

Abstract
Background
Psychological stress is a significant risk factor for hypertension and also directly affects the immune system. We have previously reported that T lymphocytes are essential for development of hypertension and that the central nervous system contributes to peripheral T lymphocyte activation and vascular inflammation in this disease, however the role of T cell activation in stress-related hypertension remains unclear.
Methods
Wild-type (WT) and T cell deficient (RAG-1−/−) mice were subjected to daily episodes of stress and blood pressure was measured. Circulating T cell activation markers and vascular infiltration of immune cells were analyzed as well as stress hormone levels and gene expression changes in the brain. The effects angiotensin II infusion in the presence of chronic stress was also studied.
Results
Repeated daily stress contributed to acute elevations in blood pressure that were associated with increased activation of circulating T cells and increased vascular infiltration of T cells. Repeated stress increased blood pressure in WT but not RAG-1−/− mice. Adoptive transfer of T cells to RAG-1−/− mice restored blood pressure elevation in response to stress. Stress-related hypertension and vascular infiltration of T cells was markedly enhanced by angiotensin II. Moreover, angiotensin II infused mice exposed to chronic stress exhibited greater blood pressure reactivity to an episode of acute stress.
Conclusions
These data demonstrate that stress-dependent hypertension triggers an inflammatory response that raises blood pressure at baseline and augments the hypertension caused by angiotensin II. These data provide insight as to how psychological stress contributes to hypertension.
Keywords: Psychological stress, Hypertension, T lymphocytes, Inflammation, vascular, Angiotensin II
INTRODUCTION
Experimental and epidemiological studies have suggested that psychological stress contributes to the development of cardiovascular disease (1,2). In particular, evidence suggests that psychological stress may promote the development of systemic hypertension and/or predict the risk for hypertension based on blood pressure responses to acute laboratory stressors. For example, in the CARDIA (coronary artery disease risk development in young adults) study individuals who exhibited enhanced cardiovascular responses to mental stress challenges had a markedly enhanced incidence of hypertension during a 13-year follow up (3). A meta-analysis of 6 studies involving more than 34,000 patients have confirmed these results (1). Moreover, work-related stress and stress arising from socioeconomic disadvantage and discrimination are associated with sustained increases in blood pressure and increased risk of coronary heart disease (4,5). This relationship between hypertension and psychological stress has been suggested to be in part caused by increased sympathetic neural activity, which leads to increased heart rate, cardiac output and blood pressure (6). Involvement of the sympathetic nervous system in response to acute stress is well documented, but the mechanisms by which stress contributes to sustained elevations in blood pressure over time are not well understood.
Despite extensive study, the etiology of most cases of human hypertension are unknown (7). There is emerging evidence suggesting that inflammation, in particular the adaptive immune response, plays a significant role in the pathogenesis of hypertension (8,9). Recent studies from our laboratory have shown that mice lacking lymphocytes (RAG1−/−) are resistant to various forms of experimental hypertension and that adoptive transfer of T cells but not B cells restores their hypertensive phenotype (7,10). These studies have also shown that hypertensive stimuli such as angiotensin II and high salt cause T cell activation and entry of activated T cells into the peripheral blood vessels and kidney. Data from our laboratory and others have shown that these cells release pro-inflammatory cytokines that promote vasoconstriction and sodium retention leading to hypertension (10–13). This concept represents a novel paradigm underlying the pathogenesis of hypertension.
The central nervous system also contributes to hypertension, in part through autonomic regulation of blood pressure, fluid water balance and vasopressin release (14). Early studies have shown that experimental lesioning of specific circumventricular organs of the forebrain, including the subfornical organ, the anteroventral third ventricle region involving the inferior aspects of the lateral terminalis, prevents many forms of experimental hypertension (15,16). Recent studies from our laboratory have further shown that these forebrain sites contribute to peripheral activation of T cells and vascular inflammation during angiotensin II-dependent hypertension (17). Interestingly, projections from these same forebrain regions extend to sites critical to the emotional stress response, including the bed nucleus of the stria terminalis (BNST) (18,19).
While the immune and central nervous systems have been established to play a role in hypertension, the manner in which these interact in the setting of psychological stress to cause hypertension remains undefined. In the present study, we therefore sought to examine the roles of T cell activation and peripheral vascular inflammation in stress-dependent hypertension and to assess whether stress augments the hypertension caused by angiotensin II.
METHODS
Animals
Three month-old male C57BL/6J and RAG−/− mice, which are void of lymphocytes, were obtained from Jackson Laboratories and housed on a normal light dark cycle. All animals were on a BL6 background.
Chronic Stress Protocol
These animals were exposed to a combination of daily restraint stress and cage-switch stress for one week. Mice were divided into stressed and non-stressed groups. On each day of stress, mice were individually restrained in a well-ventilated 50 ml conical tube for 60 minutes. Immediately following this, the mice were placed in a cage, previously inhabited by another male mouse, for 30 minutes. The mice were subsequently switched to a second dirty cage for an additional 30 minutes. The order of cage switch and restraint stress was randomized to minimize habituation to the stress protocol. In total, with the combination of both stressors, mice were exposed to approximately 2 hours of stress each day and at the end of the stress protocol they were returned to their home cages. The non-stressed control group was exposed to variable daily handling. The institutional Animal Care and Use Committee at Emory University approved all of the experimental protocols.
Blood Pressure Monitoring and Angiotensin II infusion
Blood pressure was measured either invasively in freely moving animals using radiotelemetry or non-invasively using the tail cuff method as previously described (10). When using the tail cuff method, measurements were obtained at least 16 hrs after the last stress session. When radiotelemetry was employed, the transmitters were implanted 10 days prior to the initiation of the stress protocol.
In some studies, a low dose of angiotensin II (140 ng · kg−1 · min−1) was infused using a 14-day osmotic minipump (Alzet, Model 2002) surgically implanted subcutaneously. This dose of angiotensin II was chosen because it causes minimal elevation in blood pressure in normal animals (20). As a control, mice were implanted with osmotic mini pumps containing vehicle for angiotensin II. In these studies, the stress paradigm was carried out starting 7 days after mini-pump implantation through day 14. In other studies, adoptive transfer of T cells by tail vein injection into RAG-1−/− mice was performed using methods similar to those previously described (10). In preliminary experiments, flow cytometry was employed to confirm successful reconstitution of T cells by adoptive transfer (Figure S2 in the Supplement). The stress paradigm was applied 3 weeks following adoptive transfer of T cells in subsequent experiments. See Figure S1 in the Supplement for schematic of experimental design used in the current study.
Behavioral Testing
The elevated plus maze consisted of two open arms and two enclosed arms arranged in a plus orientation. To begin each test, mice were placed in the center of the maze facing one of the open arms and allowed to freely explore the apparatus for five minutes, during which time their behavior was videotaped. Mice were returned to their home cage at the end of the 5 min test. The measure used for analysis was the percentage of time spent exploring the open arms, which was calculated by dividing the time spent in the open arms by the combined time spent in open and closed arm (21).
Neurohumoral and qPCR Quantification
Plasma serum corticosterone levels were measured by radioimmunoassay by the Emory University Biomarkers Core. Blood was collected on ice with .1 mol/L ethylenediaminetetraacetic acid (EDTA), and plasma was separated in a refrigerated centrifuge and stored at −70°C. The level of paraventricular nuclei (PVN) corticotrophin releasing hormone (CRH)-mRNA was detected by relative quantitative-RT-PCR (Applied Biosystems FAST 7500). Bilateral PVN tissue punches were performed according to the Mouse Brain Atlas by Watson and Paxinos (22) (Figure 2D). Total RNA was isolated from the PVN using Qiagen RNeasy kit. One microgram of RNA was used for reverse transcription using the Multiscribe Reverse Transcriptase kit from Applied Biosystems and cDNAs were used to determine gene expression using SYBR Green. The CRH primers were purchased from SA Biosciences, Reference Sequence No. NM 205769 and 18s house keeping control gene, Reference Sequence No. KO1364.
Figure 2. Effect of chronic stress on blood pressure, T cell activation and vascular infiltration.
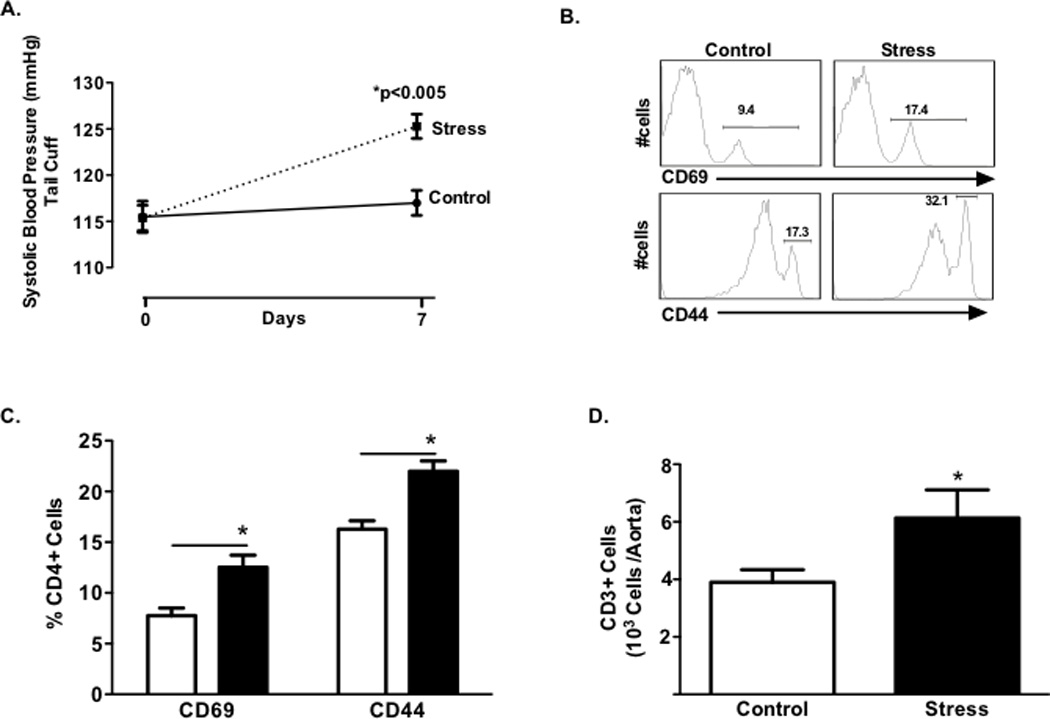
Panel A: Blood pressure at baseline and following 7 days of stress (n=18) or control no stress (n=15). Representative histograms (Panel B) and mean data (Panel C) of percentage of circulating CD4+ lymphocytes expressing the early activation marker CD69 and tissue homing marker CD44 for stressed and control mice. Total CD3+ T cells (Panel D) in aortas of stressed (n=15) and control (n=11) mice. (*P<0.05) (Dashed Line represents stress; Solid Line no stress control).
Fluorescence-Activated Cell Sorting (FACS) and Analysis of Cellular Inflammation
Mice were sacrificed 24 hours following the last stress period. Circulating T cells and inflammatory cells in vascular homogenates of the aorta were analyzed using FACS as previously described (10). Antibodies (BD Biosciences) used for staining were as follows: FITC anti-CD45 (30-F11); APC anti-CD4 (GK1.5); PerCP anti-CD8 (53–6.7); APC anti-CD3e (145-2C11); FITC CD44 (IM7); FITC CD69 (H1.2F3). After immunostaining, cells were resuspended in FACS buffer (0.5% bovine serum albumin in PBS) and analyzed immediately on a LSR-II flow cytometer with DIVA software (Becton Dickinson). Data were analyzed with FlowJo software (Tree Star, Inc.).
Data presentation and statistical analysis
Data in the manuscript are expressed as the mean ± SEM and values of P < 0.05 were considered statistically significant. Comparisons between more than 2 groups were made using ANOVA. When differences were observed, a Bonferroni post-hoc test was employed to compare specific groups. When identical measurements were made over time, we employed 2-way repeated measures ANOVA with a Bonferroni post hoc test. When 2 groups were compared we used an unpaired two-tailed Students T test
RESULTS
The Effects of Repeated Stress on Anxiogenesis, Blood Pressure and T lymphocyte Activation and Vascular Infiltration
Mice were exposed to a combination of daily restraint stress and cage-switch stress for one week. This stress paradigm elicited an anxiogenic response as evidenced by reduced time spent in open arms of the elevated plus maze (Fig. 1A), increased plasma corticosterone levels (Fig. 1B) and increased expression of CRH mRNA in the PVN (Fig. 1C). This stress paradigm also increased blood pressure, as measured with the tail cuff procedure, by 10 mmHg (Fig. 2A, p < 0.005). These measurements were obtained 24 hours following the last stress session.
Figure 1. Effect of chronic stress on neurohormonal and behavioral measures.
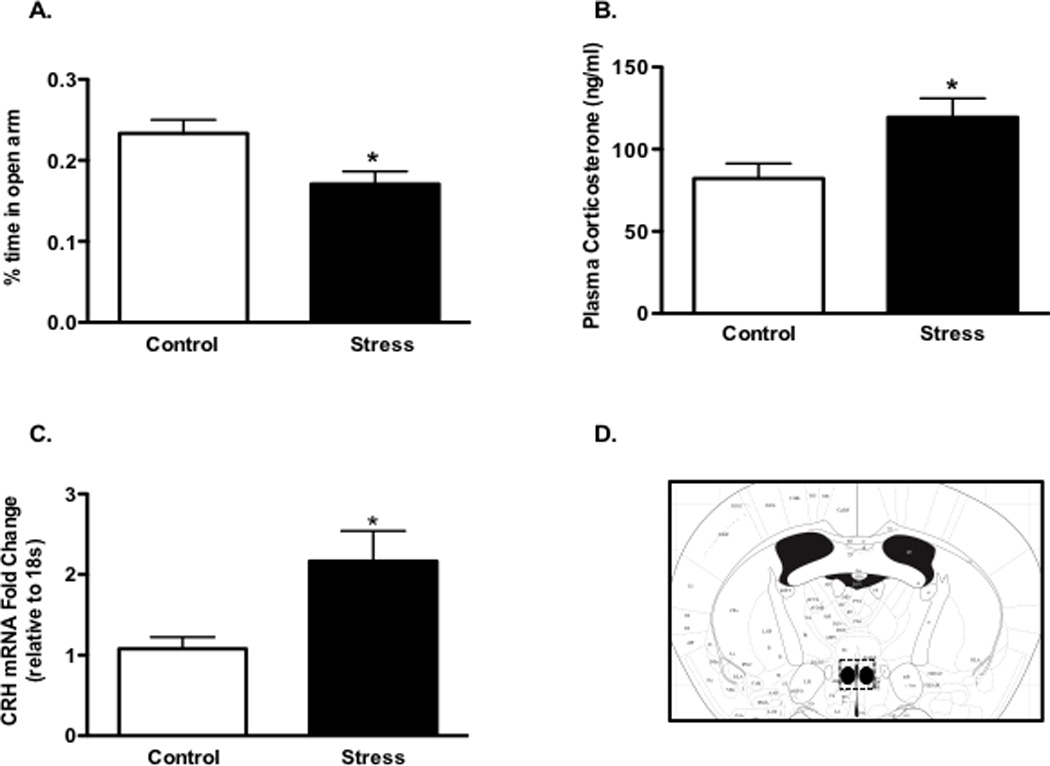
Panel A: Percent time in open arms of elevated plus maze test of stress (n=16) and control (n=12) groups. Plasma corticosterone in stress (n=19) and control (n=15) groups. Corticotropin-releasing hormone mRNA levels in the paraventricular nucleus (PVN) of stress (n=8) and control (n=9) groups (Panel C). Panel D depicts a coronal brain section with a dashed line surrounding the region of the PVN isolated for RT-qPCR. (*P<0.05).
It has been previously shown that T cell activation and vascular inflammation contribute to many forms of experimental hypertension (10–12,17). In particular, we have found that RAG1−/− mice are protected from various forms of experimental hypertension. Further, adoptive transfer of T cells restores their capacity to develop hypertension in response to stimuli such as angiotensin II, norepinephrine and high salt. We therefore sought to determine the role of T cells during stress-dependent hypertension. We found that animals exposed to one week of stress had a greater percent of circulating CD4+ cells that expressed the early T cell activation marker CD69 as well as the tissue homing marker CD44high (Fig. 2B–C, p<0.001; p<0.01). Similar to our findings in other hypertensive models, the aortas of stressed mice contained increased number of CD3+ cells, suggesting that repeated daily stress contributes to vascular T cell infiltration (Fig. 2D, p <0.05).
In comparison to wild type (WT) mice, the increase in blood pressure caused by one week of repeated stress was absent in RAG1−/− mice (Fig. 3A). Reconstitution of T cells into the RAG1−/− mice by adoptive transfer restored the hypertensive response to repeated stress (Fig. 3A, p < 0.001). These experiments indicate that T cells are required for stress-dependent hypertension.
Figure 3. Effect of chronic stress on blood pressure and neurohormonal and behavioral measures in RAG1−/− mice.
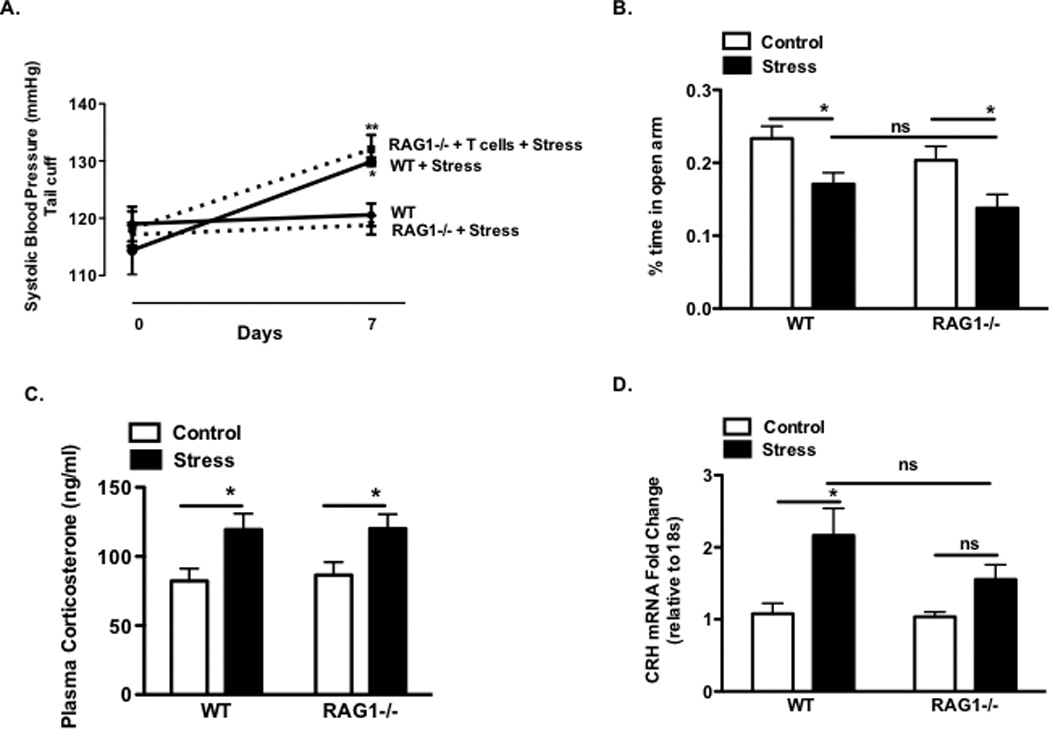
Panel A: Blood pressure measured by tail cuff at baseline and following 7 days of stress. WT control and WT + stress (n=5); RAG1−/− + stress (n=8) or RAG1−/− + T-cells + stress (n=5). Percent time in open arms of elevated plus maze test of WT (n=18–20) and RAG1−/− (n=17–20) groups (Panel C). Plasma corticosterone measurements in WT (n=28) and RAG1−/− (n=25) groups (Panel D). Corticotropin releasing hormone mRNA gene expression in the PVN of WT (n=11–12) and RAG1−/− (n=10–12) groups (Panel E). (WT+ stress vs WT *P<0.05) (RAG1−/− + T cell + stress vs RAG1−/− + stress **P<0.05).
The differences in blood pressure in RAG1−/− mice might have been related to differences in the perception of stress in these animals. To address this possibility we examined behavioral and neurohormonal indices in WT and RAG1−/− mice. Following stress, there were no differences in time spent in open arms of the elevated plus maze test (Fig. 3B; p <0.05). Moreover, corticosterone levels between the RAG1−/− and WT mice were similar in control and stressed animals (Fig. 3C; p <0.05). In contrast, the level of CRH mRNA in the PVN was significantly increased by stress in WT mice but not in RAG1−/− mice (Fig. 3D; p <0.05).
The Effects of Repeated Stress in Combination with a Low-Dose Infusion of Angiotensin II on Blood Pressure
The renin-angiotensin system is critical to blood pressure regulation, is involved in the stress response, and more recently has been shown to contribute to T lymphocyte activation and vascular inflammation (11,12,23). To evaluate whether angiotensin II exacerbates the stress-dependent elevation in blood pressure and vascular T cell infiltration, animals were infused for 14 days via osmotic mini-pump with a previously identified low pressor dose of angiotensin II (140 ng · kg−1 · min−1). Following 7 days of angiotensin II infusion we initiated the cage switch / restraint stress paradigm. As previously reported,(20) this low dose of angiotensin II caused only a mild elevation in blood pressure on days 7 and 14 (Fig. 4A, p <0.01), while repeated stress markedly increased the elevation in blood pressure to angiotensin II (Fig. 4A, p <0.001). The vehicle treated animals exposed to stress exhibited a modest increase in blood pressure following 7 days of stress, similar to our results in Figure 1A (Fig 4A), p <0.01). We then repeated these studies using radiotelemetry to assess mean arterial blood pressure in awake and freely moving animals. These measurements confirmed that angiotensin II alone led to a mild elevation in blood pressure from baseline to day 7 through day 14. Superimposition of stress upon angiotensin II increased the daytime blood pressures during some of the initial days but returned to similar levels at the end of the stress paradigm (Fig. 4C). Similarly stress did not appear to alter the final 24-hour blood pressure in the vehicle treated animals, although an unexpected decrease in nighttime blood pressure was observed on Day 11 (Fig. 4B).
Figure 4. Effect of acute and chronic stress following 7 days of low-dose angiotensin II infusion on blood pressure.
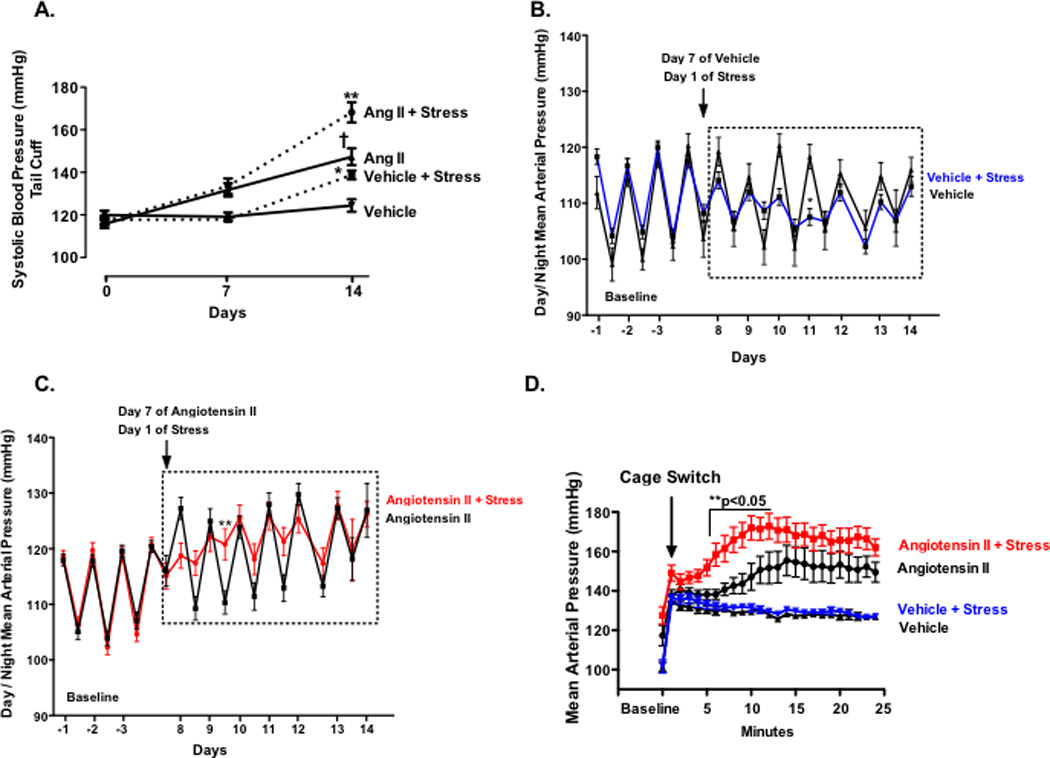
Panel A: Blood pressure at baseline and following 14 days of angiotensin II infusion and 7 days of repeated stress. Ang II + stress (n=16);Ang II (n=12); Vehicle + stress (n=13) or Vehicle (n=10). Diurnal blood pressure for Vehicle + stress (n=7) or Vehicle (n=3) and Ang II + stress (n=9);Ang II (n=7) (Panel B–C). Following seven days of daily stress, acute blood pressure response to cage switch stress (n=8–10) (Panel D). (*P<0.05 Vehicle vs Vehicle + stress; **P<0.05 Ang II vs Ang II + stress; †P<0.05 Ang II vs Vehicle).
Prior studies have shown that hypertensive individuals have increased cardiovascular reactivity to acute stressors (24,25). We therefore examined the acute pressor response to cage switch stress in the stressed and angiotensin II infused animals. As shown in Figure 4D, animals exposed to repeated stress plus angiotensin II exhibited a markedly enhanced acute pressor response compared to stressed animals treated with vehicle. These data suggest that previous exposure to chronic stress and angiotensin II augments the pressor response to acute stress.
Repeated Stress in Combination with Angiotensin II Augments Vascular T lymphocyte Infiltration and CRH gene expression in the PVN
Angiotensin II affects T cell-mediated inflammation in hypertension and has anxiogenic properties that can influence behavior and the stress response (26,27). We therefore examined the effects of stress in the presence of a low dose infusion of angiotensin II on circulating T lymphocyte activation and vascular infiltration of these cells. As shown in Figure 5A–B, neither angiotensin II nor stress affected the percent of circulating T cells expressing CD69 or CD44. However, the number of inflammatory cells infiltrating the aorta was greater in angiotensin II-infused animals that underwent repeated stress compared to either angiotensin II alone or vehicle stress (Fig. 5C–D) (p <0.01). These data suggest that angiotensin II superimposed on stress markedly augments the vascular T cell infiltration and inflammation that is associated with hypertension.
Figure 5. Effect of chronic stress on T cell activation and vascular infiltration following low-dose angiotensin II.
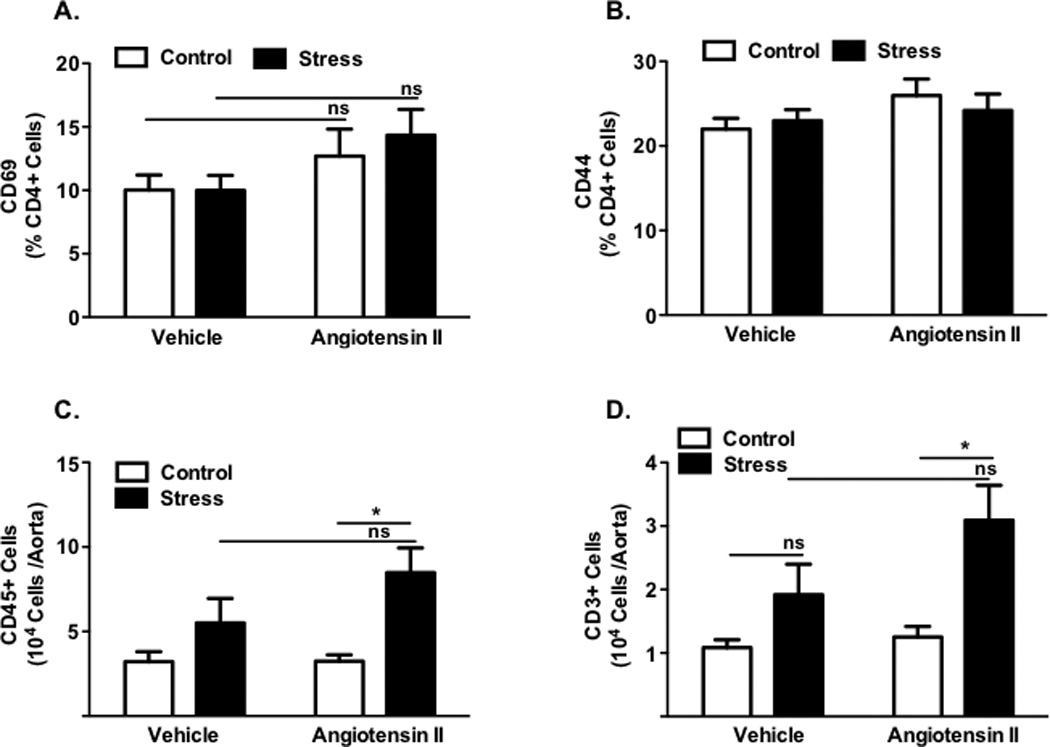
Percentage of circulating CD4+ lymphocytes expressing the early activation marker CD69 and tissue homing marker CD44 (Panels A–B) for stressed and control Ang II and vehicle mice (n=11–16). Total number of CD45+ leukocytes (Panel C) and CD3+ T cells (Panel D) in aortas of stressed and control Ang II and vehicle mice (n=10–13). (*P<0.01 Ang II vs Ang + Stress).
Further experiments were performed to examine the role of angiotensin II on behavioral and neurohormonal responses to stress. As shown in Figure 1 these studies showed that stress reduced the amount of time in the open arms of the elevated maze. Interestingly, angiotensin II alone mimicked this effect, however there was no additional effect of angiotensin II when it was superimposed on stress (Fig. 6A). As in figure 1, stress increased circulating levels of corticosterone and these were not further affected by angiotensin II (Fig. 6B; p <0.05). CRH gene expression was increased by chronic stress and this was augmented by angiotensin II infusion (Fig. 6C; p <0.01). These data suggest that angiotensin II augments some, but not all, of the behavioral and central neurohormonal responses to chronic stress.
Figure 6. Effect of chronic stress following 7 days of low-dose angiotensin II infusion on neurohormonal and behavioral measures.
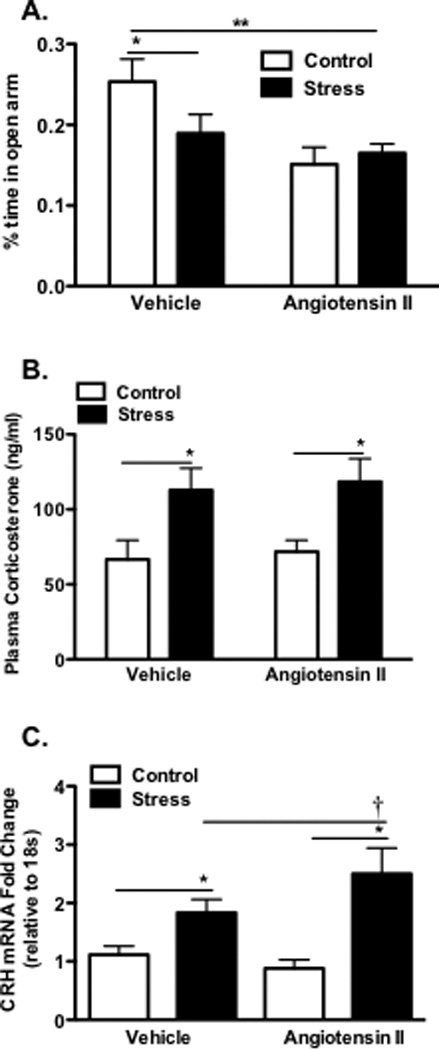
Panel A: Percent time in open arms of elevated plus maze test of vehicle (n=16–18) and Ang II (n=17–19) groups. Plasma corticosterone in vehicle (n=16–17) and Ang II (n=18–20) groups (Panel B). Corticotropin releasing hormone mRNA levels in the PVN of vehicle (n=6) and Ang II (n=7) groups (Panel C). (**P<0.05 Vehicle control vs Ang II) (*P<0.05 Control vs Stress) (†P<0.05 Vehicle + stress vs Ang II + stress).
DISCUSSION
In the current study, stress-related hypertension was associated with increased activation and vascular infiltration of T cells. In line with this, the blood pressure of RAG1−/− mice exposed to stress did not increase, but following adoptive transfer of T cells, stress-related hypertension was restored in these animals. We also showed that angiotensin II augments the anxiety, hypertension, vascular inflammation and CRH gene expression in the PVN of stressed animals. These data illustrate, for the first time, that chronic stress, angiotensin II and vascular inflammation interact to modulate hypertension.
Psychological stress can contribute to the development of hypertension and has been correlated with an excessive rise in blood pressure in patients with unstable hypertension (3,28,29). To further understand this phenomenon many have utilized various rodent models of chronic stress. It is well known that acute exposure to stressors such as restraint stress and cage switch stress elicit robust transient increases in blood pressure (30,31). On the other hand, repeated or chronic exposure to stressors has been shown to elicit changes in baseline blood pressure in some animal models of stress but not others (32–35). These divergent blood pressure results are likely attributed to differences in the nature (physical vs social stress), duration of stressor, rodent strain as well as the compensatory mechanisms involved in maintaining blood pressure homeostasis (14,36). A recent review by Navailako et al., further discusses these discrepancies and provides a nice overview of studies using animal models to examine the cardiovascular responses to chronic stress (37).
Despite these differences in the literature, a very intriguing recent study has shown a possible genetic link underlying susceptibility to the development of stress-dependent hypertension in animals and humans (38). Mice lacking the gene phosducin displayed elevated blood pressure at baseline and were more susceptible to stress-dependent elevations in blood pressure compared to wild-type controls. The authors went on to show that these effects were in part due to alterations in autonomic activity in mice with a targeted deletion of the G-protein regulator gene, phosducin. Another important factor in the genesis of stress-related hypertension most likely involves the renin-angiotensin system (26,30,39). Pharmacological inhibition of the renin-angiotensin system is a common approach for the treatment of hypertension and inhibition of this pathway can reduce the effects of acute stress in rodents (40) and more recently was associated with decreased PTSD symptoms in a clinical population (41). Low-grade systemic inflammation is also considered a contributing factor to the pathogenesis of hypertension (8,42) as well as other stress-related conditions(43). Therefore, the overall aim of the current study was to further elucidate the current understanding of the link between psychological stress, hypertension and inflammation.
The stress paradigm used in the present study produced significant anxiogenic effects as determined by increases in CRH gene expression in the PVN, elevated plasma corticosterone levels and decreased time spent in the open arms of the elevated plus maze. These findings are in keeping with the concept that the autonomic nervous system plays a major role in the cardiovascular and neuroendocrine responses to stress. This is likely due in part to activation of the HPA axis as we observed increased CRH mRNA in the PVN accompanied by increased corticosterone levels. Our findings are in line with the known role of CRH in the PVN activating the HPA axis, increasing autonomic outflow and modulating heart rate and blood pressure (44). Either electrical or chemical stimulation of the PVN can increase mean arterial pressure, heart rate and sympathetic nerve activity to the kidney (45,46). In a more recent study, Busnardo and colleagues showed that blockade of synaptic neurotransmission in the PVN prevented the increase in blood pressure and plasma corticosterone during acute restraint stress (47). These studies clearly indicate the importance of this pathway in the cardiovascular response to stress and have been suggested by others to be linked to the development of hypertension (48–50).
Our studies also demonstrate that repeated stress increases T cell activation and vascular inflammation both at baseline and in response to angiotensin II. Prior studies have shown that stress can both inhibit and activate the immune system (51,52). T lymphocytes express primarily beta 2 adrenergic receptors and these are known to modulate T cell activation and can polarize T cells to either a Th1 or Th2 cytokine profile depending on the existing cytokine milieu (53,54). Other data from our laboratory have shown that hypertension is associated with T cell activation likely involving engagement of the T cell receptor and co-stimulatory signaling characteristic of classical T cell activation (7,55). Our previous data have supported the concept that modest elevations in pressure caused by angiotensin II, excessive salt or catecholamines lead to T cell activation. It is possible that repeated bouts of pressure elevation in response to episodic stress could promote T cell activation in a similar fashion. These prior studies also indicate that various hypertensive stimuli promote entry of T cells into the vasculature and kidney and release cytokines such as TNFα and IL-17, which promote vasoconstriction, sodium reabsorption and hypertension (10,56). The T cells activated in response to stress likely play a similar role in promoting hypertension. These findings are in agreement with studies demonstrating an activated and pro-inflammatory T cell phenotype in response to stress (43,52,57–60). In line with this, clinical studies have shown that hypertensive patients have a greater percentage of circulating CD3+ and CD8+ cells in response to acute mental stress compared to normotensive controls (61).
In the current study, stress not only caused a modest elevation in resting pressure but also substantially enhanced the blood pressure response to angiotensin II. There are multiple mechanisms by which stress and angiotensin II can interact. For example, angiotensin II can enhance sympathetic nerve release of catecholamines (62,63), stimulate renin release and promote vasoconstriction (64). In addition, angiotensin II contributes to pro-inflammatory and pro-oxidative events (10,65) (66). Emotional stress can also activate the renin-angiotensin system and thus increase endogenous production of angiotensin II (39,67). Our studies indicated that angiotensin II infusion increases blood pressure reactivity to acute stress, vascular inflammation and CRF gene expression in the PVN. These effects are likely due to direct actions of infused angiotensin II, but might also reflect stress-induced production of angiotensin II. To our knowledge this is the first study to demonstrate that psychological stress augments both the central neurohormonal stress response and the peripheral vascular inflammation in response to chronic low-dose angiotensin II infusion. Pharmacological inhibition of angiotensin II is commonly used to treat hypertension and more recently has been suggested as a treatment of emotional stress and anxiety-related disorders (40,41).
In addition to measuring blood pressure using the tail cuff method we also used radiotelemetry. As shown in figure 4C, these measurements confirmed that angiotensin II alone led to a mild elevation in blood pressure from baseline. However our radiotelemetry measurements did not show that stress altered the average resting blood pressure or the blood pressure response to angiotensin II. These findings therefore differ from our non-invasive measurements of blood pressure. It should be emphasized, however, that radiotelemetry measurements are made under very different conditions. Radiotelemetry measurements are obtained throughout the day in unperturbed animals while the tail cuff measurements are obtained during a brief period of restraint that could serve as an acute stressor. Our studies suggest that the chronically stressed mice may have perceived the tail cuff procedure as an additional stressor. Studies in both humans (25) and animals (68) have shown that following repeated stress, exposure to additional acute stressors elicits an exaggerated acute pressor response which has been suggested to be due to increased cardiovascular/autonomic sensitivity. In keeping with this, we found that chronically stressed mice infused with angiotensin II exhibited an exaggerated acute pressor response to cage switch stress compared to non-stress controls.
In summary, we found that repeated daily stress promotes acute increases in blood pressure leading to increased T lymphocyte activation and vascular inflammation. In response to stress, low-dose angiotensin II augments the blood pressure, vascular T cell infiltration and CRH gene expression in the PVN. These studies indicate that stress dependent hypertension can trigger a T cell-mediated inflammatory response that in turn can contribute to the development of hypertension. Overall, these data provide new insight as to how psychological stress contributes to cardiovascular disease and hypertension.
Supplementary Material
ACKNOWLEDGEMENTS
This work was financially supported by The National Institutes of Health, the Emory University Neuroscience Initiative Scholars Program in Interdisciplinary Research and the Burroughs Wellcome Foundation.
We thank the Emory University Biomarkers and Rodent Behavioral Cores for their contribution to this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
FINANCIAL DISCLOSURES: All authors report no biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.Gasperin D, Netuveli G, Dias-da-Costa JS, Pattussi MP. Effect of psychological stress on blood pressure increase: A meta-analysis of cohort studies. Cad Saude Publica. 2009;25:715–726. doi: 10.1590/s0102-311x2009000400002. [DOI] [PubMed] [Google Scholar]
- 2.Esler M, Eikelis N, Schlaich M, Lambert G, Alvarenga M, Dawood T, et al. Chronic mental stress is a cause of essential hypertension: Presence of biological markers of stress. Clin Exp Pharmacol Physiol. 2008;35:498–502. doi: 10.1111/j.1440-1681.2008.04904.x. [DOI] [PubMed] [Google Scholar]
- 3.Matthews KA, Katholi CR, McCreath H, Whooley MA, Williams DR, Zhu S, et al. Blood pressure reactivity to psychological stress predicts hypertension in the cardia study. Circulation. 2004;110:74–78. doi: 10.1161/01.CIR.0000133415.37578.E4. [DOI] [PubMed] [Google Scholar]
- 4.Gillespie CF, Bradley B, Mercer K, Smith AK, Conneely K, Gapen M, et al. Trauma exposure and stress-related disorders in inner city primary care patients. Gen Hosp Psychiatry. 2009;31:505–514. doi: 10.1016/j.genhosppsych.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strogatz DS, Croft JB, James SA, Keenan NL, Browning SR, Garrett JM, et al. Social support, stress, and blood pressure in black adults. Epidemiology. 1997;8:482–487. doi: 10.1097/00001648-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Lambert E, Dawood T, Straznicky N, Sari C, Schlaich M, Esler M, et al. Association between the sympathetic firing pattern and anxiety level in patients with the metabolic syndrome and elevated blood pressure. J Hypertens. 2010;28:543–550. doi: 10.1097/HJH.0b013e3283350ea4. [DOI] [PubMed] [Google Scholar]
- 7.Marvar PJ, Gordon FJ, Harrison DG. Blood pressure control: Salt gets under your skin. Nat Med. 2009;15:487–488. doi: 10.1038/nm0509-487. [DOI] [PubMed] [Google Scholar]
- 8.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller DN, Kvakan H, Luft FC. Immune-related effects in hypertension and target-organ damage. Curr Opin Nephrol Hypertens. 2011;20:113–117. doi: 10.1097/MNH.0b013e3283436f88. [DOI] [PubMed] [Google Scholar]
- 10.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin ii-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–R1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1136–R1142. doi: 10.1152/ajpregu.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin ii-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 15.Brody MJ. Central nervous system and mechanisms of hypertension. Clin Physiol Biochem. 1988;6:230–239. [PubMed] [Google Scholar]
- 16.Brody MJ, Johnson AK. Role of the anteroventral third ventricle region in fluid and electrolyte balance, arterial pressure regulation, and hypertension. New York: Frontiers in Neuroendocrinology Raven Press; 1980. [Google Scholar]
- 17.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, et al. Central and peripheral mechanisms of t-lymphocyte activation and vascular inflammation produced by angiotensin ii-induced hypertension. Circ Res. 2010;107:263–270. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swanson LW, Lind RW. Neural projections subserving the initiation of a specific motivated behavior in the rat: New projections from the subfornical organ. Brain Res. 1986;379:399–403. doi: 10.1016/0006-8993(86)90799-7. [DOI] [PubMed] [Google Scholar]
- 19.Sunn N, McKinley MJ, Oldfield BJ. Circulating angiotensin ii activates neurones in circumventricular organs of the lamina terminalis that project to the bed nucleus of the stria terminalis. J Neuroendocrinol. 2003;15:725–731. doi: 10.1046/j.1365-2826.2003.00969.x. [DOI] [PubMed] [Google Scholar]
- 20.Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, et al. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:277–283. doi: 10.1161/HYPERTENSIONAHA.109.142646. 276p following 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellow S, Chopin P, File SE, Briley M. Validation of open:Closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods. 1985;14:149–167. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- 22.Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. San Diego, CA: Academic Press; 2006. [Google Scholar]
- 23.Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. 2010;10:203–207. doi: 10.1016/j.coph.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucini D, Di Fede G, Parati G, Pagani M. Impact of chronic psychosocial stress on autonomic cardiovascular regulation in otherwise healthy subjects. Hypertension. 2005;46:1201–1206. doi: 10.1161/01.HYP.0000185147.32385.4b. [DOI] [PubMed] [Google Scholar]
- 25.Nyklicek I, Bosch JA, Amerongen AV. A generalized physiological hyperreactivity to acute stressors in hypertensives. Biol Psychol. 2005;70:44–51. doi: 10.1016/j.biopsycho.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Groeschel M, Braam B. Connecting chronic and recurrent stress to vascular dysfunction: No relaxed role for the renin-angiotensin system. Am J Physiol Renal Physiol. 2011;300:F1–F10. doi: 10.1152/ajprenal.00208.2010. [DOI] [PubMed] [Google Scholar]
- 27.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, et al. Regulation of t-cell function by endogenously produced angiotensin ii. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–R216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boone JL. Stress and hypertension. Prim Care. 1991;18:623–649. [PubMed] [Google Scholar]
- 29.Kaplan MS, Nunes A. The psychosocial determinants of hypertension. Nutr Metab Cardiovasc Dis. 2003;13:52–59. doi: 10.1016/s0939-4753(03)80168-0. [DOI] [PubMed] [Google Scholar]
- 30.Davern PJ, Chen D, Head GA, Chavez CA, Walther T, Mayorov DN. Role of angiotensin ii type 1a receptors in cardiovascular reactivity and neuronal activation after aversive stress in mice. Hypertension. 2009;54:1262–1268. doi: 10.1161/HYPERTENSIONAHA.109.139741. [DOI] [PubMed] [Google Scholar]
- 31.Lee DL, Webb RC, Brands MW. Sympathetic and angiotensin-dependent hypertension during cage-switch stress in mice. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1394–R1398. doi: 10.1152/ajpregu.00306.2004. [DOI] [PubMed] [Google Scholar]
- 32.Alkadhi KA, Alzoubi KH, Aleisa AM, Tanner FL, Nimer AS. Psychosocial stress-induced hypertension results from in vivo expression of long-term potentiation in rat sympathetic ganglia. Neurobiol Dis. 2005;20:849–857. doi: 10.1016/j.nbd.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 33.Bechtold AG, Patel G, Hochhaus G, Scheuer DA. Chronic blockade of hindbrain glucocorticoid receptors reduces blood pressure responses to novel stress and attenuates adaptation to repeated stress. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1445–R1454. doi: 10.1152/ajpregu.00095.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDougall SJ, Paull JR, Widdop RE, Lawrence AJ. Restraint stress : Differential cardiovascular responses in wistar-kyoto and spontaneously hypertensive rats. Hypertension. 2000;35:126–129. doi: 10.1161/01.hyp.35.1.126. [DOI] [PubMed] [Google Scholar]
- 35.Henry JP, Liu YY, Nadra WE, Qian CG, Mormede P, Lemaire V, et al. Psychosocial stress can induce chronic hypertension in normotensive strains of rats. Hypertension. 1993;21:714–723. doi: 10.1161/01.hyp.21.5.714. [DOI] [PubMed] [Google Scholar]
- 36.Cowley AW., Jr The genetic dissection of essential hypertension. Nat Rev Genet. 2006;7:829–840. doi: 10.1038/nrg1967. [DOI] [PubMed] [Google Scholar]
- 37.Nalivaiko E. Animal models of psychogenic cardiovascular disorders: What we can learn from them and what we cannot. Clin Exp Pharmacol Physiol. 2011;38:115–125. doi: 10.1111/j.1440-1681.2010.05465.x. [DOI] [PubMed] [Google Scholar]
- 38.Beetz N, Harrison MD, Brede M, Zong X, Urbanski MJ, Sietmann A, et al. Phosducin influences sympathetic activity and prevents stress-induced hypertension in humans and mice. J Clin Invest. 2009;119:3597–3612. doi: 10.1172/JCI38433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krause EG, de Kloet AD, Scott KA, Flak JN, Jones K, Smeltzer MD, et al. Blood-borne angiotensin ii acts in the brain to influence behavioral and endocrine responses to psychogenic stress. J Neurosci. 2011;31:15009–15015. doi: 10.1523/JNEUROSCI.0892-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saavedra JM, Sanchez-Lemus E, Benicky J. Blockade of brain angiotensin ii at1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: Therapeutic implications. Psychoneuroendocrinology. 2011;36:1–18. doi: 10.1016/j.psyneuen.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khoury N, Marvar PJ, Gillespie C, Wingo A, Schwartz A, Bradley B, et al. The renin-angiotensin pathway in ptsd: Ace inhibitor and arb medications are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2011 doi: 10.4088/JCP.11m07316. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coffman TM. Under pressure: The search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409. doi: 10.1038/nm.2541. [DOI] [PubMed] [Google Scholar]
- 43.Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16:300–317. doi: 10.1159/000216188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lowry CA, Moore FL. Regulation of behavioral responses by corticotrophin-releasing factor. Gen Comp Endocrinol. 2006;146:19–27. doi: 10.1016/j.ygcen.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 45.Kannan H, Hayashida Y, Yamashita H. Increase in sympathetic outflow by paraventricular nucleus stimulation in awake rats. Am J Physiol. 1989;256:R1325–R1330. doi: 10.1152/ajpregu.1989.256.6.R1325. [DOI] [PubMed] [Google Scholar]
- 46.Li YF, Mayhan WG, Patel KP. Nmda-mediated increase in renal sympathetic nerve discharge within the pvn: Role of nitric oxide. Am J Physiol Heart Circ Physiol. 2001;281:H2328–H2336. doi: 10.1152/ajpheart.2001.281.6.H2328. [DOI] [PubMed] [Google Scholar]
- 47.Busnardo C, Tavares RF, Resstel LB, Elias LL, Correa FM. Paraventricular nucleus modulates autonomic and neuroendocrine responses to acute restraint stress in rats. Auton Neurosci. 2010;158:51–57. doi: 10.1016/j.autneu.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Hashimoto K, Makino S, Hirasawa R, Takao T, Sugawara M, Murakami K, et al. Abnormalities in the hypothalamo-pituitary-adrenal axis in spontaneously hypertensive rats during development of hypertension. Endocrinology. 1989;125:1161–1167. doi: 10.1210/endo-125-3-1161. [DOI] [PubMed] [Google Scholar]
- 49.Imaki T, Naruse M, Harada S, Chikada N, Nakajima K, Yoshimoto T, et al. Stress-induced changes of gene expression in the paraventricular nucleus are enhanced in spontaneously hypertensive rats. J Neuroendocrinol. 1998;10:635–643. doi: 10.1046/j.1365-2826.1998.00249.x. [DOI] [PubMed] [Google Scholar]
- 50.Porter K, Hayward LF. Stress-induced changes in c-fos and corticotropin releasing hormone immunoreactivity in the amygdala of the spontaneously hypertensive rat. Behav Brain Res. 2011;216:543–551. doi: 10.1016/j.bbr.2010.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: Implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- 52.Viswanathan K, Dhabhar FS. Stress-induced enhancement of leukocyte trafficking into sites of surgery or immune activation. Proc Natl Acad Sci U S A. 2005;102:5808–5813. doi: 10.1073/pnas.0501650102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Madden KS, Sanders VM, Felten DL. Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu Rev Pharmacol Toxicol. 1995;35:417–448. doi: 10.1146/annurev.pa.35.040195.002221. [DOI] [PubMed] [Google Scholar]
- 54.Swanson MA, Lee WT, Sanders VM. Ifn-gamma production by th1 cells generated from naive cd4+ t cells exposed to norepinephrine. J Immunol. 2001;166:232–240. doi: 10.4049/jimmunol.166.1.232. [DOI] [PubMed] [Google Scholar]
- 55.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the b7/cd28 t-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin ii-induced hypertension and vascular dysfunction. Hypertension. 2009 doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bosch JA, Berntson GG, Cacioppo JT, Dhabhar FS, Marucha PT. Acute stress evokes selective mobilization of t cells that differ in chemokine receptor expression: A potential pathway linking immunologic reactivity to cardiovascular disease. Brain Behav Immun. 2003;17:251–259. doi: 10.1016/s0889-1591(03)00054-0. [DOI] [PubMed] [Google Scholar]
- 58.Flint MS, Budiu RA, Teng PN, Sun M, Stolz DB, Lang M, et al. Restraint stress and stress hormones significantly impact t lymphocyte migration and function through specific alterations of the actin cytoskeleton. Brain Behav Immun. 2011;25:1187–1196. doi: 10.1016/j.bbi.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 59.Huang M, Pang X, Karalis K, Theoharides TC. Stress-induced interleukin-6 release in mice is mast cell-dependent and more pronounced in apolipoprotein e knockout mice. Cardiovasc Res. 2003;59:241–249. doi: 10.1016/s0008-6363(03)00340-7. [DOI] [PubMed] [Google Scholar]
- 60.Merlot E, Moze E, Dantzer R, Neveu PJ. Cytokine production by spleen cells after social defeat in mice: Activation of t cells and reduced inhibition by glucocorticoids. Stress. 2004;7:55–61. doi: 10.1080/1025389042000208150. [DOI] [PubMed] [Google Scholar]
- 61.Mills PJ, Farag NH, Hong S, Kennedy BP, Berry CC, Ziegler MG. Immune cell cd62l and cd11a expression in response to a psychological stressor in human hypertension. Brain Behav Immun. 2003;17:260–267. doi: 10.1016/s0889-1591(03)00055-2. [DOI] [PubMed] [Google Scholar]
- 62.Dendorfer A, Thornagel A, Raasch W, Grisk O, Tempel K, Dominiak P. Angiotensin ii induces catecholamine release by direct ganglionic excitation. Hypertension. 2002;40:348–354. doi: 10.1161/01.hyp.0000028001.65341.aa. [DOI] [PubMed] [Google Scholar]
- 63.Weiner N. Multiple factors regulating the release of norepinephrine consequent to nerve stimulation. Fed Proc. 1979;38:2193–2202. [PubMed] [Google Scholar]
- 64.Evans RG, Head GA, Eppel GA, Burke SL, Rajapakse NW. Angiotensin ii and neurohumoral control of the renal medullary circulation. Clin Exp Pharmacol Physiol. 2010;37:e58–e69. doi: 10.1111/j.1440-1681.2009.05233.x. [DOI] [PubMed] [Google Scholar]
- 65.Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin ii. Int J Biochem Cell Biol. 2003;35:881–900. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 66.Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98:121–128. doi: 10.1016/j.amjcard.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 67.Aguilera G, Kiss A, Luo X, Akbasak BS. The renin angiotensin system and the stress response. Ann N Y Acad Sci. 1995;771:173–186. doi: 10.1111/j.1749-6632.1995.tb44679.x. [DOI] [PubMed] [Google Scholar]
- 68.McDougall SJ, Lawrence AJ, Widdop RE. Differential cardiovascular responses to stressors in hypertensive and normotensive rats. Exp Physiol. 2005;90:141–150. doi: 10.1113/expphysiol.2004.028308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.