

Abstract
We previously identified a region of recurrent amplification on chromosome 22q11.21 in a subset of primary lung adenocarcinomas. Here we show that CRKL, encoding for an adaptor protein, is amplified and overexpressed in non-small cell lung cancer (NSCLC) cells that harbor 22q11.21 amplifications. Overexpression of CRKL in immortalized human airway epithelial cells promoted anchorage independent growth and tumorigenicity. Oncogenic CRKL activates SOS1-RAS-RAF-ERK and SRC-C3G-RAP1 pathways. Suppression of CRKL in NSCLC cells that harbor CRKL amplifications induced cell death. Overexpression of CRKL in EGFR mutant cells induces resistance to gefitinib by activating ERK and AKT signaling. We identified CRKL amplification in an EGFR inhibitor treated lung adenocarcinoma that was not present prior to treatment. These observations show that CRKL overexpression induces cell transformation, credential CRKL as a therapeutic target for a subset of NSCLC that harbor CRKL amplifications and implicate CRKL as an additional mechanism of resistance to EGFR-directed therapy.
Keywords: non small cell lung cancer, CRKL, cell transformation, RAP1, oncogene
Introduction
Although the prognosis for non small cell lung cancer (NSCLC) patients that present with late stage disease remains poor, somatic mutations of the epidermal growth factor receptor (EGFR) serve as predictive biomarkers for both clinical response and survival in patients who receive EGFR inhibitors (1–7). Moreover, recent work indicates that mutations or translocations of the ALK tyrosine kinase also occur in a subset of NSCLC (8, 9), and tumors that harbor such mutations are sensitive to ALK inhibitors (10, 11). Collectively, these studies suggest that identifying and characterizing genetic alterations in NSCLC will provide new targets for therapeutic strategies.
Prior work has identified 57 recurrent events of genomic gain or loss in primary NSCLC (12, 13). Asmall number of these recurrent genomic events has been found to harbor known and novel oncogenes and tumor suppressor genes, including amplification or mutation in EGFR, KRAS, MYC, MDM2, TERT, CCND1, CCNE1 and NKX2-1, and deletions of CDKN2A/B and PTEN (12, 14–17). However, for many of these recurrently amplified regions, the target gene(s) believed to drive cancer pathogenesis remains to be identified and validated (12, 14–17). For example, chromosome 22q11.21 is focally amplified in 3% of lung adenocarcinoma samples and the peak region contains 15 genes, including CRKL (v-crk sarcoma virus CT10 oncogene homolog (avian)-like) (12, 16, 17). Recurrent amplifications of 22q11.21 have not been described in squamous cell lung carcinomas (18) and small cell lung carcinomas (19).
CRKL is a member of adaptor proteins that participate in signal transduction in response to both extracellular and intracellular stimuli, such as growth factors, cytokines and the oncogenic BCR-ABL fusion protein (20, 21). CRKL consists of an N-terminal Src homology 2 (SH2) domain followed by two SH3 domains (SH2-SH3N-SH3C) (20). The SH2 domain of CRKL binds to phosphorylated Y-x-x-P motif present in many docking proteins, such as BCAR1 (also known as p130CAS), Paxillin and GAB (20, 21), whereas the SH3N domain binds to proline-rich P-x-x-P-x-K motif-containing proteins, such as Son of Sevenless (SOS), RAPGEF1 (also known as C3G) (22), p85 (23), ABL1 and BCR-ABL (24, 25). Through these interactions, CRKL facilitates the timely and localized formation of protein complexes required for signal transduction in many biological processes including cell proliferation, survival, adhesion and migration (20, 21).
CRKL and several proteins that interact with CRKL have been implicated in cancer. Overexpression of CRKL in Rat-1 fibroblast cells has been shown to promote anchorage independent growth but the signaling pathways necessary for this phenotype remains undefined (26, 27). Activating mutations of ALK have been shown to activate RAP1 through CRKL-C3G complexes in neuroblastomas (28). Moreover, expression of the oncogenic RET-PTC1 fusion protein led to increased RAP1 activity while expression of a dominant interfering RAP1N17 inhibited proliferation of papillary thyroid carcinoma cells (29). However, it remains unclear how overexpression of CRKL affects C3G-RAP1 signaling and whether RAP1 signaling plays a role in proliferation/survival and transformation of NSCLC cells.
In prior work, we and others showed that a subset of NSCLC are dependent on CRKL expression for proliferation (17, 30). Moreover, overexpression of CRKL in immortalized human lung epithelial cells promoted EGF independent proliferation (17). Here we credential CRKL as an oncogene in NSCLC that transforms human lung epithelial cells through the coordinate activation of the RAS and RAP1 pathways and that is involved in resistance to EGFR inhibitors.
Results
Amplification and overexpression of CRKL gene in NSCLC cells
In prior work, we and others found recurrent focal copy number gain at chromosome 22q11.21 involving CRKL in 3% of 371 primary lung adenocarcinomas, with another 13% of tumors exhibiting broad copy number gain spanning that region (12, 17). To identify other NSCLC cell lines that harbor copy number gain of this region, we examined a panel of 84 NSCLC cell lines that had been characterized by high-density single nucleotide polymorphism (SNP) arrays (19, 31) and identified 6 cell lines with high-level focal amplifications of 22q11.21 (Figure 1A). We confirmed that CRKL was amplified by fluorescence in situ hybridization (FISH) in three of these cell lines, HCC515, H1819 and H1755 cells (Figure 1B). In contrast, in the H1833 cell line in which we found normal 22q copy number (Figure 1A), we detected only two copies of the CRKL gene (Figure 1B). To confirm these findings, we also performed quantitative PCR to measure the copy number of CRKL and detected 12 to 18 copies of CRKL in NSCLC cell lines that harbored 22q11.21 amplification (Figure 1C).
Figure 1. Amplification and overexpression of CRKL gene in NSCLC cell lines that harbored amplification of 22q11.21.
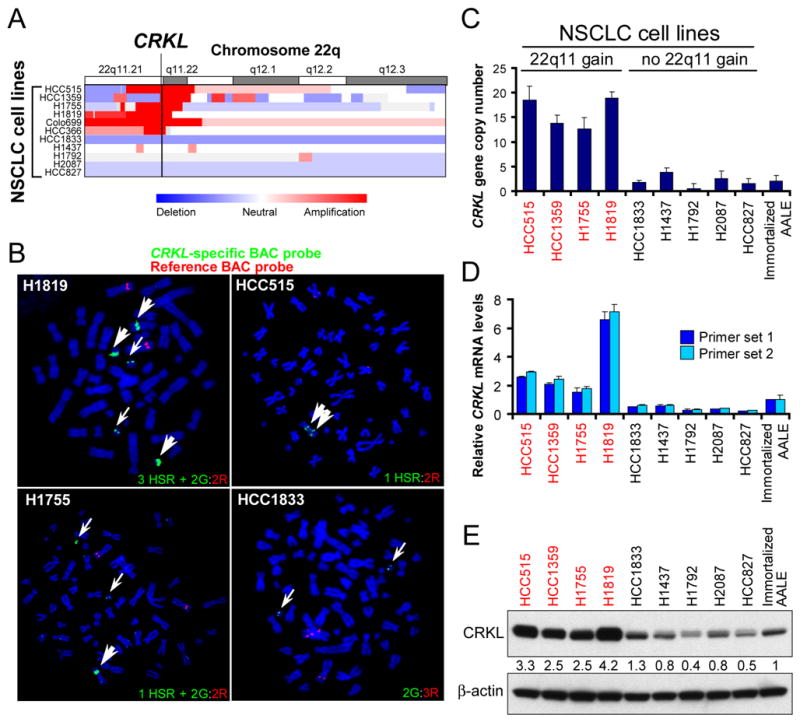
(A) SNP array colorgram showing genomic amplification of chromosome 22q11.21 in NSCLC cell lines. Regions of genomic amplification and deletion are denoted in red and blue, respectively.
(B) FISH of NSCLC cells using a CRKL-specific probe (green, G) and a reference probe (red, R). HSR denotes a homogenously staining region.
(C) Quantitative PCR analysis of CRKL copy number in NSCLC cells. CRKL copy number in NSCLC cells was normalized to LINE-1 and immortalized AALE cells using the standard curve method. Data represent mean + s.d. of triplicate measurements.
(D) Quantitative RT-PCR analysis of CRKL mRNA levels in NSCLC cell lines and immortalized AALE cells. Data represent mean + s.d. of triplicate measurements.
(E) Immunoblot of CRKL in NSCLC cell lines and immortalized AALE cells. β-actin was used as a loading control. Relative intensity of CRKL levels is determined.
To determine whether this observed gene amplification correlates with increased CRKL expression, we examined expression data from the same panel of NSCLC cell lines (31) and observed that each of the cell lines that harbored 22q11.21 amplification expressed high levels of CRKL (Figure S1). Using quantitative RT-PCR, we detected a 2-7-fold increase in CRKL mRNA levels in NSCLC cells that harbored 22q11.21 amplification compared to immortalized but non-tumorigenic human airway epithelial cells (AALE) (32) (Figure 1D). Correlating with these observed transcript levels, CRKL protein expression was also increased (2.5–4.2 fold increase) in NSCLC cells that harbored 22q.11.21 amplification (Figure 1E and S2A). We note that in addition to CRKL, 4 genes within the amplicon, PI4KA, ZNF74, THAP7 and LZTR1, were also expressed at higher levels in samples that harbored 22q11.21 amplification (Figure S1). In addition, looking at gene expression data, we found that CRKL was expressed at high levels in a subset of cell lines that did not harbor 22q11.21 amplification (Figure S1C). We confirmed that CRKL protein expression was increased in these cell lines compared to AALE cells, suggesting that mechanisms other than gene amplification may also lead to CRKL overexpression. These observations confirm that CRKL is highly amplified and overexpressed in a subset of NSCLC cells.
NSCLC cells that harbor 22q11.21 amplification are dependent on CRKL
We and others showed that a subset of NSCLC are dependent on CRKL expression for proliferation (17, 30). To examine whether NSCLC cells that harbor the 22q11.21 amplification require CRKL expression for cell proliferation or survival, we tested the effects of suppressing CRKL on the proliferation of several NSCLC cells and AALE cells. We found that CRKL suppression by two independent shRNAs substantially decreased the proliferation of NSCLC cell lines that harbored 22q11.21 gain (HCC515, HCC1359, H1755, H1819, HCC366 and Colo699) (Figure 2A and S2B) and that exhibited CRKL overexpression but not 22q11.21 gain (H1915, H2009, H28 and H1299) (Figure S2A and S2B). In contrast, suppression of CRKL in two NSCLC cell lines (H1833 and H1792) that have normal copy number at 22q11.21 and express lower levels of CRKL were relatively insensitive to CRKL suppression (p=0.0001 and p=0.0056 for shCRKL#1 and shCRKL#2, respectively, compared to cell lines that harbored 22q11.21 gain, t-test; Figure 2A). In addition, the proliferation of immortalized AALE cells, which also exhibit normal copy number at 22q11.21, were unaffected by CRKL suppression (Figure 2A).
Figure 2. Suppression of CRKL decreases the proliferation of NSCLC cells that harbor 22q11.21 amplifications.
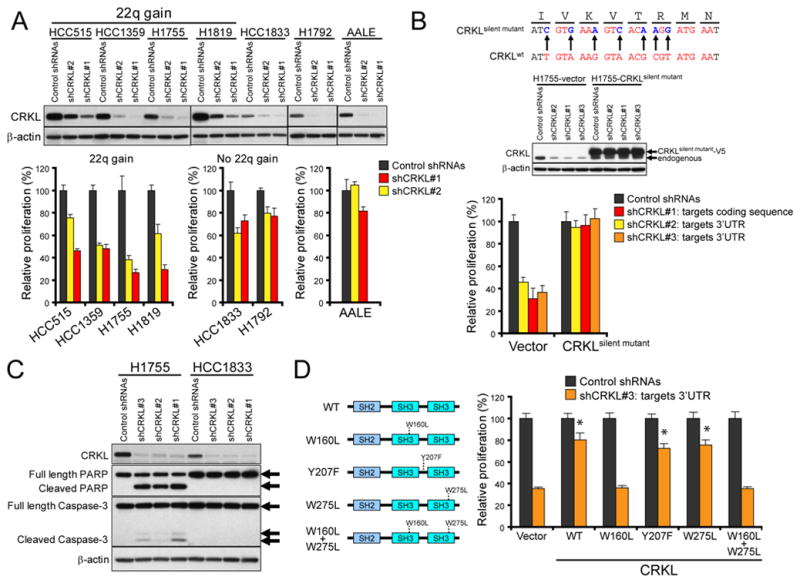
(A) Effect of CRKL suppression on relative proliferation of NSCLC cell lines and AALE cells. Top, Immunoblot of CRKL proteins in each cell line expressing control shRNAs or two distinct CRKL-specific shRNAs. Bottom, Cell proliferation 6 d after infection with indicated shRNAs for each cell line. Data represent mean + s.d of six replicate measurements.
(B) Effect of expression of shRNA-resistant CRKL on the proliferation of H1755 cells expressing CRKL-specific shRNAs. Top, Generation of a shRNA-resistant CRKL cDNA (CRKLsilent mutant). The 21-mer sequence targeted by shCRKL#1 is marked in red color. Middle, Immunoblot of CRKL 4 d after infection with indicated shRNAs in H1755 cells expressing a control vector or V5-epitope tagged CRKLsilent mutant. Bottom, Effect of CRKL suppression by CRKL-targeting shRNAs on the proliferation of H1755 cells expressing a control vector or CRKLsilent mutant. The shCRKL#2 and shCRKL#3 target the 3′UTR of endogenous CRKL mRNA.
(C) Effect of CRKL suppression on cleavage of PARP and Caspase-3 proteins in NSCLC cells. Immunoblots of PARP and Caspase-3 proteins in H1755 and HCC1833 cells expressing the indicated shRNAs.
(D) Effects of mutations on CRKL function in H1755 cells. Left, Schematic of CRKL mutants. Right, Effect of expressing wild-type or mutant CRKL on the proliferation of the H1755 cells expressing a CRKL-specific shRNA (shCRKL#3). Data represent mean + s.d. of six replicate measurements. * indicates p<0.0001 as compared to cells expressing a control vector and shCRKL#3.
To confirm that the observed effects of these shRNAs on proliferation were specific for CRKL, we constructed a shRNA-resistant CRKL mutant (CRKLsilent mutant) which contained multiple nucleotide substitutions to the targeting sequence of shCRKL#1 that do not change the amino acid sequence of CRKL (Figure 2B). We then tested whether expression of the CRKLsilent mutant rescued the decreased proliferation induced by shCRKL#1 and two other shRNAs that target the 3′ untranslated region (3′UTR) of CRKL mRNA (shCRKL#2 and shCRKL#3). We found that expression of the CRKLsilent mutant permitted H1755 cells expressing each of these CRKL-targeting shRNAs to proliferate, whereas the expression of a control vector failed to rescue such cells (Figure 2B). These observations extend prior studies (17) and show that NSCLC cells that harbor amplifications of 22q11.21 are particularly dependent on CRKL expression for proliferation, suggesting that, like other oncogenes, amplification of CRKL induces oncogene addiction (33).
To determine whether CRKL suppression induced apoptosis, we performed immunoblotting for PARP and Caspase-3. We found that suppression of CRKL in H1755 cells that harbored 22q11.21 amplifications resulted in the cleavage of PARP and Caspase-3 (Figure 2C). In contrast, no cleaved forms of PARP and Caspase-3 were detected after CRKL suppression in HCC1833 cells that did not harbor 22q11.21 gain. These findings show that suppression of CRKL induced apoptotic cell death in NSCLC cells that harbored CRKL amplifications.
We next investigated which domains were essential for CRKL-induced oncogene addiction. Specifically, we generated CRKL mutants that harbor amino acid substitutions predicted to disrupt the function of the SH3N domain (W160L), the SH3C domain (W275L) or Tyr207 phosphorylation (Y207F) (26). We then tested whether expression of these CRKL mutants rescued the decreased cell proliferation induced by CRKL suppression. We first introduced a control vector or each of these CRKL mutants into H1755 cells and then introduced a CRKL-specific shRNA (shCRKL#3) that targets the 3′UTR of CRKL mRNA (Figure S2C). We found that expression of exogenous wild-type CRKL, CRKLY207F or CRKLW275L mutants permitted cells expressing the CRKL-specific shRNA to proliferate (Figure 2D), whereas the expression of the SH3N mutants (either CRKLW160L or CRKLW160L+Y207F) or a control vector failed to rescue such cells (Figure 2D). These findings demonstrate that the SH3N domain is essential for CRKL function in NSCLC cell lines that harbor increased copy number of 22q11.21.
Suppression of CRKL in NSCLC cells inhibited tumor growth
To examine whether CRKL suppression affected tumorigenic growth of NSCLC cells that harbor CRKL amplification, we confirmed that HCC515 cells were able to form tumors after subcutaneous implantation into immunodeficient mice. To enable a noninvasive monitoring of tumor growth by luminescent imaging, we generated a polyclonal population of HCC515 cells stably expressing luciferase. We then introduced either a scrambled control shRNA or a validated CRKL-specific shRNA (shCRKL#1) in vectors where the expression of shRNA was under the control of a doxycycline-inducible promoter. Doxycycline treatment of cells that harbor the inducible shRNA (shCRKL#1) induced decreased CRKL protein levels compared to cells grown in the absence of doxycycline (Figure 3A). In contrast, doxycycline treatment failed to affect CRKL protein levels in cells expressing the scrambled control shRNA. We then injected cells expressing the inducible control shRNA or the CRKL-specific shRNA subcutaneously into opposite flanks of the same immunodeficient mice and allowed them to form tumors (65 mm3 in volume, 11 days) (Figure 3B). After 3 weeks, we observed that in mice feeding on chow containing doxycycline, tumors expressing the control shRNA grew into large tumors while tumors expressing the CRKL-specific shRNA exhibited growth arrest (Figure 3C and 3D). As expected, tumors in mice fed with normal chow grew into large tumors with comparable size (Figure 3C and 3D). To determine whether CRKL suppression induced apoptotic cell death in tumors, we performed TUNEL staining and detected a significantly increased percentage of TUNEL-positive cells in tumors expressing CRKL-specific shRNA compared to tumors expressing a control shRNA (Figure 3E). Together, these findings demonstrate that CRKL is required for tumorigenic growth of NSCLC cells that harbor CRKL amplifications.
Figure 3. Effects of suppressing CRKL on NSCLC tumorigenicity.

(A) Generation of NSCLC cell lines expressing a doxycycline (DOX)-inducible CRKL-specific shRNA. A scrambled control shRNA or a validated CRKL-specific shRNA (shCRKL#1) under the control of DOX-inducible promoter were introduced into HCC515 cells. Immunoblot of CRKL proteins in cells in the absence or presence of 200 ng/ml DOX for 96 h.
(B) Experimental scheme. Each HCC515 cell line expressing a DOX-inducible vectors encoding either shCRKL#1 or scrambled control shRNA was implanted subcutaneously into the opposite flanks of the same mouse (n=10). Tumors were allowed to grow for 11 d to attain a volume of ~65 mm3. Mice were then fed on normal chow (n=5) or DOX-containing chow (n=5) to induce shRNA expression. Tumor growth was measured by caliper and bioluminescent imaging.
(C) Effect of CRKL suppression on the growth of HCC515 xenografts. DOX-induced CRKL suppression induced growth arrest of tumors derived from cell lines as described in (A) and (B). Data represent mean ± s.d.
(D) Representative bioluminescent images of the growth of HCC515 xenografts. DOX-induced CRKL suppression induced growth arrest of tumors derived from cell lines as described in (A) and (B). Images shown are the day 11 images (before DOX) and day 32 images (with or without DOX).
(E) TUNEL staining of xenografts. Left, DOX-treated tumors expressing a control shRNA or shCRKL#1 were harvested on day 32 as described in (B) and stained for TUNEL (green) and DAPI (blue). Right, Quantification of percent of TUNEL-positive cells. Mean + s.d. are shown.
Overexpression of CRKL in immortalized human airway epithelial cells induces anchorage independent colony formation
Overexpression of CRKL promotes anchorage independent growth of Rat-1 fibroblast cells (27), but not NIH3T3 cells (34), demonstrating that transformation induced by CRKL expression is context-dependent. To dissect the mechanism and genetic context required for the transformation of NSCLC, we used human airway epithelial cells that can be rendered tumorigenic by the introduction of specific combinations of genes (AALE cells) (32) to test whether CRKL expression induces cell transformation. Specifically, we introduced a retroviral vector encoding CRKL or a control vector into AALE cells and confirmed that CRKL was expressed at levels comparable to those found in NSCLC cells that harbored CRKL amplification and overexpression (Figure 4A). We found that overexpression of CRKL in AALE cells led to >4-fold increased anchorage independent colony formation as compared to AALE cell lines expressing a control vector (Figure 4B). We also tested the ability of CRKL mutants that were predicted to disrupt the function of the SH3N domain (W160L), the SH3C domain (W275L) and Tyr207 phosphorylation (Y207F) to induce anchorage independent growth. We found that the CRKL SH3C and Tyr207 mutants (W275L, Y207F and Y207F+W275L) induced anchorage independent colony formation similarly to wild-type CRKL when overexpressed in AALE cells (Figure 4B). In contrast, the SH3N mutants (W160L, W160L+Y207F and W160L+W275L) failed to promote anchorage independent growth (Figure 4B). These findings suggest that overexpression of CRKL in AALE cells induces cell transformation that is dependent on the integrity of the SH3N domain.
Figure 4. The role of SRC-C3G-RAP1 signaling in transformation induced by CRKL.

(A) Immunoblot of CRKL in AALE cell lines overexpressing a control vector or CRKL, as compared to NSCLC cells that harbored CRKL amplifications. Relative intensity of bands is determined.
(B) Anchorage independent growth of AALE cell lines stably overexpressing a control vector, wild-type or mutant CRKL. Colony number indicates colonies greater than 0.2 mm in diameter 4 weeks after plating. Data represent mean + s.d. of six replicate determinations from two independent experiments. Wild-type CRKL and CRKL mutants that induce anchorage independent growth are marked in red.
(C) Overexpression of CRKL in AALE cells increased the phosphorylation of SRC. Immunoblot of phospho-Y419 SRC proteins in AALE cells overexpressing a control vector, CRKL or CRKLW160L mutant.
(D) Immunoblot of phospho-T185/Y187 ERK1/2 and phospho-S473 AKT in AALE cell lines overexpressing wildtype or mutant CRKL. Wild-type CRKL and CRKL mutants that induced anchorage independent growth in (B) are marked in red.
(E) Effect of CRKL overexpression on CRKL-C3G interaction and phosphorylation of C3G in AALE cells. CRKL immune complexes in AALE cells expressing indicated constructs were isolated and immunoblotting for phosphorylated C3G and total C3G proteins was performed using specific antibodies.
(F) Overexpression of CRKL in AALE cells increased RAP1 activity. Pull-down assay for GTP-bound RAP1 proteins were performed followed by immunoblotting for RAP1 proteins. Immunoblot of RAP1 proteins in total lysates before pull-down assay was used as loading control. Relative intensity of bands is determined.
(G) Effect of CRKL suppression on RAP1 activity in NSCLC cells that harbored amplification of CRKL. Pull-down assays were performed to assess the RAP1-GTP levels in NSCLC cells 4 d after infection with a control shRNA targeting GFP or a CRKL-specific shRNA (shCRKL#1) followed by immunoblotting for RAP1. Immunoblot of RAP1 proteins in total lysates was shown. Relative intensity of bands is shown.
(H) Effect of expressing RAP1 on cell proliferation of H1755 cells after CRKL suppression. Each H1755 cell line expressing a control vector, or wildtype RAP1A or RAP1B or constitutively active mutants, RAP1A63E or RAP1BV12, was infected with control shRNAs or a CRKL-specific shRNA (shCRKL#3). Cell proliferation was measured 6 d after infection with shRNA. Data represent mean + s.d. of six replicate measurements. * indicates p<0.0001 as compared to cells expressing a control vector and shCRKL#3.
(I) Effect of suppression of SRC, C3G or RAP1 on CRKL-induced anchorage independent growth. Left, Immunoblots of SRC, C3G or RAP1 proteins in CRKL-overexpressing AALE cells expressing a control shRNA targeting GFP or each gene-specific shRNA. The shRNAs that suppressed the target protein the best were marked in red color. The antibody for RAP1 detects both RAP1A and RAP1B. Right, Anchorage independent growth of AALE cells expressing indicated constructs. Colony number indicates colonies greater than 0.2 mm in diameter 4 weeks after plating. Data represent mean + s.d. of six replicate determinations from two independent experiments. * indicates p<0.001 as compared to cells expressing CRKL and shGFP.
(J) Effect of suppression of SRC, C3G or RAP1 on phospho-ERK1/2 levels in CRKL-overexpressing AALE cells. Immunoblots of phospho-ERK1/2 in CRKL-overexpressing AALE cells expressing indicated shRNAs. The shRNAs marked in red were validated in (I). (K) Effect of dasatinib treatment on anchorage independent growth of CRKL-overexpressing AALE cells. Anchorage independent growth of CRKL-overexpressing AALE cells that expressed wildtype SRC or mutant SRC or a control vector in the presence of dasatinib at indicated concentration. Colony number indicates colonies greater than 0.2 mm in diameter 4 weeks after plating. Mean + s.d. are shown. * indicates p<0.001 compared to cells expressing a control vector.
(L) Effect of dasatinib treatment on phospho-SRC and phospho-ERK1/2 in CRKL-overexpressing AALE cells. AALE cells expressing indicated constructs were exposed to dasatinib at indicated concentration for 6 h followed by immunoblotting.
CRKLinduces cell transformation by coordinately activating multiple signaling pathways
The SH3 domain of CRKL and other CRK family proteins interacts with many proline-rich motif-containing proteins, such as SOS, C3G (35, 36) and p85 (23), implicating CRKL in the regulation of several pathways including those regulated by RAS-MAPK (27, 37), C3G-RAP1 (38–42) and PI3K-AKT (23, 43, 44). Thus, we hypothesized that overexpression of CRKL induces cell transformation through coordinately activating several signaling pathways. To investigate the mechanism by which CRKL induces cell transformation in human NSCLC, we first determined whether CRKL overexpression alters the phosphorylation state of tyrosine kinases in AALE cells. Specifically, we applied a multiplex method that uses Luminex beads pre-coupled with protein-specific or phospho-specific antibodies (Figure S3) to isolate the majority of tyrosine kinases and other kinases followed by measurement of tyrosine phosphorylation levels by a phospho-tyrosine-specific antibody (4G10) (45). We detected significantly increased levels of phospho-T185/Y187 ERK1/2 and phospho-Y419 SRC proteins in AALE cells expressing wild-type CRKL compared to cells transfected with a control vector or CRKLW160L mutant that failed to induce cell transformation (p=0.0244 and p=0.0035, respectively, t-test; Figure S3). We confirmed that AALE cells stably overexpressing CRKL exhibited increased levels of phospho-Y419 SRC (Figure 4C) and phospho-T185/Y187 ERK1/2 (Figure 4D) compared to cells expressing a control vector by immunoblotting with phospho-specific antibodies. Moreover, CRKL mutants (Y207F, W275L and Y207F+W275L) that induced anchorage independent growth when overexpressed in AALE cells also led to increased levels of phospho-ERK1/2 whereas CRKL mutants (W160L, W160L+Y207F and W160L+W275L) that failed to induce anchorage independent growth failed to induce similar patterns of tyrosine phosphorylation (Figure 4D). In contrast, phospho-S473 AKT levels remained unchanged between cells overexpressing CRKL or a control vector (Figure 4D). These observations indicate that overexpression of CRKL in AALE cells results in constitutive activation of both SRC and ERK1/2 signaling.
We next investigated whether CRKL-induced SRC activation plays a role in transformation of AALE cells. CRKL and SRC have been shown to share many common substrates, such as p130CAS and Paxillin. Overexpression of CRKL in AALE cells also resulted in increased phosphorylation levels of p130CAS and Paxillin compared to cells expressing a control vector or CRKLW160L mutants (Figure S4A). Moreover, suppression of CRKL resulted in reduced phosphorylation levels of SRC and p130CAS at varied levels in NSCLC cells that harbored CRKL amplifications (Figure S4B). These results suggest that overexpression of CRKL may represent a mechanism for aberrant SRC activation and signaling in NSCLC cells.
Recently, activating point mutations of ALK have been shown to activate RAP1 through CRKL-C3G complexes in neuroblastomas (28). C3G, a guanine nucleotide exchange factor required for RAP1 activation, has been shown to form stable complexes with CRKL and other CRK family members (35, 36), and SRC has been implicated in activation of C3G-RAP1 through CRK family adaptors (46). We therefore investigated the role of RAP1 signaling in CRKL-induced transformation of AALE cells by determining whether overexpression of CRKL in AALE cells affected its interaction with C3G and phosphorylation status of C3G. We isolated CRKL immune complexes from AALE cells overexpressing CRKL and detected increased C3G bound to CRKL and, to a lesser extent, the CRKLW160L mutant, compared to cells expressing a control vector (Figure 4E). We noted that increased phosphorylation levels of C3G were detected only in CRKL immune complexes isolated from CRKL-overexpressing AALE cells by immunoblotting with phospho-specific antibody (Figure 4E). These observations suggest that overexpression of CRKL promotes the interaction with C3G and modulates C3G activity.
We next determined whether overexpression of CRKL in AALE cells also altered RAP1 activity by performing pull-down assays to assess the levels of GTP-bound RAP1. Overexpression of CRKL in AALE cells induced a substantially increased levels of RAP1-GTP (2.3-fold increase) compared to cells expressing a control vector or CRKLW160L mutant (Figure 4F). In addition, we found that suppression of CRKL in NSCLC cell lines that harbor CRKL amplifications resulted in reduction of both RAP1-GTP and total RAP1 levels (Figure 4G). Moreover, inhibition of SRC with dasatinib (0.1 μM) led to decreased RAP1-GTP levels in three NSCLC cell lines that harbored CRKL amplifications (Figure S4C). These findings indicate that CRKL regulates RAP1 activity in a SRC-dependent manner in NSCLC cells that harbored amplification of CRKL.
To examine whether CRKL-induced RAP1 activation is required for proliferation of NSCLC cells that harbored CRKL amplifications, we tested the ability of constitutively active RAP1 mutants to rescue cells from the cytotoxic effect of CRKL suppression. We transduced H1755 cells with a control lentiviral vector or lentiviral vectors expressing wild-type or constitutively active RAP1 mutants and then introduced a CRKL-specific shRNA (shCRKL#3). We found that overexpression of constitutively active form of RAP1A or RAP1B partially reversed the anti-proliferative effect of CRKL suppression (Figure 4H). In contrast, expression of wild-type RAP1A, RAP1B or a control vector failed to affect the decreased proliferation induced by CRKL suppression (Figure 4H). These findings suggest that CRKL-induced RAP1 activation in part is required for proliferation and survival of NSCLC cells that harbor CRKL amplification. The partial rescue conferred by active RAP1 also suggests that CRKL may activate additional signaling pathways beyond that involving RAP1 that are required for proliferation and survival.
Since RAP1 has been shown to modulate ERK signaling in a cell context-dependent manner (39–41), we examined whether CRKL-induced ERK activation and cell transformation requires a signaling pathway involving SRC-C3G-RAP1. Specifically, we introduced SRC-, C3G- or RAP1A/B-specific shRNAs into AALE cells overexpressing CRKL and then assessed anchorage independent growth. We found that shRNAs specific for SRC, C3G or RAP1B, but not RAP1A, substantially decreased the anchorage independent growth of CRKL-overexpressing AALE cells (Figure 4I and Figure S5A). Correlating with these observations, suppression of SRC, C3G or RAP1B in CRKL-overexpressing AALE cells resulted in decreased levels of phospho-ERK1/2 compared to cells expressing a control shRNA targeting GFP (Figure 4J). In contrast, suppression of p130CAS, one of the substrate proteins of CRKL and SRC, failed to inhibit CRKL-induced ERK activation and anchorage independent growth in AALE cells (Figure S4D). To confirm that SRC is required for CRKL-induced cell transformation, we assessed the anchorage independent growth of AALE cells overexpressing CRKL in the presence of a SRC inhibitor dasatinib. SRC inhibition significantly decreased the anchorage independent growth and phospho-ERK levels in CRKL-overexpressing AALE cells (Figure 4K and 4L). Expression of the gatekeeper SRCT341I mutant, but not wildtype SRC, conferred resistance to dasatinib and allowed CRKL-overexpressing AALE cells to grow in an anchorage independent manner in the presence of dasatinib (Figure 4K and 4L). Taken together, these findings suggest that SRC-C3G-RAP1 signaling pathway is required for sustaining CRKL-induced ERK activation and cell transformation.
CRKL-induced cell transformation requires activation of SOS1-RAS-RAF signaling
Since prior work has linked CRKL function and RAS signaling (27, 37), we determined whether overexpression of CRKL in AALE cells altered RAS activity. We found that overexpression of CRKL in AALE cells resulted in increased levels of GTP-bound, active RAS (Figure 5A). The CRKL-induced RAS activity in turn activates MAPK pathways as indicated by increased in vitro kinase activity of BRAF (Figure 5B) and increased phosphorylation levels of RAF1 (Figure 5C)). These observations suggest that CRKL augments the activation of the RAS-RAF-MAPK pathway.
Figure 5. CRKL-induced cell transformation requires SOS1-RAS-RAF.
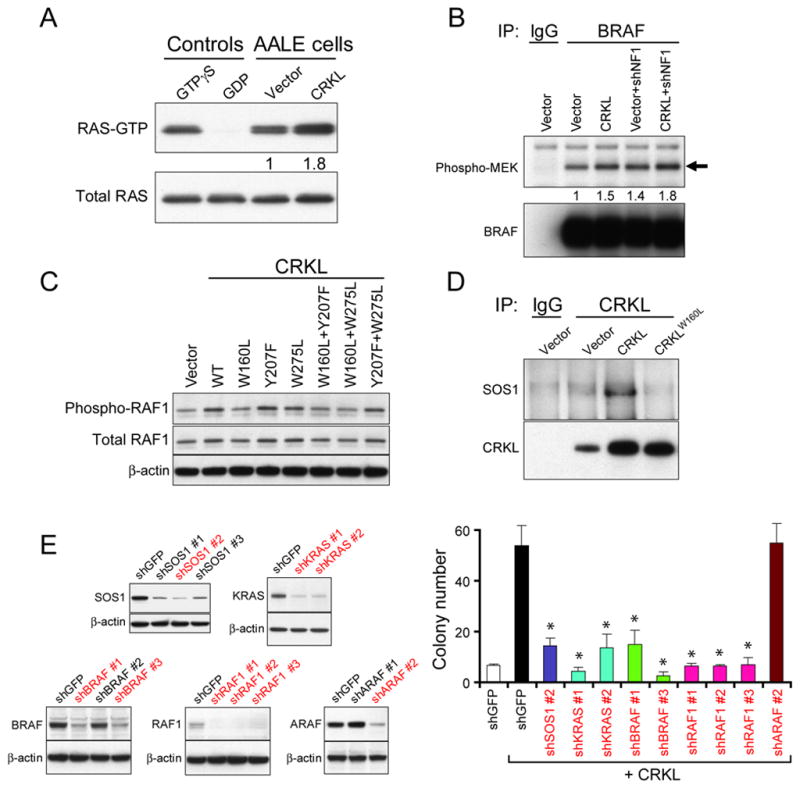
(A) Overexpression of CRKL increased RAS activity. The levels of GTP-bound RAS in AALE cells overexpressing a control vector or CRKL were measured by a pull-down assay followed by immunoblotting for RAS. Total RAS levels in total lysates were used as loading control. Positive and negative technical controls were obtained by incubating the total lysates with non-hydrolyzable analog of GTP (GTPγS) or GDP, respectively, before pull-down assays.
(B) Overexpression of CRKL increased in vitro BRAF kinase activity. The BRAF proteins in AALE cells expressing indicated constructs were isolated by immunoprecipitation. The kinase activity was assessed by incubating with substrate proteins (MEK1). Immunoblots of phospho-MEK1 and BRAF proteins in the isolated BRAF immune complexes after kinase activity assay are shown.
(C) Immunoblot of phospho-S338-RAF1 in AALE cell lines overexpressing wildtype or mutant CRKL.
(D) Interaction between CRKL and SOS1 in AALE cells overexpressing CRKL. CRKL immune complexes were isolated followed by immunoblotting for SOS1 or CRKL proteins in AALE cells expressing indicated constructs.
(E) CRKL-induced anchorage independent growth required SOS1-RAS-BRAF/RAF1 signaling. Left, Immunoblots of SOS1, KRAS, BRAF, RAF1 or ARAF proteins in CRKL-overexpressing AALE cell lines expressing a control shRNA targeting GFP or each gene-specific shRNA. ShRNAs that suppressed more than 50% of target protein levels were marked in red color. Right, Anchorage independent growth of AALE cells expressing indicated constructs. Colony number indicates colonies greater than 0.2 mm in diameter 4 weeks after plating. Data represent mean + s.d. of six replicate determinations from two independent experiments. * indicates p<0.0001 as compared to cells expressing CRKL and shGFP.
Prior work has shown that CRK family proteins interact with SOS1, a guanine nucleotide-exchange factor, that is a well-known RAS activator (22, 38, 47). We therefore determined whether overexpression of CRKL affects its interaction with SOS1 proteins in AALE cells. We isolated CRKL immune complexes and detected the presence of SOS1 proteins in AALE cells overexpressing CRKL, but not in cells expressing a control vector or CRKLW160L mutant (Figure 5D). The observation that CRKL promoted the interaction with SOS1 proteins via the SH3 domain suggests that overexpression of CRKL may recruit SOS1 proteins to membrane and thereby facilitating RAS activation.
To examine whether CRKL-induced cell transformation requires activation of the RAS-RAF signaling pathway, we suppressed SOS1, KRAS or different RAF members with RNAi in CRKL-overexpressing AALE cells and then evaluated anchorage independent growth. Suppression of SOS1, KRAS, BRAF or RAF1, but not ARAF, significantly inhibited the anchorage independent growth of CRKL-overexpressing AALE cells (Figure 5E and Figure S5B). These findings suggest that CRKL induced cell transformation requires activation of SOS1-RAS-BRAF/RAF1 signaling pathways.
CRKL cooperates with loss of NF-1 to induce tumorigenicity in human lung epithelial cells
Expression of oncogenic alleles of H- or K-RAS confers the ability to form anchorage independent colonies and tumors to AALE cells (32) (Figure 6A). In contrast, overexpression of CRKL alone in AALE cells induced anchorage independent growth but failed to allow tumor formation in AALE cells. In prior work, we and others have found that the co-expression of activated alleles of members of known RAS effector pathways, such as MEK and AKT or MEK and IKBKE substitute for RAS and induce tumorigenicity (48–51). We therefore searched for potential candidates that would cooperate with CRKL to promote tumorigenicity in the context of lung epithelial cells.
Figure 6. Overexpression of CRKL promotes tumor formation.
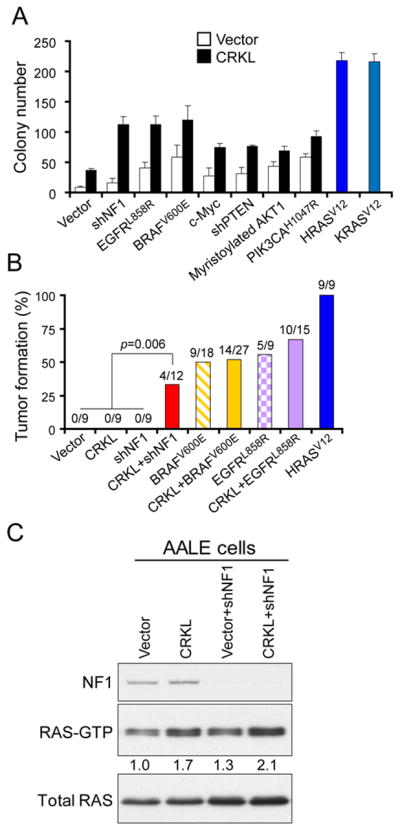
(A) Overexpression of CRKL promotes anchorage independent growth of AALE cells. Anchorage independent growth of AALE cell lines stably expressing the indicated constructs is shown. Colony number indicates colonies greater than 0.2 mm in diameter 4 weeks after plating. Data represent mean + s.d. of six replicate determinations from two independent experiments.
(B) Overexpression of CRKL cooperated with NF1 suppression in inducing tumor formation of AALE cells. AALE cell line expressing indicated constructs was implanted subcutaneously into immunodeficient mice (2×106 cells per injection site). The frequency of tumor formation and the number of tumor formation/number of injections within 5 months after implantation are depicted.
(C) Pull-down assay for RAS-GTP levels. AALE cell lines expressing indicated constructs were subjected to pull-down assay for RAS-GTP levels followed by immunoblotting for RAS. Relative intensity of RAS-GTP levels was determined. Immunoblot of RAS proteins in the total lysates was used as loading control. Immunoblot of NF1 proteins is also shown.
We introduced constitutively active alleles or shRNAs into AALE cells that activated the receptor tyrosine kinase signaling (EGFRL858R), MAPK signaling (BRAFV600E), PI3K-AKT signaling (myristoylated AKT1, PIK3CAH1047R or PTEN-shRNA), c-MYC or RAS signaling (NF1-shRNA). We then introduced CRKL or a control vector into these cell lines and tested their ability to grow in anchorage independent manner. We observed that CRKL overexpression in combination with each of these alleles promoted anchorage independent growth beyond that induced by CRKL alone (Figure 6A). In particular, overexpression of CRKL together with NF1 suppression showed a pronounced synergy in promoting anchorage independent growth (p=0.0006 and p=0.0003 compared to cells overexpressing CRKL or NF1-shRNA alone, respectively, t-test; Figure 6A). These results suggest that overexpression of CRKL promotes anchorage independent growth in many different contexts.
Since co-expression of CRKL and EGFRL858R, BRAFV600E or NF1-shRNA led to the most significant increase in anchorage independent growth among all combinations tested, we examined whether overexpression of CRKL together with these alleles promotes tumorigenicity in immunodeficient mice. We found that the AALE cells overexpressing EGFRL858R or BRAFV600E were tumorigenic (Figure 6B), suggesting that activation of EGFR or MAPK signaling suffice to induce tumorigenicity. Overexpression of CRKL in the setting of expressing EGFR or BRAF failed to increase the rate of tumor formation. Interestingly, shRNA-mediated suppression of NF1 in AALE cells failed to induce subcutaneous tumors in immunodeficient mice (Figure 6B). In contrast, overexpression of CRKL together with NF1 suppression conferred the ability to form tumors in 4 of 12 injection sites (33%) in AALE cells (p=0.006, Fisher’s exact test; Figure 6B). In consonance with these findings, the RAS-GTP levels (Figure 6C) and in vitro BRAF kinase activity (Figure 5B) were found to be highly increased in AALE cells that co-expressing CRKL and NF1-shRNA, indicating a robust activation of RAS-RAF signaling. These observations further demonstrate the transforming capability of CRKL overexpression, which participates in promoting tumor formation in a context-dependent manner.
To investigate whether CRKL amplification and NF1 loss co-occur in primary NSCLC tumors and cell lines, we re-analyzed mutation and copy number data generated in Tumor Sequencing Project (TSP) (12, 52). Among 188 primary lung adenocarcinomas, 1 of the 3 samples that harbor focal amplification of CRKL was found to harbor an inactivating mutation of NF1. No mutations were detected in the other oncogenes that were analyzed by sequencing in the remaining 2 CRKL-amplified tumor samples. In cell lines, we examined the expression of NF1 protein in 4 CRKL-amplified cell lines and found that HCC1359 cells failed to express NF1 compared to immortalized AALE cells (data not shown). These observations suggest that CRKL amplification and NF1 loss co-occur in a subset of NSCLCs and thus may cooperate in cell transformation.
Overexpression of CRKL decreases sensitivity to EGFR inhibitor treatment
We noticed a mutual exclusion between amplifications of CRKL and amplifications and mutations of EGFR in the 371 primary lung adenocarcinomas (12, 52) (Figure S6A). Specifically, we found that tumors that harbor focal high-level CRKL amplifications did not harbor amplifications of EGFR gene (p<0.0001, Fisher’s exact test, Figure S6A). Similarly, when we analyzed a panel of NSCLC cell lines (31, 53) by co-occurrence of genome alterations, we observed that the 6 cell lines that harbored CRKL amplifications appeared to form a subgroup distinct from the 12 cell lines that harbored mutations and/or high-level amplifications of EGFR gene (p<0.0001, Fisher’s exact test, Figure S6B). These observations suggest that amplifications of CRKL and EGFR may share some proliferation/survival pathways.
To investigate whether CRKL overexpression promoted proliferation and survival of NSCLC cells in response to EGFR inhibition, we determined whether overexpression of CRKL in a gefitinib-sensitive cell line, HCC827, that harbored the EGFRE746-A750del mutation (54) but exhibited normal copy number of 22q21.22 (Figure 1A) and low expression levels of CRKL (Figure 1C), affected the sensitivity to gefitinib. We observed that HCC827 cells overexpressing CRKL became resistant to gefitinib (IC50 = 2.2 μM) compared to cells expressing a control vector (IC50 = 0.01 μM) (Figure 7A).
Figure 7. Overexpression of CRKL induces EGFR inhibitor resistance in NSCLC cells that harbor EGFR mutations.

(A) Effect of overexpressing CRKL on gefitinib sensitivity in HCC827 cells that harbored EGFR mutation. Cell proliferation was measured 72 h after treatment with gefitinib. Data represent mean ± s.d. of triplicate measurements relative to untreated cells.
(B) Effect of overexpressing CRKL on phosphorylation levels of EGFR, ERK, AKT and SRC in response to gefitinib. HCC827 cells expressing a control vector or CRKL were exposed to gefitinib at indicated concentrations for 6 h and cell lysates were collected for immunoblotting with indicated specific antibodies.
(C) Effect of overexpressing CRKL on its interaction with SOS1 proteins. HCC827 cells expressing a control vector or CRKL were exposed to 1 μM gefitinib for 6 h. After isolating CRKL immune complexes from cell extracts, immunoblotting for SOS1 and CRKL proteins was performed.
(D) Effect of SOS1 suppression on CRKL-induced gefitinib resistance. Cell proliferation of each HCC827 cell line expressing indicated constructs 3 d after treatment with gefitinib. Data represent mean ± s.d. of six replicate determinations relative to untreated cells for each cell line..
(E) Effect of SOS1 suppression on phospho-ERK levels in CRKL-expressing HCC827 cells in response to gefitinib. Cells expressing indicated constructs were exposed to 1 μM gefitinib for 6 h followed by immunoblotting with indicated specific antibodies.
(F) Effect of overexpressing CRKL on the interaction of p85 with CRKL and ERBB3 proteins. HCC827 cells expressing a control vector or CRKL were either untreated or treated with 1 μM gefitinib for 6 h. After isolating p85 immune complexes, immunoblotting for CRKL, phospho-Y1289 ERBB3, total ERBB3 and p85 proteins was performed.
(G) Effect of GDC-0941 on CRKL-induced gefitinib resistance. Cell proliferation of CRKL-expressing HCC827 cells 3 d after treatment with gefitinib alone or in combination with GDC-0941 at indicated concentrations. Data represent mean ± s.d. of triplicate measurements.
(H) Effect of GDC-0941 on phospho-AKT levels in CRKL-expressing HCC827 cells in response to gefitinib. Cells were exposed to 1 μM gefitinib alone or in combination with GDC-0941 at indicated concentrations for 6 h followed by immunoblotting with specific antibodies.
(I) Acquired amplification of CRKL in an EGFR inhibitor-resistant lung tumor. FISH analysis of a NSCLC obtained from a patient prior to EGFR inhibitor therapy (left) or after acquiring drug resistance (right) using a CRKL-specific probe (green) and chromosome 22 telomere-specific probe (red). Nuclei were stained with DAPI (blue).
We next examined whether CRKL-induced ERK and SRC activation observed in transformation of AALE cells were involved in EGFR inhibitor resistance in HCC827 cells induced by CRKL overexpression. We observed that, upon gefitinib treatment, the phosphorylation of EGFR was completely suppressed in HCC827 cells that were either overexpressing CRKL or a control vector (Figure 7B). In contrast, phosphorylation of ERK1/2 persisted at substantially higher levels in HCC827 cells that overexpressed CRKL compared to cells expressing a control vector in the presence of gefitinib (Figure 7B). Overexpression of CRKL in HCC827 cells also markedly elevated phosphorylation levels of SRC compared to control vector-expressing cells that were unaffected by gefitinib (Figure 7B). These results demonstrate that overexpression of CRKL in HCC827 cells activates persistent ERK and SRC signaling upon gefitinib treatment.
To determine whether SRC activation contributes to CRKL-induced EGFR inhibitor resistance, we examined whether SRC inhibition suppressed growth of CRKL-overexpressing HCC827 cells. Cells were exposed to dasatinib alone or in combination with gefitinib. Interestingly, we observed that cells overexpressing CRKL also exhibited resistance to dasatinib treatment alone compared to cells expressing a control vector (Figure S7A). In addition, combined treatment failed to decrease the relative proliferation of CRKL-overexpressing cells compared to gefitinib treatment alone (Figure S7B and S7C). Moreover, activation of SRC or RAP1 by overexpression of SRC or constitutively active RAP1 alleles in HCC827 cells failed to induce resistance to gefitinib (Figure S8). Together, these results suggest that SRC activation is not required for CRKL-induced gefitinib resistance.
We next investigated whether overexpression of CRKL affects its interaction with SOS1, we isolated CRKL immune complexes from HCC827 cells treated with or without an EGFR inhibitor and examined SOS1 interactions. We detected an increase in the abundance of SOS1 associated with CRKL in HCC827 cells overexpressing CRKL (Figure 7C), and this interaction was further increased upon gefitinib treatment. These results suggest that overexpression of CRKL activates MAPK pathways by interacting with SOS1 in response to gefitinib.
To determine whether SOS1 was required for CRKL-induced EGFR inhibitor resistance, we introduced two SOS1-specific shRNAs or a control shRNA into CRKL-overexpressing HCC827 cells and measured the gefitinib sensitivity. Although suppression of SOS1 alone led to reduction in proliferation (Figure S9A), suppression of SOS1 in combination with gefitinib resulted in significant reduction in relative proliferation of CRKL-overexpressing cells compared to cells treated with gefitinib alone (Figure 7D). In contrast, expression of the control shRNA targeting GFP failed to alter gefitinib resistance (Figure 7E). In consonance with these observations, suppression of SOS1 in CRKL-overexpressing HCC827 cells led to reduced phospho-ERK1/2 levels compared to cells expressing shGFP upon gefitinib treatment (Figure 7E). These results suggest that SOS1-dependent MAPK signaling in part contributes to CRKL-induced gefitinib resistance.
As MET amplification has been shown to induce gefitinib resistance in EGFR mutant tumors by activating ERBB3-dependent PI3K-AKT signaling (54), we examined whether overexpression of CRKL in HCC827 cells affects phosphorylation of AKT in response to gefitinib. We observed that, upon gefitinib treatment, phosphorylation of AKT persisted at substantially higher levels in HCC827 cells that overexpressed CRKL compared to cells expressing a control vector (Figure 7B). These results suggest that like MET, overexpression of CRKL in HCC827 cells led to a persistent AKT signaling in response to gefitinib.
Since CRKL has been shown to interact with p85 regulatory subunit of PI3K (23), we hypothesized that overexpression of CRKL promotes its interaction with p85 proteins to induce persistent PI3K/AKT signaling that is resistant to gefitinib treatment. We isolated the CRKL immune complexes from HCC827 cells that overexpressed CRKL or a control vector and detected a markedly increased interaction with p85 proteins in cells overexpressing CRKL (Figure 7C). Consistently, when we isolated endogenous p85 immune complexes, we also detected increased interaction between p85 and CRKL proteins in cells that overexpressed CRKL (Figure 7F). Gefitinib treatment failed to affect this enhanced interaction between CRKL and p85 proteins (Figure 7C and 7F). In contrast, overexpression of CRKL in HCC827 cells reduced the interaction between p85 and phospho-ERBB3 or ERBB3 proteins under normal culture condition (Figure 7F). Upon gefitinib treatment, the interaction between p85 and ERBB3 was completely disrupted in cells that either overexpressed CRKL or a control vector (Figure 7F). Together, these results suggest that overexpression of CRKL promotes its interaction with p85 proteins to induce persistent ERBB3-independent PI3K/AKT signaling in the presence of an EGFR inhibitor.
To determine whether CRKL-induced EGFR inhibitor resistance involves PI3K/AKT activation, we examined whether treatment with the PI3K inhibitor GDC-0941 suppressed growth of CRKL-overexpressing HCC827 cells in response to gefitinib. Cells were exposed to GDC-0941 alone or in combination with gefitinib. Combined treatment with GDC-0941 and gefitinib resulted in substantial decrease in relative proliferation of CRKL-overexpressing HCC827 cells compared to gefitinib treatment alone (Figure 7G). The observed decrease in proliferation correlates with a decreased phosphorylation levels of AKT in CRKL-overexpressing HCC827 cells after combined treatment with LY294002 and gefitinib (Figure 7H). Similar findings were also observed with another PI3K inhibitor, LY294002 (Figure S9B-D). In contrast, combined treatment with a MET inhibitor PHA665752 and gefitinib failed to alter the relative proliferation of CRKL-overexpressing HCC827 cells (Figure S9E). These observations show that activation of PI3K-AKT signaling contributes to CRKL-induced EGFR inhibitor resistance.
CRKL amplification in EGFR inhibitor resistant lung cancer
We next examined whether amplification of CRKL occurs in EGFR-mutant tumors that developed acquired resistance following EGFR inhibitor treatment by performing fluorescence in situ hybridization (FISH) on 11 EGFR inhibitor-resistant tumors. We identified copy number gain of CRKL (with an average of 5.7±0.3 copies per cell) (revised Figure 7I) in one EGFR inhibitor resistant sample that was not present in the sample obtained prior to EGFR inhibitor treatment (average of 2.2±0.2 copies of CRKL per cell). Since we also detected increased signal using a chromosome 22 telomere-specific probe (5.7±0.3 copies per cell), we concluded that the amplification event likely involves the gain of a large fraction of the chromosome 22. This patient had shown a prolonged response to erlotinib for 2 years but showed signs of tumor regrowth while still receiving the treatment. Although 8 of 11 tumors had acquired the known resistance alteration EGFR T790M mutation, several laboratories have reported that other known resistance mechanisms may occur concomitantly with the EGFR T790M mutation, including the MET amplification (54) and CTNNB1 (β catenin) mutations (55). These observations indicate that the copy number gain of CRKL detected in the EGFR inhibitor-resistant tumor represents a new genetic event after treatment and implicate amplification of CRKL as a mechanism of acquired resistance to EGFR inhibitor treatment in a subset of NSCLCs.
Discussion
Amplification of CRKL defines a new subset of NSCLCs
In prior work, we and others identified a recurrent high-level and focal amplification peak on chromosome 22q11.21 in 3% of primary lung adenocarcinomas (n=371) (12). An additional 13% of tumors exhibited broad copy number gain spanning that region (12, 17). We also observed a mutual exclusion relationship between amplification of CRKL and EGFR in both primary lung adenocarcinomas and DFCI-84 NSCLC cell line collection. Herein we extend prior work that showed that NSCLC cells that harbor 22q11.21 amplification containing the CRKL gene depend on CRKL expression for proliferation to a larger panel of NSCLC cell lines and show that suppression of CRKL induces apoptosis. Suppression of CRKL in established tumors derived from NSCLC cells with amplification of CRKL induced tumor regression in vivo. Overexpression of CRKL in immortalized human airway epithelial cells induces anchorage independent growth and cooperates with suppression of NF1 to induce tumorigenicity in vivo. Indeed, we identified evidence that amplification of CRKL and loss of NF1 co-occur in a subset of NSCLC. These observations credential CRKL as a NSCLC oncogene. Although CRKL amplifications occur in a relatively small fraction of NSCLC, the finding that a similar fraction of NSCLC that harbor translocations involving ALK respond to treatment with crizotinib indicates that targeting genetic alterations present even in a subset of NSCLC may have clinical importance.
Amplification of CRKL induces cell transformation by activating RAS and RAP1 signaling
Using both biochemical and genetic approaches, we have found that malignant transformation induced by CRKL involves both RAS and RAP signaling. Using a multiplex luminex assay, we identified ERK and SRC as the downstream targets of CRKL. We further confirmed that CRKL activates RAS-RAF-ERK and SRC-RAP1 signaling by forming complexes with SOS1 and C3G, respectively. Our findings that expression of constitutively active RAP1 partially rescued NSCLC cells from proliferation inhibition induced by CRKL suppression define an important role of RAP1 signaling in proliferation and survival of NSCLCs with CRKL amplifications.
A prior report using a cell-free assay system showed that recombinant RAP1 stimulates BRAF kinase activity at a comparable level to K-RAS (42). RAP1 was also able to enhance KRAS-stimulated BRAF kinase activity in an additive manner (42). In consonance with these findings, we found that RAP1 activation contributes to CRKL-induced MAPK activation and anchorage independent growth in AALE cells. These findings suggest that CRKL induces transformation of human airway epithelial cells through its ability to coordinate with activation of both RAS and RAP1 pathways, resulting in robust activation of MAPK signaling.
Overexpression of CRKL induces resistance to small molecule EGFR inhibitors
Although most NSCLCs with EGFR mutations initially respond to treatment with an EGFR inhibitor, the majority of these tumors ultimately develop resistance to the treatment. In about half of these cases, resistance is due to the occurrence of a secondary mutation (T790M) in EGFR. Prior study showed that other genetic events, such as amplification of MET or mutation of PIK3CA, may contribute to drug resistance in some of the remaining cases and also co-occur with secondary EGFR mutations possibly due to the selection of drug-resistant clones harboring different resistance mechanisms (54, 55). These alterations promote cell growth by activating PI3K-AKT signaling in the presence of EGFR inhibitor. Here we report that overexpression of CRKL in EGFR mutant cells induces resistance to EGFR inhibitor by activating both SOS1-dependent MAPK and p85-dependent PI3K-AKT signaling. When we examined samples derived from tumors resistant to EGFR inhibitor treatment, we found CRKL amplifications in a patient whose tumors exhibited a clinical response to EGFR inhibitor therapy but subsequently developed acquired resistance. Since CRKL amplification was not identified in a pretreatment sample, these observations implicate CRKL amplifications as another mechanism of resistance to EGFR inhibitor therapy beyond secondary EGFR mutations, MET amplifications, and PIK3CA mutations (55).
CRKL as a novel therapeutic target in NSCLCs that harbored amplifications of CRKL
The statistical analysis of copy number alterations in 3131 cancer specimens derived from 27 histological types identified the 22q11.21 amplicon as one of the top 12 most commonly amplified regions in multiple cancer types (GISTIC q-value=3.71×10−26), including NSCLCs, melanoma, ovarian and colorectal cancers (19). CRKL is located at the center of the minimal region of amplification at 22q11.21 (19). These findings suggest that CRKL acts as an oncogene in other cancers that harbor CRKL amplifications and that strategies to inhibit CRKL signaling may be useful in several cancer types.
We found that suppression of CRKL has no effect on proliferation of AALE cells but selectively induced apoptotic cell death in NSCLC cells that harbored CRKL amplifications, suggesting that targeting CRKL may have a high therapeutic index. Using both RNAi and complementation approach, we further confirmed that disruption of the SH3N domain abolished the ability of CRKL to rescue NSCLC cells from shCRKL-induced apoptosis. Our findings not only credential CRKL as a therapeutic target but also demonstrate that blocking the function of the SH3N domain is sufficient to modulate CRKL activity in NSCLCs that harbored CRKL amplifications.
Materials and Methods
Plasmids
Human CRKL (obtained from CCSB ORFeome collection) was cloned into pWzl-blast and pLenti6.3-blast-C-terminal V5 epitope tagged vectors. The CRKL mutants (CRKLW160L, CRKLY207F, CRKLW275L, CRKLW160L+Y207F, CRKLW160L+W275L, CRKLY207F+W275L, CRKLsilent mutant) were generated using Quikchange Site-Directed Mutagenesis kit (Stratagene). Human RAP1A, RAP1AQ63E (provided by Gromoslaw Smolen, Massachusetts General Hospital), RAP1B (Origene) and RAP1BG12V were cloned into BamHI and BsrGI sites of pLenti6.3-blast. pLenti6.3-blast-SRC and SRCT351I have been described (45). pLenti6.2-blast-LacZ control vector was provided by Guo Wei, Dana-Farber Cancer Institute. pMKO.1-puro-shNF1, pBabe-puro-HRASV12, KRASV12 and myristoylated AKT1 have been described (51). pLKO.1-puro-shRNA constructs were obtained from The RNAi Consortium. The sequences targeted by CRKL-specific shRNAs are as follows: shCRKL#1 (TRCN0000006378), 5′-GCTCTGCTCTACCATGTTTAA-3′; shCRKL#2 (TRCN0000006380) 5′-CGTGAAAGTCACAAGGATGAA-3′; shCRKL#3 (TRCC0007470203), 5′-GCCTACTGAGTAGCTTTCATT-3′. A pool of 85 control shRNAs targeting reporter genes (GFP, RFP, Luciferase and LacZ) was used to generate control lentiviruses (Control shRNAs) (30). The sequences targeted by shGFP or scrambled control shRNA are 5′-ACAACAGCCACAACGTCTATA-3 ′ (TRCN0000072181) and 5′-GTGGACTCTTGAAAGTACTAT-3′, respectively. Other shRNA constructs used are as follows: shARAF#1 (TRCN0000000570), shARAF#2 (TRCN0000000567), shBCAR1#1 (TRCN0000115983), shBCAR#2 (TRCN0000115984), shBRAF#1 (TRCN0000006289), shBRAF#2 (TRCN0000006290), shBRAF#3 (TRCN0000006292), shRAF1#1 (TRCN0000001066), shRAF1#2 (TRCN0000001068), shRAF1#3 (TRCN0000001065), shKRAS#1 (TRCN0000033263), shKRAS#2 (TRCN000003326), shRAP1A#1 (TRCN0000029784), shRAP1A#2 (TRCN0000029787), shRAP1B#1 (TRCN0000029176), shRAP1B#2 (TRCN0000029177), shSOS1#1 (TRCN0000048145), shSOS#2 (TRCN0000048144), shSOS1#3 (TRCN0000048146), shSRC#1 (TRCN0000195339), shSRC#2 (TRCN0000038150), shC3G#1 (TRCN0000048128), shC3G#2 (TRCN0000048129), shC3G#3 (TRCN0000048130) and shC3G#4 (TRCN0000048131).
Cell culture and virus production
HCC515, H1819, HCC1833, H2087 and HCC827 cells were maintained in DMEM (Mediatech) supplemented with 10% fetal bovine serum (FBS, Sigma). HCC1359, H1755, H1437 and H1792 cells were maintained in RPMI1640 (Mediatech) supplemented with 10% FBS. Immortalized human lung airway epithelial (AALE) cells (32) were maintained in SABM supplemented with SAGM SingleQuots (Lonza). Retroviruses were produced by transfecting 293T packaging cells with pBabe/pWzl/pMKO and pCL-Ampho plasmids (51). Lentiviruses were produced by transfecting 293T packaging cells with a three-plasmid system (30). To generate stable cell lines, cells were selected in media containing 2 μg/ml of puromycin for 2 d or 10 μg/ml of blasticidin for 4 d.
Chemicals
Gefitinib, dasatinib and GDC-0941 were purchased from Selleck Chemicals. PHA665752 was purchased from Tocrus Biosciences. LY294002 was purchased from EMD Biosciences.
Cell proliferation assay
Cells were seeded into 96-well plates for 24 h. Six replicates infections were performed for control shRNAs or each gene-specific shRNA in the presence of 4 μg/ml of polybrene for 24 h followed by selection in media containing 2 μg/ml of puromycin. The ATP content was measured at 5 d by using CellTiter-Glo luminescent cell viability assay (Promega). Data represent mean + s.d. of 6 replicate measurements.
For the rescue experiments, H1755 (1.8×105) cells were incubated with lentiviruses expressing LacZ control, CRKL, RAP1, RAP1A63E, RAP1B or RAP1BV12 in 24-well plates in the presence of 4 μg/ml polybrene, and spin-infected at 2,000 rpm for 2 h at 37°C. Cells were then trypsinized and re-plated at a density of 1,500 cells/well of 96-well plates for 24 h.
For measuring IC50, HCC827 cells (1.5×103) expressing CRKL or a control vector were seeded into 96-well plates for 24 h and then incubated with gefitinib for 72 h. The ATP content was measured by using CellTiter-Glo luminescent cell viability assay (Promega). Data represent mean ± s.d. relative to untreated cells for each cell line. For the RNAi experiments, HCC827 cells overexpressing CRKL or a control vector were infected with lentiviruses encoding indicated shRNAs for 24 h, and then selected with 2 μg/ml of puromycin for 2 d. Cells were re-plated into 96-well plates for 24 h before treatment with gefitinib for 72 h.
Luminex immunosandwich assay
AALE (1.5×105) cells were plated onto each well of 6-well plates for 24 h. Triplicate transfections with 3 μg of pLenti-LacZ, CRKL or CRKLW160L were performed using Fugene 6 (Roche) for 16 h. After the incubation, medium was replaced with fresh medium and cells were cultured for another 32 h before collecting cell lysates. Luminex immunosandwich assay was performed as described (45).
Anchorage independent growth assay
Growth in soft agar was determined by plating 5×104 cells in triplicate in 0.4% Noble agar (51). Colonies greater than 0.2 mm in diameter were counted 4 weeks after plating. Data represent mean + s.d. of 6 replicate measurements from 2 independent experiments.
Tumorigenicity assay
Tumor xenograft experiments were performed as described (51). AALE cells expressing indicated constructs were trypsinized and collected in DMEM supplemented with 10% FBS. Cells (8×106) were resuspended in 400 μl of 1× PBS and mixed with 400μl of Matrigel™-Basement Membrane Matrix, LDEV-free (BD Biosciences). 200 μl of the cell mixture (containing 2×106 cells) was injected subcutaneously into 6-week-old male BALB/c nude mice (Charles River). Tumor injection sites were monitored for 5 months.
For tumor growth studies, HCC515 cells were infected with lentiviruses expressing Luciferase followed by selection in media containing 10 μg/ml of blasticidin for 5 d. Luciferized HCC515 cells were transduced with lentiviruses encoding pLKO-Tet-On-scrambled control shRNA or pLKO-Tet-On-shCRKL#1 and selected in 2 μg/ml of puromycin to generate stable cell lines. 5×106 cells were resuspended in 200 μl of PBS and injected subcutaneously into 6-week-old male BALB/c nude mice (Charles River). Tumor size was measured by a caliper twice weekly. Non-invasive bioluminescent imaging was performed at 11 and 32 d after implantation. Mice were given a single intraperitoneal injection of a mixture of luciferin (50 mg/kg), ketamine (150 mg/kg) and xylazine (12 mg/kg) in sterile water. After 5 min, mice were placed in a chamber and photons were captured for a time period of 120 s by using IVIS imaging camera (Xenogen). Images were generated by using LIVING IMAGE 2.60.1 software.
Immunoblotting
Cell lysates were prepared by scraping cells in lysis buffer [50 mM Tris (pH 8), 150 mM NaCl, 1% Nonidet P40, 0.5% Sodium Deoxycholate and 0.1% SDS] containing complete protease inhibitors (Roche) and phosphatase inhibitors (10 mM Sodium Fluoride and 5 mM Sodium Orthovanadate). Protein concentration was measured by using BCA Protein Assay kit (Pierce). An equal amount of protein (30 μg) was separated by NuPAGE Novex Bis-Tris 4–12% gradient gels (Invitrogen) and then transferred onto a polyvinylidene difluoride membrane (Amersham). The membrane was then incubated with primary antibodies for 1 h at room temperature. Phospho-specific antibodies against phospho-S473 AKT (#9271), phospho-Y1068 EGFR (#3777), phospho-Y1289 ERBB3 (#4791), phospho-T202/Y204 ERK1/2 (#4370), phospho-Y118 Paxillin (#2541), phospho-S338 RAF1 (#9427) and phospho-Y416 SRC (#2113), were purchased from Cell Signaling. Antibodies against ERK1/2 (#4695), ARAF (#4432), RAF1 (#9422), Caspase-3 (#9662) and PARP (#9532) were purchased from Cell Signaling Technology. Antibodies against BRAF (sc-5284), phospho-Y504 C3G (sc-12926), C3G (sc-869), CRKL (sc-319), KRAS (sc-30), SOS1 (sc-256) and SRC (sc-19) were from Santa Cruz Biotechnology. Antibodies against phospho-Y249 p130CAS (#558401) and p130CAS (#610272) were from BD Bioscience. Antibodies against p85 (#06-496), RAS (#05-516), RAP1 (#07-916), phospho-MEK (#07-461) and phospho-tyrosine (4G10) were from Millipore. Antibody specific for NF-1 was provided by Karen Cichowski, Brigham and Women’s Hospital. After incubation with the appropriate horseradish peroxidase-linked secondary antibodies (Bio-Rad), signals were visualized by enhanced chemiluminescence plus Western blotting detection reagents (Amersham). Expression of β-actin was also assessed as an internal loading control by using a specific antibody (sc-8432-HRP, Santa Cruz). Intensities of bands were quantified by LabWorks image analysis software (UVP).
Immunoprecipitation
Cell lysates were collected by scrapping cells in immunoprecipitation buffer [20 mM Tris (pH 8), 100 mM NaCl, 2 mM EDTA and 0.5% NP-40] containing protease and phosphatase inhibitors. 1 mg of protein extracts was incubated with 2 μg of anti-CRKL antibody (sc-319, Santa Cruz), 5 μg of anti-p85 antibody (#06-496, Millipore) or normal Rabbit IgG (sc-2070, Santa Cruz) for 2 h at 4°C followed by incubation with 30 μl of Protein A/G-PLUS agarose beads (sc-2003, Santa Cruz) for 1 h at 4°C. Beads were collected by centrifugation at 2,000 ×g for 3 min at 4°C and resuspended in 1 ml of immunoprecipitation buffer for washing. After repeated washing and centrifugation for 4 times, beads were boiled for 5 min in 1× NuPAGE LDS sample buffer (Invitrogen).
Pull-down assay for GTP-bound RAS or RAP1 was performed using RAS and RAP1 activation assay kits (Millipore), respectively. For measuring in vitro BRAF kinase activity, 500μg of protein extracts was incubated with 2 μg of anti-BRAF antibody (sc-5284, Santa Cruz) or normal Mouse IgG (sc-2025, Santa Cruz) for 2 h at 4°C followed by incubation with 20 μl of Protein A/G-Plus agarose beads for 1 h at 4°C. After washing with immunoprecipitation buffer for 5 times, beads were incubated with 1.34 μg MEK substrate proteins (#17-359, Millipore) in the presence of 37.5 mM MgCl2 and 250 μM ATP for 30 min at 30°C. Beads were boiled in sample buffer for 5 min and supernatants were resolved for immunoblotting for phospho-MEK1 (#07-461, Millipore).
Real-Time Quantitative Reverse-Transcription PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) and 1 μg of total RNA was used to synthesize the first-strand cDNA using Oligo(dT)20/random hexamer primer cocktails and Superscript III reverse transcriptase (Invitrogen). Quantitative PCR reactions were performed using SYBR green PCR Master Mix (Applied Biosystems). The primer sequences used are as follows: CRKL-primer set 1 (forward: 5′-CTGTCGGTGTCCGAGAACTC-3′; reverse: 5′-ATTGGTGGGCTTGGATACCTG-3′), CRKL-primer set 2 (forward: 5′-AAGCCCACCAATGGGATCTG-3′; reverse: 5′-ACTCCACCACTGTTCTTCAGG-3′) and GAPDH (forward: 5′-CCTGTTCGACAGTCAGCCG; reverse: CGACCAAATCCGTTGACTCC). Triplicate reactions were performed separately on the same cDNA samples by using the ABI 7900HT real time PCR instrument (Applied Biosystems). The mean cycle threshold (Ct) was used for the comparative Ct analysis method (ABI User Bulletin #2).
Quantitative PCR for gene copy number
The standard curve method was used to determine the copy number of CRKL in NSCLC and AALE cells. Genomic DNA was extracted using DNeasy blood and tissue kit (Qiagen). The primer sequences for detecting CRKL were 5′-TTGACAGGCACTGGCTTAGA-3′ and 5′-GGCACTCCACCACTGTTCTT-3′. The primer sequences for detecting LINE-1 were 5′-AAAGCCGCTCAACTACATGG-3′ and 5′-TGCTTTGAATGCGTCCCAGAC-3′. The standard curve was generated by PCR of serially diluted genomic DNA of AALE cells (50, 10, 2, 0.4 and 0.08 ng). Triplicate reactions were performed using 2 ng of genomic DNA extracted from NSCLC cells. The gene copy number was normalized to LINE-1 and normal reference DNA of AALE cells.
Fluorescence in situ hybridization (FISH)
BAC RP11-505B16 clone containing CRKL (Invitrogen) was labeled with digoxigenin (Roche) and BAC RP11-47N6 clone containing 22q13.2 as a reference (Invitrogen) was labeled with biotin using BioPrime labeling mix (Invitrogen). Labeled DNA was precipitated at −80°C for 2 h with glycogen (20 μg/μl), pelleted by centrifugation at 18,000 ×g for 15 min at 4°C, air-dried for 10 min, and resuspended in 50 μl of hybridization buffer (50% deionized formamide, 10% dextran sulfate, 2× SSC).
Slides containing metaphase chromosomes were pretreated with 1:25 Digest-All III (Invitrogen) at 37°C for 6 min, and fixed in 10% buffered formalin for 1 min. Slides were dehydrated for 2 min each in 70%, 90% and 100% ethanol. Probes were prepared by mixing 2 μl of each labeled probe, 1 μl Cot-1 DNA (1 mg/ml; Invitrogen), and 11 μl of hybridization buffer. Probes were applied to air-dried slides and covered with coverslips. Slides were incubated at 72°C for 5 min to denature probes. Hybridization was performed for 18 h at 37°C in a dark humid chamber. After hybridization, slides were washed in 0.5× SSC at 72°C for 5 min and rinsed in PBS containing 0.025% Tween-20 at room temperature. Slides were blocked with CAS-Block containing 10% normal goat serum (Invitrogen) and incubated with FITC-anti-digoxigenin (Roche) and Alexa Fluor 594 Streptavidin (Invitrogen). Slides were washed in PBS containing 0.025% Tween-20 and counterstained with DAPI (Invitrogen). Images were captured using Zeiss Axio Observer Z1 microscope and AxioVision imaging software (Zeiss).
Tumor specimens from erlotinib-treated patients were obtained from Dana-Farber Cancer Institute, Brigham and Women’s Hospital and Massachusetts General Hospital. All samples were analyzed under IRB-approved protocols. The presence of EGFR mutations in each specimen was confirmed as described (54, 55). FISH analysis of tumor specimens using CRKL-specific probe (BAC RP11-505B16) and chromosome 22 telomere-specific probe (Abbott Molecular) were performed as described (54).
Supplementary Material
Statement of Significance.
These studies credential CRKL as an oncogene in a subset of NSCLC. CRKL overexpression induces cell transformation and resistance to EGFR inhibitor treatment and suggest that therapeutic interventions targeting CRKL may confer a clinical benefit in a defined subset of NSCLCs.
Acknowledgments
Grant support: This work was supported in part by grants from the U.S. National Institutes of Health (R33 CA128625, U54 CA112962, R01 CA135257, R01 CA114465, P50 CA090578). W.C.H. and M.M. are consultants for Novartis Pharmaceuticals. P.A.J. is a consultant for Roche, Genentech, Astra-Zeneca, Boehringer Ingelheim and Pfizer.
We thank Karen Cichowski, Heidi Greulich, Guo Wei and Gromoslaw Smolen for reagents and cell lines. We thank Ronny Drapkin, Lynette Sholl, Craig Mermel, Rameen Beroukhim, Milan Chheda, Susan Moody and David Barbie for helpful discussions. We thank Qi Wang, Laura Johnson, and Sapana Thomas for technical assistance.
References
- 1.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–67. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 3.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 4.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 5.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 8.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 9.Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–53. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–95. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 12.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–12. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KC, Lakshmi B, et al. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci U S A. 2007;104:16663–8. doi: 10.1073/pnas.0708286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwei KA, Kim YH, Girard L, Kao J, Pacyna-Gengelbach M, Salari K, et al. Genomic profiling identifies TITF1 as a lineage-specific oncogene amplified in lung cancer. Oncogene. 2008;27:3635–40. doi: 10.1038/sj.onc.1211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–70. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 17.Kim YH, Kwei KA, Girard L, Salari K, Kao J, Pacyna-Gengelbach M, et al. Genomic and functional analysis identifies CRKL as an oncogene amplified in lung cancer. Oncogene. 2010;29:1421–30. doi: 10.1038/onc.2009.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–42. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–71. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- 21.Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feller SM, Knudsen B, Hanafusa H. Cellular proteins binding to the first Src homology 3 (SH3) domain of the proto-oncogene produce c-Crk indicate Crk-specific signaling pathways. Oncogene. 1995;10:1465–73. [PubMed] [Google Scholar]
- 23.Sattler M, Salgia R, Shrikhande G, Verma S, Pisick E, Prasad KVS, et al. Steel factor induces tyrosine phosphorylation of CRKL and binding of CRKL to a complex containing c-Kit, phosphatidylinositol 3-kinase, and p120CBL. J Biol Chem. 1997;272:10248–53. doi: 10.1074/jbc.272.15.10248. [DOI] [PubMed] [Google Scholar]
- 24.Nichols GL, Raines MA, Vera JC, Lacomis L, Tempst P, Golde DW. Identification of CRKL as the constitutively phosphorylated 39-kD tyrosine phosphoprotein in chronic myelogenous leukemia cells. Blood. 1994;84:2912–8. [PubMed] [Google Scholar]
- 25.Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD, Druker BJ. Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J Biol Chem. 1994;269:22925–8. [PubMed] [Google Scholar]
- 26.Senechal K, Heaney C, Druker B, Sawyers CL. Structural requirements for function of the Crkl adapter protein in fibroblasts and hematopoietic cells. Mol Cell Biol. 1998;18:5082–90. doi: 10.1128/mcb.18.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Senechal K, Halpern J, Sawyers CL. The CRKL adaptor protein transforms fibroblasts and functions in transformation by the BCR-ABL oncogene. J Biol Chem. 1996;271:23255–61. doi: 10.1074/jbc.271.38.23255. [DOI] [PubMed] [Google Scholar]
- 28.Schonherr C, Yang HL, Vigny M, Palmer RH, Hallberg B. Anaplastic lymphoma kinase activates the small GTPase Rap1 via the Rap1-specific GEF C3G in both neuroblastoma and PC12 cells. Oncogene. 2010;29:2817–30. doi: 10.1038/onc.2010.27. [DOI] [PubMed] [Google Scholar]
- 29.De Falco V, Castellone MD, De Vita G, Cirafici AM, Hershman JM, Guerrero C, et al. RET/papillary thyroid carcinoma oncogenic signaling through the Rap1 small GTPase. Cancer Res. 2007;67:381–90. doi: 10.1158/0008-5472.CAN-06-0981. [DOI] [PubMed] [Google Scholar]
- 30.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci USA. 2008;105:20380–5. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–40. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundberg AS, Randell SH, Stewart SA, Elenbaas B, Hartwell KA, Brooks MW, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21:4577–86. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- 33.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 34.de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 1997;14:507–13. doi: 10.1038/sj.onc.1200885. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka S, Morishita T, Hashimoto Y, Hattori S, Nakamura S, Shibuya M, et al. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc Natl Acad Sci USA. 1994;91:3443–7. doi: 10.1073/pnas.91.8.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gotoh T, Hattori S, Nakamura S, Kitayama H, Noda M, Takai Y, et al. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol Cell Biol. 1995;15:6746–53. doi: 10.1128/mcb.15.12.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nosaka Y, Arai A, Miyasaka N, Miura O. CrkL mediates Ras-dependent activation of the Raf/ERK pathway through the guanine nucleotide exchange factor C3G in hematopoietic cells stimulated with erythropoietin or interleukin-3. J Biol Chem. 1999;274:30154–62. doi: 10.1074/jbc.274.42.30154. [DOI] [PubMed] [Google Scholar]
- 38.Smit L, van der Horst G, Borst J. Sos, Vav, and C3G participate in B cell receptor-induced signaling pathways and differentially associate with Shc-Grb2, Crk and Crk-L adaptors. J Biol Chem. 1996;271:8564–9. doi: 10.1074/jbc.271.15.8564. [DOI] [PubMed] [Google Scholar]
- 39.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 40.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, et al. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 1998;392:622–6. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 41.Bouschet T, Perez V, Fernandez C, Bockaert J, Eychene A, Journot L. Stimulation of the ERK pathway by GTP-loaded Rap1 requires the concomitant activation of Ras, protein kinase C, and protein kinase A in neuronal cells. J Biol Chem. 2003;278:4778–85. doi: 10.1074/jbc.M204652200. [DOI] [PubMed] [Google Scholar]
- 42.Ohtsuka T, Shimizu K, Yamamori B, Kuroda S, Takai Y. Activation of brain B-Raf protein kinase by Rap1B small GTP-binding protein. J Biol Chem. 1996;271:1258–61. doi: 10.1074/jbc.271.3.1258. [DOI] [PubMed] [Google Scholar]
- 43.Akagi T, Shishido T, Murata K, Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci USA. 2000;97:7290–5. doi: 10.1073/pnas.140210297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stam JC, Geerts WJ, Versteeg HH, Verkleij AJ, van Bergen en Henegouwen PM. The v-Crk oncogene enhances cell survival and induces activation of protein kinase B/Akt. J Biol Chem. 2001;276:25176–83. doi: 10.1074/jbc.M009825200. [DOI] [PubMed] [Google Scholar]
- 45.Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, Beroukhim R, et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol. 2009;27:77–83. doi: 10.1038/nbt.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuyama T, Ogita H, Kawakatsu T, Fukuhara T, Yamada T, Sato T, et al. Involvement of the c-Src-Crk-C3G-Rap1 signaling in the nectin-induced activation of Cdc42 and formation of adherens junctions. J Biol Chem. 2005;280:815–25. doi: 10.1074/jbc.M411099200. [DOI] [PubMed] [Google Scholar]
- 47.Matsuda M, Hashimoto Y, Muroya K, Hasegawa H, Kurata T, Tanaka S, et al. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Mol Cell Biol. 1994;14:5495–500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–57. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171–83. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464–74. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–79. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 52.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 54.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 55.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.