

Abstract
Thiazolidinediones (TZDs), known as peroxisome proliferator-activated receptor (PPAR) agonists, are used to treat type 2 diabetes. However, ∼5% of patients experience the treatment-limiting side effect of edema. Studies have implicated activation of the epithelial sodium channel (ENaC) as a cause of TZD-induced fluid retention, although there have been conflicting reports. The goal of this study was to resolve the role of PPARγ in control of ENaC isoforms in the kidney. Herein, we demonstrate in mice that rosiglitazone (RGZ), a PPARγ ligand, increases body weight and abdominal fat pad fluid content and reduces hematocrit. Seven days of RGZ decreases ENaCα and ENaCβ mRNA and ENaCγ protein expression in the kidney cortex, and acute treatment for 5 h with pioglitazone, another potent TZD, does not increase renal ENaC isoform mRNA or protein expression. Pioglitazone also decreases ENaCα and ENaCγ mRNA expression in a cortical collecting duct cell line. As no direct transcriptional studies had been conducted, we examined the PPARγ-dependent regulation of ENaC. Pioglitazone represses ENaCγ promoter activity, and this repression is partially relieved by inhibition of protein synthesis. Chromatin immunoprecipitation assays revealed that repression is associated with a decrease in histone H4K5 acetylation at the proximal ENaCγ promoter. In summary, TZDs do not increase ENaC mRNA expression in the kidney, and in fact repress the ENaCγ promoter via an indirect transcriptional mechanism.
Keywords: thiazolidinediones, edema, ENaC promoter
thiazolidinediones (TZDs), such as pioglitazone and rosiglitazone (RGZ), have been used over the last decade to treat type 2 diabetes. Despite favorable effects on glycemic control, these agents are associated with fluid retention in at least 5% of patients (reviewed in Ref. 35), and as such are currently contraindicated in people with congestive heart failure (CHF). The severity of weight gain (29) and the risk of developing CHF (30) increase in patients treated with TZDs in combination with insulin therapy.
TZDs are agonists for a subfamily of nuclear receptors known as peroxisome proliferator-activated receptors (PPARs) (23). There are three PPAR isoforms (α, β, γ), and TZDs are high-affinity ligands for PPARγ (23). PPARγ is expressed in the medullary (12) and cortical collecting ducts, the distal convoluted tubule (16), and glomerular (13, 56) and vascular smooth muscle cells (27). Evidence suggests that PPAR agonists alter the conformation of PPARs, enabling them to modify gene expression at the transcriptional level and control a wide range of signaling pathways and metabolic processes. PPARs can positively or negatively regulate transcription by binding directly to the response element in a gene as a heterodimer with the retinoid X receptor (RXR). An alternative method of negative regulation occurs via an indirect mechanism–by association of PPARs with other sequence-specific transcription factors, a process referred to as transrepression (10).
Due to adverse side effects, there has been significant interest in elucidating the causes of PPARγ ligand-induced fluid retention. Edema is dependent on PPARγ in the collecting duct, as two studies have elegantly demonstrated that knockout of PPARγ in the collecting duct attenuates TZD-induced edema (11, 59). Investigators have proposed that fluid retention is related to enhanced activity of the epithelial sodium channel (ENaC) in the distal nephron, although conflicting reports challenge this hypothesis (32, 52). ENaC is composed of three subunits (α, β, γ) and acts as the rate-limiting step for Na reabsorption across many epithelia. Serum and glucocorticoid-regulated kinase (Sgk-1), a serine/threonine protein kinase expressed in the kidney, increases ENaC protein abundance in the cell membrane. Thus, Sgk-1 has also been proposed as a factor in fluid retention (1, 16), although this remains unclear.
The goal of this study, therefore, was to understand the role of PPARγ in regulating ENaC isoforms in the kidney. Herein, we demonstrate that 7-day TZD treatment potently induces fluid retention in mice, and this is associated with declines in both cortical ENaCα and ENaCβ mRNA, as well as a decline in the cortical protein expression of the 85-kDa ENaCγ subunit. In correlation with in vivo findings, in M1 cells, a murine cortical collecting duct cell line (49), PPARγ ligands significantly decrease ENaCα and ENaCγ mRNA expression. We then performed transcriptional studies including chromatin immunoprecipitation (ChIP) studies to determine that PPARγ ligands do not increase ENaC in the kidney, and in fact transcriptionally repress ENaCγ expression via an indirect pathway.
METHODS
Animals.
Mice were housed and handled in accordance with Veterans Affairs and National Institutes of Health guidelines and approved by the Institutional Laboratory Animal Care and Use Committee at the Veterans Affairs San Diego Healthcare System (VASDHS). Male 129/Sv mice were derived from a breeding colony at the VASDHS Veterinary Medical Unit. Mice were housed in a 12:12-h light-dark cycle in standard rodent cages with free access to food. Mice were fed a repelleted standard diet (1% K+, 0.4% Na+, Harlan Teklad TD.7001). RGZ (320 mg/kg) (52, 59) was added to this diet and repelleted. For short-term PPARγ agonist studies, mice were injected with 25 mg/kg ip pioglitazone (Takeda Pharmaceuticals) or vehicle.
Assessment of weight and fluid gain and urinary electrolytes.
Body weight was determined before RGZ treatment and daily during the 7 days of RGZ feeding. Hematocrit was measured using a microcapillary reader (2201, International Equipment). A radioimmunoassay detected plasma aldosterone (DSL-8600, Diagnostic Systems Laboratories, Beckman Coulter, Webster, TX) (38). Intra-abdominal fat pads were removed and the fluid content was determined by heating samples in an oven at 70°C over 7 days. Urine was obtained by picking-up the mice and eliciting reflex urination at 5 h after pioglitazone injection. Urinary Na+ and K+ were measured by flame photometry (Cole-Parmer Instrument, Vernon Hills, IL).
Cells.
M1 cortical collecting duct cells (49) were obtained from American Type Culture Collection and grown in DMEM/F-12 media with 5% FBS (Atlanta Biologicals), 1% penicillin/streptomycin, and 1% glutamine (Invitrogen, Carlsbad, CA). Cells were treated for 24 h with pioglitazone (Takeda Pharmaceuticals) and RNA analysis was performed. Cells were also treated with the PPARγ antagonist GW9662 (Cayman Chemical) (22).
Reverse transcription and real-time PCR.
Kidneys were divided into cortical and medullary sections and stored in RNAlater (Qiagen). RNA was purified with the RNeasy Mini Kit (Qiagen). RNA from cells in culture was prepared with TRIzol (Invitrogen) and purified with the RNeasy Mini Kit. DNase digestion was performed with Turbo DNase (Ambion, Austin, TX) and cDNA was prepared with the Superscript II First Strand Synthesis System (Invitrogen). For quantitation, Table 1 describes the primers (11) used with Power SYBR Green PCR Master Mix (10 min at 95°C with 50 cycles of 15 s at 95°C and 1 min at 60°C) in a Chromo4 real-time PCR Detector (MJ Research and Bio-Rad). Amplification efficiencies were normalized against RPL19 to obtain a “relative fold induction,” and relative fold increases were calculated using the Pfaffl technique of relative quantification, which accounts for real-time efficiencies (37). Each experiment was performed at least three times in triplicate.
Table 1.
Real-time PCR primers
| Target | Forward | Reverse |
|---|---|---|
| SGK1 | ACATCTACCTTCTGTGGCACGC | TCATACAGGACAGCCCCAAGAC |
| ENaCα | TGCACCTTCGGCATGATGTA | ACCAGCTTGTCCGAATTGAGG |
| ENaCβ | CCCACCCAGCAACTAGTGAA | GGCTCTTTGTACAAGGCCTAA |
| ENaCγ | ATGCTTCCAAACGAAGATGG | AGTTGGGGTGTTGCTGGTAG |
| RPL19 | TGCTCAGGCTACAGAAGAGGCTTG | GGAGTTGGCATTGGCGATTTC |
ENaC, epithelial sodium channel.
Western blotting.
Kidneys and M1 cells were homogenized in a buffer with 10 mM triethanolamine and 250 mM sucrose with protease inhibitors. Homogenates were initially spun for 5 min at 3,000 g, 4°C and then the supernatant was respun at 17,000 g for 90 min at 4°C for membrane preparations. Samples were run on NuPAGE Bis-Tris gels (Invitrogen) and transferred onto nitrocellulose membranes (Invitrogen). Immunoblotting was performed overnight with the following primary antibodies as previously described: anti-ENaCα (25), anti-ENaCβ (7), anti-ENaCγ (25) (E4902, Sigma), and actin (Santa Cruz Biotechnology) and detection was performed with ECL Plus Detection Reagents (GE Healthcare, Piscataway, NJ).
Plasmids and transient transfections.
The murine ENaCα promoter (−3031 to −232, based on ATG start site) was a kind gift from Dr. André Dagenais, Centre Hospitalier de L'Université de Montréal (4). The human ENaCγ promoters (−2926 and −1248) were kind gifts from Dr. Christie P Thomas, University of Iowa Health Care (50). We created a 439-bp fragment (−439 to −1, +1 is the ATG) of the proximal human ENaCγ promoter using pGL3-hENaCγ-2926 as a template and the following primers: 5′-GTGGCCTGGCGGGGTACCCCCTGCTG-3′ and 5′-CTAGCATCCCGCTCGAGCGGCACCGCG-3′, with PFU Ultra DNA Polymerase (Agilent Technologies). The cloned truncated promoter was ligated using T4 DNA Ligase (Invitrogen) into pGL3-Luc to generate hENaCγ-Luc-p439. All plasmids were sequenced by the UCSD Cancer Center Sequencing Core.
M1 cells were plated in a 24-well plate in 5% FBS/DMEM/F12. Cells were transfected in Opti-MEM with Lipofectamine 2000 reagent 1:4 (DNA:Lipofectamine) and 500 ng/well of the luciferase reporter plasmids, 150 ng/well of PPARγ (pCMX-PPARγ) or the empty vector pCMX, and 100 ng/well of a Renilla luciferase reporter vector (pRL-CMV, Promega) as an internal control for transfection efficiency. Four hours posttransfection, the cells were treated with control (DMSO) or PPARγ ligand at varying concentrations in 0.1% FBS media. The next day, luciferase activity was measured as we previously described (MicroLumatPlus LB 96V, Berthold Technologies) (3). Experiments were repeated at least three times and in triplicate. Reporter activity was corrected by cotransfected CMV-Renilla (expressed as luciferase activity/renilla activity and depicted as relative fold induction). Results of each experiment for each reporter (AOX)3-TK-Luc, mENaCα-p3051-Luc, and hENaCγ-p2926-Luc control (no pioglitazone, no PPARγ) were normalized to 1.
Cycloheximide experiments.
M1 cells were plated at 0.5 × 106 cells per well (6-well plate) in 5% FBS/DMEM/F/12 media, and the next morning the media were replaced with 0.1% FBS media with or without 75 ng/ml cycloheximide. Thirty minutes later, cells were treated with control (DMSO) or pioglitazone. RNA was harvested and analyzed after 24 h.
ChIP.
ChIP assays were performed on M1 cells treated with pioglitazone (5 μM) or control (DMSO) for 16 h (10 × 106 cells/condition) per the manufacturer's instructions (EZ ChIP, Millipore) and as previously described (43). The cells were lysed, cross-linked DNA was sonicated to the desired shear of 100–1,000 bp (45% amplitude for 10 s, 8 times), and samples were precleared. Complexes were immunoprecipitated with 4 μg of the following antibodies: Pol II [N-20; Santa Cruz Biotechnology (SCBT), 899x], anti-acetyl histone H3 (Lys9/14; Millipore 06–599), anti-acetyl histone H4 (Lys5; Millipore 07–327), and normal rabbit IgG (SCBT, 2027) was the negative control. Crosslinks were reversed and DNA was isolated. DNA was amplified by quantitative PCR as previously described (43) using primers for the proximal hENaCγ promoter region and into the first 100 bp of transcription: 5′-GGAGCGTCTCCAGACTGTAC-3′ and 5′-ACGCACCACGCCCTGTGGCT-3′.
Statistics.
Data are reported as means ± SE. Data were compared with the Student's (2-tailed) t-test or ANOVAs with Tukey correction. P ≤ 0.05 was considered statistically significant.
RESULTS
RGZ increases body weight and fluid retention in mice.
Mice were treated for 7 days with RGZ. Basal body weight was not different between the control and RGZ-fed mice (31.6 ± 1.3 vs. 31.1 ± 1.4 g; n = 6/group; not significant). RGZ significantly increased body weight after just 1 day of treatment, an effect sustained through all 7 days of feeding (Fig. 1A). Hematocrit was significantly lower and fluid content of abdominal fat significantly greater in the RGZ-fed mice (Fig. 1, B and C). These RGZ-induced changes occurred in the absence of significant differences in plasma aldosterone levels (Fig. 1D).
Fig. 1.

Rosiglitazone (RGZ) increases body weight and fluid retention in mice. A: mice fed RGZ showed a significant increase in body weight after 1 day of feeding. B: hematocrit was significantly lower. C: fluid content of abdominal fat was significantly greater in the RGZ-fed mice. D: these RGZ-induced changes occurred in the absence of significant differences in plasma aldosterone levels. *P < 0.05 vs. control diet.
RGZ decreases cortical mRNA expression of ENaCα and ENaCβ.
PPARγ ligands generally alter gene expression at the transcriptional level; therefore, we evaluated the mRNA expression of ENaC isoforms in the cortical and medullary sections of the kidneys from the mice described above. In the cortex, there was a significant decline in ENaCα and ENaCβ mRNA expression (Fig. 2A). In the medulla, there were no significant changes in ENaC isoform expression (Fig. 3A). Also, in the cortex and medulla, there were no significant changes in mRNA expression of Sgk-1 associated with TZD feeding.
Fig. 2.
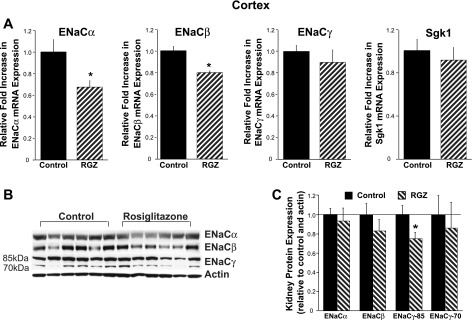
RGZ decreases epithelial sodium channel (ENaC)α and ENaCβ mRNA expression in kidney cortex. A: real-time PCR analysis of renal cortical sections from mice fed RGZ for 7 days showed a significant decrease in ENaCα and ENaCβ mRNA expression compared with standard diet (control) mice. B and C: cortical expression of the 85-kDa subunit of ENaCγ was reduced in the RGZ-fed mice. *P < 0.05 vs. control.
Fig. 3.
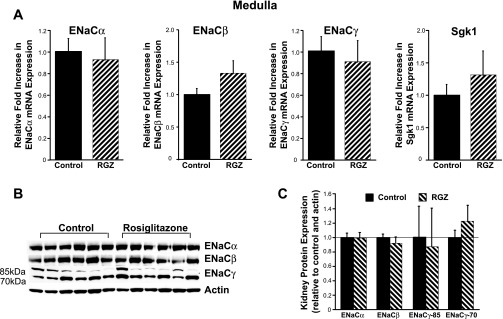
RGZ does not significantly alter ENaC isoform mRNA expression in kidney medulla. A: real-time PCR analysis. B and C: Western blotting of renal medullary sections from mice fed RGZ for 7 days showed no significant alterations in ENaC isoform expression compared with standard diet (control) mice.
Next, we evaluated protein expression of ENaC isoforms in membrane preparations from the control and RGZ-fed mice. In the cortex and medulla, there was no evidence of increases in ENaC isoform expression associated with RGZ feeding. Interestingly, there was a significant decline in the uncleaved ENaCγ subunit (85 kDa) in the cortical preparations (Fig. 2, B and C). In the medulla, similar to the mRNA findings, there were no significant changes in ENaC isoform expression (Fig. 3, B and C). Interestingly, in the medulla, independent of RGZ treatment, there appeared to be higher expression of the cleaved form of ENaCγ (70 kDa) compared with the uncleaved subunit (Fig. 3B). It is unclear whether there is more proteolytic cleavage of ENaCγ in the medulla compared with the cortex, as this has not been previously described.
Acute treatment with pioglitazone decreases urinary Na/K ratios without increasing ENaC isoform expression.
Next, we assessed the effects of acute application of another PPARγ agonist pioglitazone (25 mg/kg body wt ip). Five hours posttreatment, spontaneous urine samples were collected and kidneys were harvested. Figure 4A demonstrates a trend for pioglitazone to lower urinary [Na] without an effect on K concentrations. Notably, the ratio of urinary [Na] to [K] concentration was significantly lower in the group administered pioglitazone (Fig. 4A). Similar to the results obtained from the longer treatment course, there were no significant increases in cortical ENaC isoform (Fig. 4B) or Sgk-1 mRNA expression in the pioglitazone-treated group. There was no increase in ENaC isoform protein expression; however, there was a trend for a reduction in the 85-kDa ENaCγ subunit (P = 0.056), similar to the findings in the cortex in mice fed for 7 days with RGZ. Additionally, renal protein membrane expression of other major Na transport systems, including NHE3, NKCC2, NCC, and Na-K-ATPase, was not significantly enhanced by pioglitazone (Vallon V and Cunard R, unpublished observations).
Fig. 4.

Acute pioglitazone treatment decreases urinary Na/K excretion without increasing ENaC expression. A: five-hour treatment (25 mg/kg body wt ip) demonstrated a trend for pioglitazone (pioglit) to lower urinary [Na] without an effect on [K] excretion. The ratio of urinary [Na] to [K] concentration was significantly lower in the group administered pioglitazone; *P < 0.05 vs. control. There was no significant increase in cortical ENaC or Sgk-1 expression in the pioglitazone-treated group (B). C and D: whole kidney protein expression of ENaC isoforms was not increased in the pioglitazone-fed mice, and there was a trend for a reduction in the 85-kDa subunit of ENaCγ (P = 0.056). Ponceau staining verified equivalent loading of protein in each lane.
Pioglitazone decreases ENaCα and ENaCγ mRNA expression in M1 cells.
Next, we evaluated whether PPARγ agonists alter ENaC isoforms in murine cortical collecting duct cells (M1 cells). In accordance with in vivo findings, there was a significant decline in ENaCα mRNA expression (Fig. 5A). Interestingly, we also saw a decrease in ENaCγ mRNA; this finding differs from previous studies showing that TZDs increase ENaCγ expression in cultured collecting duct cells (11). Contrary to our in vivo findings, pioglitazone (Fig. 5) increased Sgk-1 mRNA expression in M1 cells. There were no changes in Na-K-ATPaseα and Na-K-ATPaseβ mRNA expression associated with PPARγ ligand treatment (Borsting E and Cunard R, unpublished observations). In the M1 cells, the membrane expression of ENaCα and ENaCγ did not correlate directly with the mRNA findings, suggesting that the increase in Sgk-1 expression in the M1 cells might have altered ENaCα and ENaCγ protein expression. For unclear reasons, we were not able to successfully immunoblot ENaCβ.
Fig. 5.

Pioglitazone decreases ENaCα and ENaCγ expression in M1 cells. M1 cortical collecting duct cells were treated for 24 h with pioglitazone (pioglit) or control (DMSO). Real-time PCR analysis showed that ENaCα and ENaCγ mRNA expression significantly decreased, and Sgk-1 mRNA expression significantly increased with pioglitazone 10-μM treatment compared with the control. *P < 0.05 vs. control. The membrane protein expression of ENaCα and ENaCγ did not correlate directly with the mRNA findings.
Pioglitazone decreases ENaCγ promoter activity in M1 cells.
When PPAR ligands bind to PPARs, they induce conformational changes that activate PPAR to modulate target genes, recruit co-activators, or dissociate from co-repressors. Activated PPARs can bind to specific DNA sequences in gene promoters known as PPAR response elements (PPREs) and in turn drive transcription of the gene, a process termed transactivation. The next set of studies was designed to confirm that M1 cells can support PPARγ-dependent transactivation, as studies suggested that PPARγ ligands activate ENaCγ expression (11). In Fig. 6, M1 cells were cotransfected with acyl-CoA oxidase (AOX)3-TK-Luc (a PPRE vector) (40) and PPARγ (pCMX-PPARγ) or the empty vector pCMX. In a dose-dependent manner, pioglitazone drove the PPRE reporter, only when PPARγ was transfected, demonstrating that M1 cells can support PPARγ-dependent transactivation.
Fig. 6.

M1 cells can support peroxisome proliferator-activated receptor (PPAR)γ-dependent transactivation. M1 cells were cotransfected with acyl-CoA oxidase (AOX)3-TK-Luc [a PPAR response element reporter vector (PPRE)] (40) and PPARγ (pCMX-PPARγ) or the empty vector pCMX. In a dose-dependent manner, pioglitazone drove the PPRE reporter only when PPARγ was transfected into the system. *P < 0.05 vs. control (no PPARγ). **P < 0.05 vs. 10 nM pioglitazone + PPARγ.
We then examined whether PPARγ directly affects the ENaCα or ENaCγ promoter. Promoter activity was assayed via transient transfection of M1 cells with luciferase reporter plasmids. The mENaCα-p3051 luciferase reporter has ∼3,000 bp of the proximal murine ENaCα promoter. Figure 7 demonstrates that pioglitazone does not significantly alter ENaCα promoter activity. Next, using various truncation constructs of the ENaCγ promoter, we evaluated whether TZDs repress ENaCγ transcriptional activity. The hENaCγ-p2926 promoter construct was repressed by PPARγ, even without addition of pioglitazone (Figs. 8A and 9C), suggesting that the promoter effects are ligand independent or that M1 cells produce endogenous PPARγ agonists (43). Both the hENaCγ-p2926 and hENaCγ-p1248 reporter constructs were inhibited by high concentrations of pioglitazone.
Fig. 7.

Pioglitazone does not alter ENaCα promoter activity in M1 cells. M1 cells were transfected with an ENaCα luciferase reporter and PPARγ or the empty vector pCMX and then treated with varying concentrations of pioglitazone (pioglit). Pioglitazone did not activate or repress the ENaCα promoter construct.
Fig. 8.
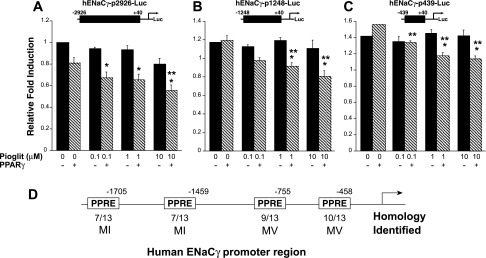
ENaCγ promoter activity is repressed by PPARγ. M1 cells were transfected with ENaCγ promoter construct plasmids [containing 2926 (A) or 1248 (B) or 439 bp (C) of the proximal promoter] plus PPARγ or the empty vector pCMX, and they were treated with pioglitazone. In hENaCγ-p2926-Luc, there was a trend (P = 0.067) for PPARγ to repress ENaCγ promoter activity, independent of ligand addition (see also Fig. 9C). In all 3 reporter constructs, pioglitazone repressed ENaCγ promoter activity. D: representation of the proximal ENaCγ promoter showing multiple candidate PPREs. *P < 0.05 vs. control (no PPARγ). **P < 0.05 vs. control, + PPARγ.
Fig. 9.

PPARγ antagonist partially relieves pioglitazone-dependent repression of ENaCγ expression. M1 cells were pretreated with GW9662 (10 μM) or control (DMSO) for 30 min, followed by pioglitazone (pioglit) or control (DMSO) for 24 h. A: GW9662 is unable to relieve pioglitazone-dependent repression of ENaCα mRNA expression. *P < 0.05 vs. pioglit 0, GW9662 0. B: treatment with GW9662 relieved repression of ENaCγ mRNA expression. *P < 0.05 vs. pioglit 0, GW9662 0. NS, not significant. C: pioglitazone repressed activity of the hENaCγ-p2926 and D: hENaCγ-p439, but this effect was attenuated by GW9662 in hENaCγ-p439. *P < 0.05 vs. pioglit 0, GW9662 0, and pCMX.
Using MacVector (MV, version 7.2.3) and MatInspector (MI, Genomatix, Germany), we and others identified multiple candidate PPREs, 5′-AGGTCANAGGTCA-3′ (17, 33), in the ENaCγ promoter and intronic regions (11). Figure 8D depicts several potential PPREs we identified in the ENaCγ promoter. Therefore, we generated a 439-bp truncation of the proximal hENaCγ promoter (hENaCγ-p439) that deleted the candidate PPREs. Surprisingly, deleting all of our candidate PPREs did not abolish the PPARγ ligand-dependent repression (Fig. 8C) and pioglitazone repressed the truncated promoter construct only when PPARγ was cotransfected into the cells.
PPARγ antagonist partially relieves pioglitazone-dependent repression of ENaCγ expression.
To resolve PPARγ-dependent vs. -independent mechanisms, we investigated the effect of the PPARγ antagonist GW9662 (22) on pioglitazone-induced repression of ENaCα and ENaCγ mRNA expression in M1 cells. Figure 9A demonstrates that GW9662 is unable to relieve pioglitazone-induced repression of ENaCα expression. However, GW9662 did relieve pioglitazone-induced repression of ENaCγ expression (Fig. 9B). GW9662 attenuated pioglitazone-induced repression of the ENaCγ (hENaCγ p439) truncation construct when it was cotransfected with PPARγ (Fig. 9D), but it had no significant effect on the longer ENaCγ promoter luciferase reporter (Fig. 9C).
Pioglitazone-induced repression of ENaCα and ENaCγ requires protein synthesis.
PPARs negatively regulate gene expression through direct repression or transrepression. Direct repression involves the sequence-specific binding of PPAR with its heterodimeric partner RXR directly to the gene. Another mechanism of repression involves transrepression, which is the indirect association or tethering of PPARs to other sequence-specific transcription factors (10). We treated M1 cells with 75 ng/ml cycloheximide (CHX), an inhibitor of protein synthesis. As expected, in the control group (no CHX) pioglitazone reduced ENaCα and ENaCγ mRNA expression. However, when cells were treated with CHX, there was a significant increase in ENaCα and ENaCγ, without any changes in ENaCβ expression. These findings suggest that ENaCα and ENaCγ expression is actively repressed in M1 cells and that the synthesis of other factors is required for gene repression (Fig. 10). There is a trend for pioglitazone to reduce ENaCα and ENaCγ expression in the CHX-treated cells, suggesting that there are both direct and indirect mechanisms causing PPARγ ligand-dependent repression of ENaC expression.
Fig. 10.
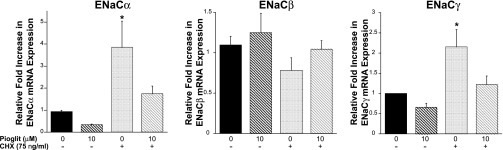
Pioglitazone-induced repression of ENaCα and ENaCγ requires protein synthesis. M1 cells were treated with or without 75 ng/ml cycloheximide (CHX) and also with control (DMSO) or pioglitazone (10 μM). In the control group (no CHX) pioglitazone reduced ENaCα and ENaCγ mRNA expression. However, with CHX, there was a significant increase in ENaCα and ENaCγ, without any changes in ENaCβ expression. When pioglitazone was added to the CHX-treated cells, there was a trend for a reduction in ENaCα and ENaCγ expression compared with the CHX-treated control cells. *P < 0.05 vs. control (no CHX).
Repression of the ENaCγ promoter is associated with a decrease in H4 acetylation at the proximal promoter.
In our final study, we explored whether chromatin modifications at the proximal ENaCγ promoter are involved in regulation of the gene. Chromatin structure and modification are critical components in transcriptional control. Transcription can be regulated by co-activators or co-repressor complexes that recognize and interact with specific histone modifications. In the case of histone H4 acetylation, acetylation of H4K5, 8, and 12 has been linked to recruitment of the transcriptional initiation factor-positive transcription elongation factor b (p-TEFb) (15). Thus, hyperacetylated chromatin is associated with transcriptionally active genes.
We first performed ChIP assays with a PPARγ antibody; however, we did not see evidence of direct binding of PPARγ to response elements in the proximal ENaCγ promoter (Borsting E and Cunard R, unpublished observations). This is not unexpected because most of the direct binding sites for PPARs are far removed from transcriptional start sites (10). Additionally, we performed ChIP with an antibody to receptor-interacting protein-140 (RIP140), a coregulator and corepressor that binds many nuclear receptors including the PPARs. However, there was no evidence of significant binding of RIP140 to the proximal promoter (Borsting E and Cunard R, unpublished observations).
ChIP assays with an acetyl histone H4K5 antibody showed a significant decrease in binding to the proximal ENaCγ promoter and into the first 100 bp of transcription with pioglitazone compared with the control (Fig. 11). Regulation of the promoter is associated more with histone H4K5 acetylation than histone H3K9/14 acetylation as we saw higher signals of H4 compared with H3 and no significant difference in H3 acetylation between the control and treatment groups. These findings suggest that the promoter becomes hypoacetylated with pioglitazone treatment and are consistent with a TZD-induced decrease in transcriptional activity.
Fig. 11.

Repression of the ENaCγ promoter is associated with a decrease in histone H4K5 acetylation at the proximal promoter. M1 cells were treated with pioglitazone (5 μM) or control (DMSO) for 16 h and chromatin immunoprecipitation studies were performed using antibodies for Pol II, histone H4 acetlyation (H4), and histone H3 acetylation (H3) with anti-rabbit IgG as the negative control. Real-time PCR analysis was performed with primers for the proximal promoter and into the first 100 bp of transcription. *P < 0.05 vs. IgG control. **P < 0.05 vs. H4 control.
DISCUSSION
TZD-induced fluid retention is a significant adverse effect that has limited the clinical utility and potentially contributes to the increased mortality and morbidity associated with the use of PPARγ ligands (31). Studies in mice with conditional knockout of PPARγ have supported the hypothesis that expression of PPARγ contributes to the edema (11, 59); however, controversy remains regarding the precise mechanisms underlying this treatment-limiting side effect. Several studies have suggested that PPARγ-dependent fluid retention is related to upregulation of ENaC expression and function (11, 16); however, to date, no direct transcriptional studies have been performed (47) and investigators have not shown that PPARγ ligand treatment is associated with increases in kidney ENaCγ mRNA or protein expression (11). Furthermore, growing evidence suggests that PPARγ agonists do not directly enhance ENaC activity or expression (32, 36, 45, 52, 54), and mice with knockdown of ENaCα in the collecting duct still develop edema in response to TZD treatment (52). In our studies, we were consistently able to show fluid retention associated with TZDs; however, this was not associated with renal upregulation of ENaC isoform mRNA and protein subunits as previously posited. Therefore, the following study was undertaken to evaluate the transcriptional regulation of ENaC subunits.
Our first set of our studies supports previous work demonstrating a rapid increase in fluid retention associated with TZD feeding. This was not associated with a reduction in plasma aldosterone levels (19, 45, 52), which could influence ENaC expression (28). In the renal cortex, we observed declines in ENaCα and ENaCβ mRNA expression despite a lack of significant alterations in medullary ENaC mRNA expression. Our studies suggest that PPARγ ligands could regulate ENaC mRNA expression differentially in the cortex than the medulla. Intrarenal heterogeneity of ENaC mRNA regulation has been previously observed and may be related to the diverse roles that each renal segment plays in Na and K balance (48). Early studies demonstrated the highest expression of PPARγ in the medullary collecting ducts (12, 57); however, more recent studies confirmed expression of PPARγ1 in the cortex (42). We propose that regional differences in TZD activity may be influenced by the relative amounts of PPARγ expressed in various renal segments.
In vivo membrane protein expression of ENaC isoforms was not increased (39, 45, 52). ENaC activity is highly regulated on multiple levels, including transcriptional, posttranscriptional, translational, and posttranslational processing, membrane trafficking, and regulation of the channel-opening probability (14, 18, 38, 51, 52). Given the complexity of ENaC expression, it is not surprising that there is not a direct correlation of the mRNA and protein expression. However, we did observe a downregulation of ENaCγ protein in the cortex of the RGZ-fed mice, which is similar to findings of Riazi et al. (39) in RGZ-fed lean Zucker rats.
To avoid potential chronic compensatory changes in ENaC expression related to volume expansion, and to evaluate the acute effects of another PPARγ ligand, we treated mice for 5 h with pioglitazone. We observed a rapid decline in urinary Na/K ratio in response to pioglitazone that was primarily the consequence of lower urinary Na concentrations, whereas urinary K was not changed. This pattern may reflect effects of pioglitazone on other Na-retaining segments where Na reabsorption is not directly linked to K secretion, i.e., the proximal tubule (6, 34, 41) or a proposed 23-pS nonselective cation channel in the inner medulla (52). Moreover, these acute physiological changes were not associated with increases in ENaC isoform or Sgk-1 expression.
Sgk-1 (21, 55) phosphorylates and inactivates the ubiquitin ligase Nedd4–2 (5, 44), thereby increasing ENaC abundance in the cell membrane (2, 20, 21). Sgk-1 also has effects on ENaC opening probability (53). Early studies suggested that PPARγ ligands induce mRNA expression of Sgk-1 (1, 16, 44). We observed in vivo that TZDs do not significantly alter Sgk-1 mRNA expression in the cortical or medullary kidney, nor did we observe an increase in the abundance of ENaC isoforms in membrane preparations. Moreover, knockout of Sgk-1 in mice attenuated but did not fully account for volume retention due to pioglitazone feeding (1). However, these investigators observed increased Sgk-1 mRNA and protein expression in the pioglitazone-fed mice (1), indicating the potential influence of different experimental conditions. Unfortunately, these investigators did not evaluate renal expression of ENaC isoforms (1). Our own studies did not reveal an effect of Sgk-1 knockout in mice on pioglitazone- or RGZ-induced fluid retention or body weight increase (Vallon V, unpublished observation). Interestingly, in M1 cells we saw augmentation of Sgk-1 mRNA expression associated with PPARγ agonists. However, in mpkCCD cells, PPARγ ligands did not increase Sgk-1 mRNA or activity (32, 54). Moreover, treatment with RGZ or pioglitazone for 10 days did not significantly alter the opening probability or number of ENaC channels per patch in isolated, split-open cortical collecting ducts of wild-type mice (52). Therefore, we do not support the hypothesis that in vivo PPARγ ligands increase Sgk-1 activity to enhance ENaC activity.
To evaluate the mechanisms underlying PPARγ-dependent alterations in ENaC mRNA expression, and given the previous observations of PPARγ-dependent augmentation of ENaCγ expression (11), we searched for an appropriate model system that could support PPARγ-dependent transactivation (Fig. 6) (40). The model system also needed to demonstrate a PPARγ ligand-dependent reduction in ENaCα mRNA expression, to correlate with our in vivo findings. Interestingly, in the M1 cells pioglitazone also decreased ENaCγ mRNA expression, which is similar to preliminary findings of Soodvilai et al. (46) in inner medullary collecting duct (IMCD) cells. It is intriguing that in our in vivo studies, the only decline in ENaC isoform protein expression was in the cortical 85-kDa ENaCγ subunit. These findings suggest that our animal studies might not have been sensitive enough nor had the sufficient power to demonstrate a cortical decline in ENaCγ mRNA expression. To correlate our mRNA findings with functional studies, Nofziger and colleagues (32) observed a reduction in basal Na transport in M1 cells associated with application of GW7845, a potent PPARγ ligand.
In M1 cells, PPARγ ligands potently reduce ENaCα mRNA expression. However, there was no direct effect of PPARγ ligands on 3,000 bp of the proximal murine ENaCα promoter and GW9662 did not relieve repression of ENaCα mRNA expression (Fig. 9). These findings suggest that in M1 cells, inhibition of ENaCα may occur independent of PPARγ expression and may not be mediated by a direct transcriptional effect on the proximal promoter. PPARs can exert effects at genomic regions distant from the transcriptional start site or affect posttranscriptional processing of genes (9, 26). Figure 10 demonstrates that inhibition of protein synthesis increases ENaCα mRNA expression. Indeed, elegant studies in IMCD cells reveal that in basal conditions, ENaCα is repressed and derepression is associated with epigenetic modifications of the ENaCα gene (58, 60, 61).
Next, we observed PPARγ ligand-dependent repression of all of the ENaCγ promoter constructs, suggesting that repression is independent of the putative PPREs (Fig. 8D). However, activity of hENaCγ-p439-Luc was ∼40% greater than hENaCγ-p2926-Luc and cotransfection of PPARγ with hENaCγ-p2926-Luc repressed promoter activity independent of ligand addition (Figs. 8A and 9C). GW9662 did not relieve ligand-independent, PPARγ-dependent repression (Fig. 9C), arguing against the existence of endogenous ligands. These findings suggest that sequences between 2926 and 1248 in the human ENaCγ promoter may be important for ligand-independent, PPARγ-dependent repression. In a ligand-independent manner, PPARγ can bind to PPREs and repress gene expression. Administration of ligand can relieve repression and activate gene expression, a mechanism Guan et al. (11) proposed in their studies of TZD-induced activation of ENaCγ. This mechanism is not operational in our cellular system, as we have never observed TZD-induced activation of ENaCγ expression and there was no evidence of direct binding of PPARγ to the ENaCγ promoter. In contrast, our studies showed that TZD-induced repression of the ENaCγ proximal promoter and the first 100 bp of transcription are associated with a significant decrease in histone H4K5 acetylation. These findings are consistent with reduced transcriptional activity upon treatment with TZDs.
In summary, we showed that fluid retention in response to PPARγ ligand treatment does not require enhanced expression of ENaC in the kidney. In fact, we report that pioglitazone reduces ENaCα and ENaCβ mRNA and ENaCγ protein expression in kidney cortex. Moreover, we demonstrate that there are likely multiple mechanisms underlying PPARγ ligand-dependent repression of ENaCγ expression including an indirect transcriptional mechanism.
GRANTS
These studies were performed with the support of the Department of Veterans Affairs Career Development Transition Award and Merit Award and the University of California, San Diego Senate Grant awarded to R. Cunard and grants provided by the National Institutes of Health (R01HL094728, R01DK28602, and P30DK079337) and American Heart Association (GRNT3440038) to V. Vallon. The studies were also supported by a grant from Takeda Pharmaceuticals to R. Cunard and V. Vallon.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.B., V.P.-C.C., V.V., and R.C. performed experiments; E.B., V.P.-C.C., V.V., and R.C. analyzed data; E.B., C.K.G., V.V., and R.C. interpreted results of experiments; E.B. and R.C. drafted manuscript; E.B., C.K.G., V.V., and R.C. edited and revised manuscript; E.B., C.K.G., V.V., and R.C. approved final version of manuscript; C.K.G., V.V., and R.C. conception and design of research; V.V. and R.C. prepared figures.
ACKNOWLEDGMENTS
We thank Dr. André Dagenais, Centre Hospitalier de L'Université de Montréal for the murine ENaCα promoter and Dr. Christie P. Thomas, University of Iowa Health Care for the human ENaCγ promoters (−2926 and −1248). We also thank Nazila Sabri for technical work.
REFERENCES
- 1. Artunc F, Sandulache D, Nasir O, Boini KM, Friedrich B, Beier N, Dicks E, Potzsch S, Klingel K, Amann K, Blazer-Yost BL, Scholz W, Risler T, Kuhl D, Lang F. Lack of the serum and glucocorticoid-inducible kinase SGK1 attenuates the volume retention after treatment with the PPARgamma agonist pioglitazone. Pflügers Arch 456: 425–436, 2008. [DOI] [PubMed] [Google Scholar]
- 2. Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA 96: 2514–2519, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cunard R, Eto Y, Muljadi JT, Glass CK, Kelly CJ, Ricote M. Repression of IFN-gamma expression by peroxisome proliferator-activated receptor gamma. J Immunol 172: 7530–7536, 2004. [DOI] [PubMed] [Google Scholar]
- 4. Dagenais A, Denis C, Vives MF, Girouard S, Masse C, Nguyen T, Yamagata T, Grygorczyk C, Kothary R, Berthiaume Y. Modulation of α-ENaC and α1-Na+-K+-ATPase by cAMP and dexamethasone in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 281: L217–L230, 2001. [DOI] [PubMed] [Google Scholar]
- 5. Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4–2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20: 7052–7059, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Endo Y, Suzuki M, Yamada H, Horita S, Kunimi M, Yamazaki O, Shirai A, Nakamura M, Iso ON, Li Y, Hara M, Tsukamoto K, Moriyama N, Kudo A, Kawakami H, Yamauchi T, Kubota N, Kadowaki T, Kume H, Enomoto Y, Homma Y, Seki G, Fujita T. Thiazolidinediones enhance sodium-coupled bicarbonate absorption from renal proximal tubules via PPARgamma-dependent nongenomic signaling. Cell Metab 13: 550–561, 2011. [DOI] [PubMed] [Google Scholar]
- 7. Ergonul Z, Frindt G, Palmer LG. Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol 291: F683–F693, 2006. [DOI] [PubMed] [Google Scholar]
- 8. Frindt G, Palmer LG. Na channels in the rat connecting tubule. Am J Physiol Renal Physiol 286: F669–F674, 2004. [DOI] [PubMed] [Google Scholar]
- 9. Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14: 121–141, 2000. [PubMed] [Google Scholar]
- 10. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol 10: 365–376, 2010. [DOI] [PubMed] [Google Scholar]
- 11. Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med 11: 861–866, 2005. [DOI] [PubMed] [Google Scholar]
- 12. Guan Y, Zhang Y, Davis L, Breyer MD. Expression of peroxisome proliferator-activated receptors in urinary tract of rabbits and humans. Am J Physiol Renal Physiol 273: F1013–F1022, 1997. [DOI] [PubMed] [Google Scholar]
- 13. Guan Y, Zhang Y, Schneider A, Davis L, Breyer RM, Breyer MD. Peroxisome proliferator-activated receptor-γ activity is associated with renal microvasculature. Am J Physiol Renal Physiol 281: F1036–F1046, 2001. [DOI] [PubMed] [Google Scholar]
- 14. Hamm LL, Feng Z, Hering-Smith KS. Regulation of sodium transport by ENaC in the kidney. Curr Opin Nephrol Hypertens 19: 98–105, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138: 129–145, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hong G, Lockhart A, Davis B, Rahmoune H, Baker S, Ye L, Thompson P, Shou Y, O'Shaughnessy K, Ronco P, Brown J. PPARgamma activation enhances cell surface ENaCalpha via upregulation of SGK1 in human collecting duct cells. FASEB J 17: 1966–1968, 2003. [DOI] [PubMed] [Google Scholar]
- 17. Ijpenberg A, Jeannin E, Wahli W, Desvergne B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J Biol Chem 272: 20108–20117, 1997. [DOI] [PubMed] [Google Scholar]
- 18. Kashlan OB, Kleyman TR. ENaC structure and function in the wake of a resolved structure of a family member. Am J Physiol Renal Physiol 301: F684–F696, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khan O, Riazi S, Hu X, Song J, Wade JB, Ecelbarger CA. Regulation of the renal thiazide-sensitive Na-Cl cotransporter, blood pressure, and natriuresis in obese Zucker rats treated with rosiglitazone. Am J Physiol Renal Physiol 289: F442–F450, 2005. [DOI] [PubMed] [Google Scholar]
- 20. Lang F, Henke G, Embark HM, Waldegger S, Palmada M, Bohmer C, Vallon V. Regulation of channels by the serum and glucocorticoid-inducible kinase–implications for transport, excitability and cell proliferation. Cell Physiol Biochem 13: 41–50, 2003. [DOI] [PubMed] [Google Scholar]
- 21. Lang F, Huang DY, Vallon V. SGK, renal function and hypertension. J Nephrol 23, Suppl 16: S124–S129, 2010. [PMC free article] [PubMed] [Google Scholar]
- 22. Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, Plunket KD, Shenk JL, Stimmel JB, Therapontos C, Willson TM, Blanchard SG. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry 41: 6640–6650, 2002. [DOI] [PubMed] [Google Scholar]
- 23. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 270: 12953–12956, 1995. [DOI] [PubMed] [Google Scholar]
- 24. Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflügers Arch 458: 111–135, 2009. [DOI] [PubMed] [Google Scholar]
- 25. Lourdel S, Loffing J, Favre G, Paulais M, Nissant A, Fakitsas P, Creminon C, Feraille E, Verrey F, Teulon J, Doucet A, Deschenes G. Hyperaldosteronemia and activation of the epithelial sodium channel are not required for sodium retention in puromycin-induced nephrosis. J Am Soc Nephrol 16: 3642–3650, 2005. [DOI] [PubMed] [Google Scholar]
- 26. Luconi M, Cantini G, Serio M. Peroxisome proliferator-activated receptor gamma (PPARgamma): is the genomic activity the only answer? Steroids 75: 585–594, 2010. [DOI] [PubMed] [Google Scholar]
- 27. Marx N, Schonbeck U, Lazar MA, Libby P, Plutzky J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res 83: 1097–1103, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mudaliar S, Chang AR, Aroda VR, Chao E, Burke P, Baxi S, Griver KA, O'Connor DT, Henry RR. Effects of intensive insulin therapy alone and with added pioglitazone on renal salt/water balance and fluid compartment shifts in type 2 diabetes. Diabetes Obes Metab 12: 133–138, 2010. [DOI] [PubMed] [Google Scholar]
- 30. Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, Le Winter M, Porte D, Semenkovich CF, Smith S, Young LH, Kahn R. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Circulation 108: 2941–2948, 2003. [DOI] [PubMed] [Google Scholar]
- 31. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 356: 2457–2471, 2007. [DOI] [PubMed] [Google Scholar]
- 32. Nofziger C, Chen L, Shane MA, Smith CD, Brown KK, Blazer-Yost BL. PPARγ agonists do not directly enhance basal or insulin-stimulated Na+ transport via the epithelial Na+ channel. Pflügers Arch 451: 445–453, 2005. [DOI] [PubMed] [Google Scholar]
- 33. Palmer CN, Hsu MH, Griffin HJ, Johnson EF. Novel sequence determinants in peroxisome proliferator signaling. J Biol Chem 270: 16114–16121, 1995. [DOI] [PubMed] [Google Scholar]
- 34. Panchapakesan U, Pollock C, Saad S. Review article: importance of the kidney proximal tubular cells in thiazolidinedione-mediated sodium and water uptake. Nephrology (Carlton) 14: 298–301, 2009. [DOI] [PubMed] [Google Scholar]
- 35. Patel C, Wyne KL, McGuire DK. Thiazolidinediones, peripheral oedema and congestive heart failure: what is the evidence? Diab Vasc Dis Res 2: 61–66, 2005. [DOI] [PubMed] [Google Scholar]
- 36. Pavlov TS, Levchenko V, Karpushev AV, Vandewalle A, Staruschenko A. Peroxisome proliferator-activated receptor gamma antagonists decrease Na+ transport via the epithelial Na+ channel. Mol Pharmacol 76: 1333–1340, 2009. [DOI] [PubMed] [Google Scholar]
- 37. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD, Vallon V. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J 24: 2056–2065, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riazi S, Khan O, Tiwari S, Hu X, Ecelbarger CA. Rosiglitazone regulates ENaC and Na-K-2Cl cotransporter (NKCC2) abundance in the obese Zucker rat. Am J Nephrol 26: 245–257, 2006. [DOI] [PubMed] [Google Scholar]
- 40. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391: 79–82, 1998. [DOI] [PubMed] [Google Scholar]
- 41. Saad S, Agapiou DJ, Chen XM, Stevens V, Pollock CA. The role of Sgk-1 in the upregulation of transport proteins by PPAR-γ agonists in human proximal tubule cells. Nephrol Dial Transplant 24: 1130–1141, 2009. [DOI] [PubMed] [Google Scholar]
- 42. Sato K, Sugawara A, Kudo M, Uruno A, Ito S, Takeuchi K. Expression of peroxisome proliferator-activated receptor isoform proteins in the rat kidney. Hypertens Res 27: 417–425, 2004. [DOI] [PubMed] [Google Scholar]
- 43. Selim E, Frkanec JT, Cunard R. Fibrates upregulate TRB3 in lymphocytes independent of PPAR alpha by augmenting CCAAT/enhancer-binding protein beta (C/EBP beta) expression. Mol Immunol 44: 1218–1229, 2007. [DOI] [PubMed] [Google Scholar]
- 44. Snyder PM, Olson DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4–2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277: 5–8, 2002. [DOI] [PubMed] [Google Scholar]
- 45. Song J, Knepper MA, Hu X, Verbalis JG, Ecelbarger CA. Rosiglitazone activates renal sodium- and water-reabsorptive pathways and lowers blood pressure in normal rats. J Pharmacol Exp Ther 308: 426–433, 2004. [DOI] [PubMed] [Google Scholar]
- 46. Soodvilai S, Aoyagi K, Yang T. PPARgamma stimulates paracellular transport and inhibits ENaC in primary inner medullary collecting duct cells. J Am Soc Nephrol 20: 123A, 2009.19092126 [Google Scholar]
- 47. Staels B. Fluid retention mediated by renal PPARgamma. Cell Metab 2: 77–78, 2005. [DOI] [PubMed] [Google Scholar]
- 48. Stokes JB, Sigmund RD. Regulation of rENaC mRNA by dietary NaCl and steroids: organ, tissue, and steroid heterogeneity. Am J Physiol Cell Physiol 274: C1699–C1707, 1998. [DOI] [PubMed] [Google Scholar]
- 49. Stoos BA, Naray-Fejes-Toth A, Carretero OA, Ito S, Fejes-Toth G. Characterization of a mouse cortical collecting duct cell line. Kidney Int 39: 1168–1175, 1991. [DOI] [PubMed] [Google Scholar]
- 50. Thomas CP, Doggett NA, Fisher R, Stokes JB. Genomic organization and the 5′ flanking region of the gamma subunit of the human amiloride-sensitive epithelial sodium channel. J Biol Chem 271: 26062–26066, 1996. [DOI] [PubMed] [Google Scholar]
- 51. Thomas CP, Itani OA. New insights into epithelial sodium channel function in the kidney: site of action, regulation by ubiquitin ligases, serum- and glucocorticoid-inducible kinase and proteolysis. Curr Opin Nephrol Hypertens 13: 541–548, 2004. [DOI] [PubMed] [Google Scholar]
- 52. Vallon V, Hummler E, Rieg T, Pochynyuk O, Bugaj V, Schroth J, Dechenes G, Rossier B, Cunard R, Stockand J. Thiazolidinedione-induced fluid retention is independent of collecting duct alphaENaC activity. J Am Soc Nephrol 20: 721–729, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J Gen Physiol 120: 191–201, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilson SM, Mansley MK, Getty J, Husband EM, Inglis SK, Hansen MK. Effects of peroxisome proliferator-activated receptor gamma agonists on Na+ transport and activity of the kinase SGK1 in epithelial cells from lung and kidney. Br J Pharmacol 159: 678–688, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, Jansen M, Schlunz M, Klingel K, Loffing J, Kauselmann G, Bosl MR, Lang F, Kuhl D. Impaired renal Na+ retention in the sgk1-knockout mouse. J Clin Invest 110: 1263–1268, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang HC, Ma LJ, Ma J, Fogo AB. Peroxisome proliferator-activated receptor-gamma agonist is protective in podocyte injury-associated sclerosis. Kidney Int 69: 1756–1764, 2006. [DOI] [PubMed] [Google Scholar]
- 57. Yang T, Michele DE, Park J, Smart AM, Lin Z, Brosius FC, III, Schnermann JB, Briggs JP. Expression of peroxisomal proliferator-activated receptors and retinoid X receptors in the kidney. Am J Physiol Renal Physiol 277: F966–F973, 1999. [DOI] [PubMed] [Google Scholar]
- 58. Zhang D, Yu ZY, Cruz P, Kong Q, Li S, Kone BC. Epigenetics and the control of epithelial sodium channel expression in collecting duct. Kidney Int 75: 260–267, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang H, Zhang A, Kohan DE, Nelson RD, Gonzalez FJ, Yang T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc Natl Acad Sci USA 102: 9406–9411, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem 281: 18059–18068, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest 117: 773–783, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]