leeuwenburgh C. Cellular mechanisms of cardioprotection by calorie restriction: state of the science and future perspectives. Clin Geriatr Med. 2009;25:715–32. ix. doi: 10.1016/j.cger.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 12.Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, Iwanaga Y, Yoshida Y, Kosugi R, Watanabe-Maeda K, Machida Y, Tsuji S, Aburatani H, Izumi T, Kita T, Shioi T. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation. 2009;120:1695–703. doi: 10.1161/CIRCULATIONAHA.109.871137. [DOI] [PubMed] [Google Scholar]
- 13.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Shirasawa T, Mizushima N, Otsu K. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6 doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 14.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–16. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–14. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 16.Pasdois P, Parker JE, Griffiths EJ, Halestrap AP. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem J. 2011;436:493–505. doi: 10.1042/BJ20101957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finkel T. Signal Transduction by Mitochondrial Oxidants. J Biol Chem. 2011 doi: 10.1074/jbc.R111.271999. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol. 2007;292:C1983–C1992. doi: 10.1152/ajpcell.00285.2006. [DOI] [PubMed] [Google Scholar]
- 19.Grivennikova VG, Kareyeva AV, Vinogradov AD. What are the sources of hydrogen peroxide production by heart mitochondria? Biochim Biophys Acta. 2010;1797:939–44. doi: 10.1016/j.bbabio.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J, Marzetti E, Seo AY, Kim JS, Prolla TA, Leeuwenburgh C. The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech Ageing Dev. 2010;131:487–93. doi: 10.1016/j.mad.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yakes FM, Van HB. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–5. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 23.Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood) 2002;227:671–82. doi: 10.1177/153537020222700901. [DOI] [PubMed] [Google Scholar]
- 24.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–65. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly DP. Cell biology: Ageing theories unified. Nature. 2011;470:342–3. doi: 10.1038/nature09896. [DOI] [PubMed] [Google Scholar]
- 26.Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74:121–33. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 27.Leeuwenburgh C, Wagner P, Holloszy JO, Sohal RS, Heinecke JW. Caloric restriction attenuates dityrosine cross-linking of cardiac and skeletal muscle proteins in aging mice. Arch Biochem Biophys. 1997;346:74–80. doi: 10.1006/abbi.1997.0297. [DOI] [PubMed] [Google Scholar]
- 28.Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–8. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- 29.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–21. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 30.Mohamed SA, Hanke T, Erasmi AW, Bechtel MJ, Scharfschwerdt M, Meissner C, Sievers HH, Gosslau A. Mitochondrial DNA deletions and the aging heart. Exp Gerontol. 2006;41:508–17. doi: 10.1016/j.exger.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 31.Liu VW, Zhang C, Nagley P. Mutations in mitochondrial DNA accumulate differentially in three different human tissues during ageing. Nucleic Acids Res. 1998;26:1268–75. doi: 10.1093/nar/26.5.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly Y, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 33.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van RH, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 34.Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–44. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang D, Mott JL, Farrar P, Ryerse JS, Chang SW, Stevens M, Denniger G, Zassenhaus HP. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc Res. 2003;57:147–57. doi: 10.1016/s0008-6363(02)00695-8. [DOI] [PubMed] [Google Scholar]
- 36.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van RH, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 37.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–97. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–14. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(S2):1542–52. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy. 2011;7:673–82. doi: 10.4161/auto.7.7.14733. [DOI] [PubMed] [Google Scholar]
- 41.Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–9. doi: 10.1016/j.ceb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mortimore GE, Schworer CM. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature. 1977;270:174–6. doi: 10.1038/270174a0. [DOI] [PubMed] [Google Scholar]
- 43.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. 2008;15:344–57. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–44. [PubMed] [Google Scholar]
- 48.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, Liu LF. Autophagy regulates ageing in C. elegans. Autophagy. 2007;3:93–5. doi: 10.4161/auto.3636. [DOI] [PubMed] [Google Scholar]
- 50.Matecic M, Smith DL, Pan X, Maqani N, Bekiranov S, Boeke JD, Smith JS. A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet. 2010;6:e1000921. doi: 10.1371/journal.pgen.1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, Karin M. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–8. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takacs-Vellai K, Orosz L, Kovacs AL, Csikos G, Sass M, Vellai T. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008;4:330–8. doi: 10.4161/auto.5618. [DOI] [PubMed] [Google Scholar]
- 53.Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, Michaud M, Madeo F, Tavernarakis N, Kroemer G. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124:161–70. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–81. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ. SNARE Proteins Are Required for Macroautophagy. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tong J, Yan X, Yu L. The late stage of autophagy: cellular events and molecular regulation. Protein Cell. 2010;1:907–15. doi: 10.1007/s13238-010-0121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992-–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Talloczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kadandale P, Kiger AA. Role of selective autophagy in cellular remodeling: “self-eating” into shape. Autophagy. 2010;6:1194–5. doi: 10.4161/auto.6.8.13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7:297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal. 2011;14:1919–28. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem. 2007;282:5617–24. doi: 10.1074/jbc.M605940200. [DOI] [PubMed] [Google Scholar]
- 67.Green DR, Galluzzi L, Kroemer G. Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science. 2011;333:1109–12. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–21. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 71.Chen D, Gao F, Li B, Wang H, Xu Y, Zhu C, Wang G. Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J Biol Chem. 2010;285:38214–23. doi: 10.1074/jbc.M110.101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy. 2010;6 doi: 10.4161/auto.6.4.11553. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–7. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 74.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 75.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–23. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–46. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gottlieb RA, Carreira RS. Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol Cell Physiol. 2010;299:C203–C210. doi: 10.1152/ajpcell.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12:503–35. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sachs HG, Colgan JA, Lazarus ML. Ultrastructure of the aging myocardium: a morphometric approach. Am J Anat. 1977;150:63–71. doi: 10.1002/aja.1001500105. [DOI] [PubMed] [Google Scholar]
- 82.Karbowski M, Kurono C, Wozniak M, Ostrowski M, Teranishi M, Nishizawa Y, Usukura J, Soji T, Wakabayashi T. Free radical-induced megamitochondria formation and apoptosis. Free Radic Biol Med. 1999;26:396–409. doi: 10.1016/s0891-5849(98)00209-3. [DOI] [PubMed] [Google Scholar]
- 83.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 84.Arnheim N, Cortopassi G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat Res. 1992;275:157–67. doi: 10.1016/0921-8734(92)90020-p. [DOI] [PubMed] [Google Scholar]
- 85.de Grey AD. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays. 1997;19:161–6. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- 86.Kowald A. The mitochondrial theory of aging: do damaged mitochondria accumulate by delayed degradation? Exp Gerontol. 1999;34:605–12. doi: 10.1016/s0531-5565(99)00011-x. [DOI] [PubMed] [Google Scholar]
- 87.Jung T, Bader N, Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- 88.Terman A, Gustafsson B, Brunk UT. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol Aspects Med. 2006;27:471–82. doi: 10.1016/j.mam.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 89.Terman A. Garbage catastrophe theory of aging: imperfect removal of oxidative damage? Redox Rep. 2001;6:15–26. doi: 10.1179/135100001101535996. [DOI] [PubMed] [Google Scholar]
- 90.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–87. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 91.Sybers HD, Ingwall J, DeLuca M. Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct Metab. 1976;12:453–63. [PubMed] [Google Scholar]
- 92.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–61. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–89. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 94.Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res. 2011;90:234–42. doi: 10.1093/cvr/cvr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903–7. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–46. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 98.Finckenberg P, Eriksson O, Baumann M, Merasto S, Lalowski MM, Levijoki J, Haasio K, Kyto V, Muller DN, Luft FC, Oresic M, Mervaala E. Caloric Restriction Ameliorates Angiotensin II-Induced Mitochondrial Remodeling and Cardiac Hypertrophy. Hypertension. 2011 doi: 10.1161/HYPERTENSIONAHA.111.179457. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 99.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cao DJ, Hill JA. Titrating autophagy in cardiac plasticity. Autophagy. 2011;7 doi: 10.4161/auto.7.9.16176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59. doi: 10.1146/annurev-physiol-021909-135757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–70. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 103.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 104.Loos B, Lochner A, Engelbrecht AM. Autophagy in heart disease: a strong hypothesis for an untouched metabolic reserve. Med Hypotheses. 2011;77:52–7. doi: 10.1016/j.mehy.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 105.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 106.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 107.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–56. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 108.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–63. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 109.Spinale FG, Coker ML, Thomas CV, Walker JD, Mukherjee R, Hebbar L. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: relation to ventricular and myocyte function. Circ Res. 1998;82:482–95. doi: 10.1161/01.res.82.4.482. [DOI] [PubMed] [Google Scholar]
- 110.Akazawa H, Komazaki S, Shimomura H, Terasaki F, Zou Y, Takano H, Nagai T, Komuro I. Diphtheria toxin-induced autophagic cardiomyocyte death plays a pathogenic role in mouse model of heart failure. J Biol Chem. 2004;279:41095–103. doi: 10.1074/jbc.M313084200. [DOI] [PubMed] [Google Scholar]
- 111.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 112.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 113.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 114.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–8. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–57. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 116.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 117.Speakman JR, Mitchell SE. Caloric restriction. Mol Aspects Med. 2011;32:159–221. doi: 10.1016/j.mam.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 118.Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–9. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- 119.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–7. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 120.Hancock CR, Han DH, Higashida K, Kim SH, Holloszy JO. Does calorie restriction induce mitochondrial biogenesis? A reevaluation. FASEB J. 2011;25:785–91. doi: 10.1096/fj.10-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, Dunn WA., Jr. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–92. doi: 10.1089/rej.2006.0535. [DOI] [PubMed] [Google Scholar]
- 123.Shinmura K, Tamaki K, Sano M, Murata M, Yamakawa H, Ishida H, Fukuda K. Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J Mol Cell Cardiol. 2011;50:117–27. doi: 10.1016/j.yjmcc.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 124.Meyer TE, Kovacs SJ, Ehsani AA, Klein S, Holloszy JO, Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol. 2006;47:398–402. doi: 10.1016/j.jacc.2005.08.069. [DOI] [PubMed] [Google Scholar]
- 125.Landi F, Zuccala G, Gambassi G, Incalzi RA, Manigrasso L, Pagano F, Carbonin P, Bernabei R. Body mass index and mortality among older people living in the community. J Am Geriatr Soc. 1999;47:1072–6. doi: 10.1111/j.1532-5415.1999.tb05229.x. [DOI] [PubMed] [Google Scholar]
- 126.Ingram DK, Zhu M, Mamczarz J, Zou S, Lane MA, Roth GS, deCabo R. Calorie restriction mimetics: an emerging research field. Aging Cell. 2006;5:97–108. doi: 10.1111/j.1474-9726.2006.00202.x. [DOI] [PubMed] [Google Scholar]
- 127.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–93. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 128.Minor RK, Smith DL, Jr., Sossong AM, Kaushik S, Poosala S, Spangler EL, Roth GS, Lane M, Allison DB, de CR, Ingram DK, Mattison JA. Chronic ingestion of 2-deoxy-D-glucose induces cardiac vacuolization and increases mortality in rats. Toxicol Appl Pharmacol. 2010;243:332–9. doi: 10.1016/j.taap.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy. 2011;7:1254–5. doi: 10.4161/auto.7.10.16740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le CD, Shaw J, Navas P, Puigserver P, Ingram DK, de CR, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Barger JL, Kayo T, Pugh TD, Prolla TA, Weindruch R. Short-term consumption of a resveratrol-containing nutraceutical mixture mimics gene expression of long-term caloric restriction in mouse heart. Exp Gerontol. 2008;43:859–66. doi: 10.1016/j.exger.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 133.Petrovski G, Gurusamy N, Das DK. Resveratrol in cardiovascular health and disease. Ann N Y Acad Sci. 2011;1215:22–33. doi: 10.1111/j.1749-6632.2010.05843.x. [DOI] [PubMed] [Google Scholar]
- 134.Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by resveratrol: A novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103–12. doi: 10.1093/cvr/cvp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–21. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]