

Abstract
HIV-infected individuals, even with antiretroviral therapy, often display cognitive, behavioral and motor abnormalities and have decreased dopamine (DA) levels. Minocycline prevents encephalitis and neurodegeneration in SIV models, suggesting that it might also protect against nigrostriatal dopaminergic system dysfunction. Using an SIV/macaque model of HIV-associated CNS disease, we demonstrated that striatal levels of DA were significantly lower in macaques late in infection and that levels of the metabolite DOPAC also tended to be lower. DA levels declined more than its metabolites, indicating a dysregulation of DA production or catabolism. Minocycline treatment beginning at 12 but not 21 days postinoculation prevented striatal DA loss. DA decline was not due to direct loss of dopaminergic projections to the basal ganglia as there was no difference in tyrosine hydroxylase, dopamine transporter, vesicular monoamine transporter 2 or synaptophysin between minocycline-treated and untreated macaques. SIV-infected macaques had significantly higher monoamine oxidase (MAO) activity than uninfected macaques, although MAO activity was not affected by minocycline. Oxidative/nitrosative stress was examined by nitrotyrosine staining in the deep white matter and was lower in SIV-infected, minocycline-treated macaques compared with untreated macaques. These data suggest that minocycline, which has antioxidant activity, has a protective effect on DA homeostasis when administered at an appropriate time in SIV neuropathogenesis.
Keywords: Minocycline, Dopamine, SIV, HIV, Oxidative stress, Monoamine oxidase
Introduction
Globally, an estimated 33 million people are infected with HIV (UNAIDS 2010). In the US, before highly active antiretroviral therapy (HAART) was available, approximately 20% of HIV-infected individuals suffered from frank dementia and an additional 35% experienced more minor neurocognitive impairment (Ances and Ellis 2007). In the post-HAART era, while fewer people are progressing to AIDS and HIV-associated dementia (HAD), the prevalence of clinically milder forms of neurocognitive impairment is increasing, making the current estimate of HIV-associated neurocognitive disorders (HAND) approximately 50% (Ances and Ellis 2007; Heaton et al. 2011). With HIV-infected individuals living longer due to HAART, HAND is becoming an increasing burden on HIV-infected individuals and on the healthcare system.
In the CNS, HIV replicates in cells of macrophage lineage, which form a reservoir for viral persistence (Clements et al. 2005; Nath and Sacktor 2006). HIV causes neurological damage both by direct toxicity of viral proteins (e.g.- Tat, gp120, Vpr) and indirectly by activating macrophages, microglia, and astrocytes, leading to toxic chemokine and cytokine production, generation of reactive oxygen species (ROS), and eventually neuronal dysfunction (Kaul and Lipton 2006; Rumbaugh and Nath 2006; Steiner et al. 2006).
While no region of the brain is completely exempt from the effects of viral infection, the basal ganglia region is a hot spot of virus replication and HIV-associated neuropathology (Navia et al. 1986; Kumar et al. 2007). There is a wealth of data demonstrating nigrostriatal dysfunction in HIV infection and in animal models of HIV CNS disease (Berger and Arendt 2000; Nath et al. 2000; Koutsilieri et al. 2002; Ferris et al. 2008). Clinically, affected individuals exhibit cognitive, behavioral and motor deficits that are indicative of subcortical involvement (Berger and Arendt 2000; Koutsilieri et al. 2002; McArthur et al. 2005). Neurochemical analysis of brain and cerebrospinal fluid (CSF) in the terminal stages of infection supports the clinical findings of subcortical dysfunction. Demented AIDS patients have lower levels of dopamine (DA) in the caudate than seronegative controls (Sardar et al. 1996). Kumar and colleagues systematically examined DA levels throughout the brains of HIV-infected individuals who died of AIDS/HIV-related complications and found pronounced deficits in the caudate, putamen, globus pallidus, and substantia nigra (SN) and demonstrated that DA deficits in certain regions correlated with neuropsychological impairment (Kumar et al. 2009; Kumar et al. 2011). Moreover, dopamine levels also are reduced in the putamen of SIV-infected macaques early in asymptomatic infection, indicating that DA loss may be progressive (Scheller et al. 2005). Similar results have been demonstrated in CSF, with DA and HVA levels decreased in more advanced stages of HIV infection (Berger et al. 1994). However, findings of decreased DA are not unequivocal as there is also evidence of increased DA tone/release (Gelman et al. 2006; Ferris et al. 2008; Scheller et al. 2010).
Both viral proteins and glial activation are implicated in HIV-associated nigrostriatal dysfunction. HIV infection results in decreased levels of tyrosine hydroxylase (TH), the rate-limiting enzyme for DA production in both the caudate and the SN (Gelman et al. 2006; Silvers et al. 2006). The HIV transactivating protein Tat inhibits TH transcription in PC-12 cells (Zauli et al. 2000). Macrophage and microglial activation also have been correlated with nigrostriatal damage (Itoh et al. 2000; Scheller et al. 2005). Additionally, rodent models show that direct injection of viral proteins (gp120 and Tat) damages SN neurons (Zauli et al. 2000; Nosheny et al. 2006) and induces oxidative stress (Mattson et al. 2005; Agrawal et al. 2010).
Dopaminergic neurons are particularly sensitive to neuroinflammatory environments and oxidative stress (Barnum and Tansey 2010). Even under basal conditions, maintaining oxidative homeostasis in dopaminergic neurons is challenging (Berg et al. 2004). Dopamine can auto-oxidize, producing highly reactive dopamine quinones and superoxide. Additionally, metabolism of DA by monoamine oxidase (MAO) produces the byproduct H2O2, which is particularly toxic to dopaminergic neurons (Agrawal et al. 2010).
Minocycline, a second generation tetracycline derivative, decreases macrophage/microglial activation, oxidative stress, and cytochrome c release from mitochondria, scavenges ROS, and diminishes damaging MAPK signaling (Jordan et al. 2007; Kim and Suh 2009). Throughout the last decade, minocycline has proven protective in several animal models of neurodegeneration, including multiple sclerosis, Parkinson’s disease, and ischemic and traumatic brain injury (Kim and Suh 2009). Recently, minocycline was demonstrated to reduce CNS inflammation in the two most prominent SIV models of HIV-associated CNS disease. Minocycline reduced viral load in the basal ganglia and protected against neuroinflammation in a SIV pigtailed macaque model (Zink et al. 2005) and protected against decline of neuronal integrity and glial activation measured using in vivo proton magnetic resonance spectroscopy and post mortem immunohistochemistry in a rhesus/CD8 depletion model (Ratai et al. 2010; Campbell et al. 2011). Given minocycline’s ability to protect the CNS from various insults and the vulnerability of the nigrostriatal DA system in HIV/SIV infection, we used the accelerated, consistent SIV/macaque model (Zink et al. 2005; Clements et al. 2008) to evaluate minocycline’s ability to protect the nigrostriatal dopamine system.
Early minocycline treatment administered at a critical neuroimmunological juncture protected macaques from SIV-induced striatal DA decline; however later treatment did not. Additionally, minocycline-treated macaques tended to show less oxidative/nitrosative damage. These studies emphasize the importance of appropriately timing neurotherapeutic intervention with respect to neuropathogenesis.
Materials and methods
Animal infection and treatment
Juvenile pigtailed macaques (Macaca nemestrina) were either mock inoculated or co-inoculated intravenously with the immunosuppressive swarm SIV/DeltaB670 and the neurovirulent clone SIV/17E-Fr (Zink et al. 1999). Of the SIV-infected macaques, groups of 6 or 9 were euthanized at 4, 7, 10, 14, 21, or 56 days post inoculation (dpi). An additional 28 macaques were euthanized at the end stage of disease in this model, when all macaques have AIDS and the majority have encephalitis, approximately 3 months postinoculation (pi; Clements et al. 2008). SIV encephalitis, similar to HIV encephalitis, is characterized by infiltrating macrophages, perivascular cuffing, multinucleated giant cells, microglial nodules and gliosis (Zink et al. 1999; Clements et al. 2008). An additional 17 macaques were dually inoculated and administered daily minocycline treatment (4 mg/kg/day, divided into 2 oral doses) beginning either at 12 (n=6) or 21 (n=11) dpi until euthanasia at approximately 3 months pi. Prior to euthanasia, CSF samples were obtained on days 4, 7, 10, 14, 21, 28, and every 2 weeks thereafter for analysis of viral load and inflammatory markers. At euthanasia, animals were perfused with sterile saline to remove vascular blood prior to tissue storage. The brain was sliced into 0.5 cm coronal sections using a deli meat slicer and sections of basal ganglia, thalamus, parietal cortex, midbrain at the level of the pons, cerebellum, and medulla were snap frozen or fixed then paraffin-embedded. The Johns Hopkins Animal Care and Use Committee approved all animal studies. Animals were treated humanely in accordance with federal guidelines.
HPLC analysis of neurotransmitters
The HPLC method was adapted from (Pletnikov et al. 2000). Briefly, biopsy punches (~50-100 mg) from fresh frozen sections of the striatum (caudate or putamen) were weighed then homogenized on ice by sonication in 1 mL of a solution of perchloric acid (0.1 M) and dihydroxybenzylamine (3.59 mM; Sigma; St. Louis, MO). Homogenates were centrifuged at 16,000g for 10 min at 4°C to separate cell debris from the soluble fraction. Supernatants were then filtered by transferring to 0.22 μm Ultrafree-MC centrifugal filter units (Millipore; Bellerica, MA) and centrifuged for 2 min at 16,000g. Fifteen μL of the filtrate were injected onto a reversed phase HPLC column (Econosphere C18 5 μm 4.6 mm) using a Waters 717plus Autosampler, while the remainder was frozen at −80°C. The mobile phase was sodium acetate (54 mM), heptane sulfonic acid (54 mM), and EDTA (0.4 mM, pH 3.75) with acetonitrile (added 150 mL to 2,760 mL mobile phase; Sigma). Chromatographic data was recorded using an HP 3395 integrator (Hewlett-Packard; Palo Alto, CA). DA, DOPAC, and HVA peaks were identified on chromatograms by comparing retention times to known standards. Amounts of DA, DOPAC, and HVA were calculated by comparing peak heights in samples to peak heights of external standards. Ratios of DOPAC/DA and HVA/DA were calculated for each macaque. The Mann–Whitney test was used to compare groups for all measurements in this study.
Immunoblotting for markers of dopamine-producing neurons
Basal ganglia tissue (~100 mg) was homogenized in 500 μL lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 25 mM dithiothreitol, 0.2 mM phenylmethanesulfonyl fluoride) using a pellet pestle then passed several times through a 20 gauge needle. The sample then was incubated on ice for 20 min and centrifuged at 16,000g for 20 min at 4°C. The supernatant was collected and protein concentration determined using the BioRad Protein Assay kit. Ten to 20 μg of protein were solubilized and resolved under reducing conditions using 10% bis-tris gels (BioRad; Hercules, CA) and MOPS SDS running buffer (Invitrogen; Carlsbad, CA) then transferred to PVDF membranes. Membranes were blocked using 5% nonfat dry milk in TBST (20 mM Tris, 137 mM NaCl, pH 7.5, 0.1% Tween-20) and probed using primary antibodies against tyrosine hydroxylase (1/1000, Chemicon; Bellerica, MA), dopamine transporter (1/1000, Chemicon), vesicular monoamine transporter 2 (1/500, Novus; Littleton, CO), synaptophysin (1/2000, Dako; Glostrup, Denmark), or GAPDH (1/5000, Santa Cruz Biotechnology; Santa Cruz, CA). Membranes were washed in TBST then incubated with appropriate HRP or, when possible, fluorescently labeled secondary antibody. Results were visualized using either chemiluminescence and film or fluorescence and a Typhoon 9210 phosphorimager (Amersham; Piscataway, NJ). A serial dilution of sample was run on each blot to ensure samples were within linear range of detection. Immunoblots were quantitated using IQTL 7.0 software (Amersham). Samples were normalized to arbitrary units of serially diluted sample to normalize for blot-to-blot variation. Proteins of interest were normalized to GAPDH for total protein loading. Lanes with undetectable bands were set at 0 for quantitation purposes. All westerns were performed in duplicate or triplicate and the graphs show the average of the replicates. One exception to this quantitation method was dopamine transporter (DAT), which had the greatest variability and for which a suitable linear range could not be established. For the DAT westerns, instead of normalizing all samples to the serially diluted sample linear range, samples were normalized to the average of the detectable SIV-negative samples. Therefore, the quantitation of DAT shown was the result of one representative experiment.
Measurement of MAO activity
MAO activity was measured in basal ganglia homogenates using a luminescent method. Seventy-five μg of protein in triplicate per macaque were used in the MAO-Glo Assay (Promega; Madison, WI) as described by the manufacturer, with the exception that incubation with substrate was increased from 20 min to 4 h. Luminescent signal was detected using a Fluoroskan Ascent FL plate reader. MAO activity, measured in relative light units (RLU), was background corrected using a buffer-only control. Heat-inactivated samples yielded similar background values to buffer-only controls. There is potential that this assay could detect amine oxidases other than MAO. To confirm that the signal from macaque basal ganglia homogenates was due to MAO and not other amine oxidases, basal ganglia homogenate was assayed in the presence of the specific MAO A and B inhibitors clorgyline and deprenyl, which completely inhibited the luminescent signal.
Immunohistochemistry for nitrotyrosine
Nitrotyrosine (Y-NO) was measured as a general marker of oxidative/nitrosative stress in deep white matter, the area of the highest concentration of inflammatory lesions (Navia et al. 1986; Williams and Hickey 2002; Gelman 2007). Immunohistochemistry was performed on Streck-fixed, paraffin-embedded coronal sections from macaque brains. Slides were deparaffinized and rehydrated by heating for 10 min at 60°C, cleared in Histoclear (National Diagnostics; Atlanta, GA), then hydrated in a graded series of ethanol and water. Slides were pretreated by heating with 0.01 M sodium citrate, pH 6.0, prior to immunostaining. Between each immunostaining step, slides were washed with IHC buffer (PBS-0.05% Tween-20). Endogenous peroxidases were blocked using 3% hydrogen peroxide in methanol then nonspecific labeling was blocked with Power Block (Biogenex; San Ramon, CA). Primary antibody (mouse anti-nitrotyrosine, 1/300; Millipore) was applied for 1 h, followed by biotinylated anti-mouse secondary antibodies and streptavidin-conjugated HRP (Biogenex) for 20 min each. DAB chromagen (Biogenex) was applied for 10 min. For ease of quantitation, slides were not counterstained. The area fraction stained by Y-NO in the deep white matter was quantified as previously described with minor modifications (Follstaedt et al. 2008). For each slide, a series of 20 adjacent images at a resolution of 1,280×1,024 pixels, over-lapping by 15%, were acquired for each slide with a 20 × objective calibrated at 0.3 μm/pixel using a Nikon Eclipse 90i microscope (Nikon; Melville, NY) equipped with a DS-Ri1 camera (Nikon). A composite image for each slide was compiled from the 20 adjacent images using Nikon Elements AR 3.10 software (average area 2.07 mm2). The threshold for immunopositivity was set and applied identically to each slide. In this manner, the images were binarized such that each pixel was classified as stained (1) or unstained (0), providing a quantitative measure of the total area occupied by stained pixels. The amount of area stained by Y-NO was expressed as a fraction of the total area measured.
Immunohistochemistry for CD68, MHC class II, and GFAP
Immunohistochemical staining of CD68 (a marker of macrophage/microglial activation), MHC class II (a marker of macrophage/microglial and endothelial cell activation), and glial fibrillary acidic protein (GFAP; a marker of astrocyte activation), was performed and quantitated in deep white matter as described previously (Zink et al. 1999; Zink et al. 2001; Clements et al. 2002).
qRT-PCR measurement of SIV viral loads in CSF and brain
Viral and tissue RNA were isolated from macaque CSF and basal ganglia and SIV viral loads were determined by qRT-PCR as previously described (Witwer et al. 2009) using the following primers and probes: forward primer (SGAG21) 5′-GTCTGCGTCATCTGGTGCATTC-3′; reverse primer (SGAG22) 5′-CACTAGGTGTCTCTGCACTATCTGTT TTG-3′, probe (pSGAG23) 5′-(FAM) CTTCCTCAGT GTGTTTCACTTTCTCTTCTG-(BHQ_1)-3′. Statistical analyses involving viral loads were performed using log-transformed values.
Results
DA levels in the striatum decreased over the course of SIV infection
We examined DA and DA metabolite levels in the striatum during SIV infection using tissue from juvenile pigtailed macaques that were mock inoculated (SIV-) or dually inoculated with SIV/DeltaB670 and SIV/17E-Fr and euthanized at various times after inoculation. Striatal DA levels were significantly lower in the late stage of infection compared to uninfected controls (p=0.002; Fig. 1a). The DA metabolite DOPAC was also significantly lower in late stage infection (p=0.031; Fig. 1b). However, levels of the metabolite HVA, while lower, did not reach statistical significance (p=0.112; Fig. 1c). The ratios of the metabolites DOPAC and HVA to DA were calculated as a measure of DA turnover. The ratio of HVA/DA was significantly higher in SIV-infected macaques late in infection (p=0.017; Fig. 1e), but the ratio of DOPAC/DA was not (p=0.160; Fig. 1d). The observed decline in DA levels could be due to reduced DA production and/or increased DA catabolism. Changes in DA and its metabolites were not found in the early stages of infection (4-21 dpi).
Fig. 1.

DA levels in the striatum were lower in late stage SIV infection. Macaques were mock inoculated (SIV-) or inoculated with SIV, then euthanized at various days postinoculation (dpi). Levels of DA (a), DOPAC (b), and HVA (c) in the striatum were determined using HPLC with EC detection and ratios of DOPAC/DA (d) and HVA/DA (e) were calculated. Amounts are expressed as ng neurotransmitter or metabolite per mg of tissue. Levels of DA and its metabolite DOPAC were significantly lower in late stage SIV infection (p=0.002 and p= 0.031, respectively). Levels of the metabolite HVA were not significantly lower in SIV infection (p=0.112). The ratio of DOPAC/DA was not significantly different in SIV infection (p=0.160), but the ratio of HVA/DA was significantly higher in late SIV infection (p=0.017). Bars represent medians. Statistical comparisons were made using the Mann–Whitney test
Early minocycline treatment prevented the decline in striatal DA levels
Having established that our SIV model of HIV CNS disease recapitulated DA loss found in AIDS patients, we examined whether or not minocycline, which is protective in a number of animal models of neurodegenerative diseases, would prevent this loss. SIV-infected macaques were either untreated or treated orally with minocycline beginning at 12 (n=6) or 21 (n=11) dpi and euthanized at approximately 3 months pi. Levels of DA in the striatum of untreated macaques at 3 months pi were lower than levels in uninfected macaques (p=0.003; Fig. 2a). DOPAC and HVA levels were lower, but not significantly so (p=0.074 and 0.183, respectively; Fig. 2b, c). DOPAC/DA ratios tended to be higher in SIV infection, but this difference was not significant (p=0.082; Fig. 2d). HVA/DA ratios were significantly higher (p=0.020; Fig. 2e). This suggested that levels of the metabolites DOPAC and HVA tended to decline less than DA in late stage SIV infection.
Fig. 2.

Minocycline treatment initiated early in infection prevented late-stage SIV-induced striatal DA loss. Uninfected, SIV-infected untreated, and SIV-infected macaques treated with minocycline beginning at either 12 or 21 dpi were euthanized approximately 3 months postinoculation. Levels of DA (a), DOPAC (b), and HVA (c) were measured in the striatum and ratios of DOPAC/DA (d) and HVA/DA (e) were calculated. DA levels were lower in SIV-infected, untreated macaques compared with uninfected controls (p=0.003). Levels of the metabolites DOPAC and HVA were lower in SIV-infected, untreated macaques, but not significantly so (p=0.074 and 0.183, respectively). DOPAC/DA ratios tended to be higher in SIV-infected, untreated macaques, but this difference wasn’t significant (p=0.082). HVA/DA ratios were significantly higher in SIV-infected, untreated macaques (p=0.020). Minocycline treatment initiated early at 12 dpi prevented SIV-induced striatal DA loss (p<0.001) and prevented an increase in the ratios of DOPAC/DA and HVA/DA (p=0.004 and <0.001, respectively). Minocycline treatment initiated later at 21 dpi did not protect DA levels, nor did it protect the ratios of the metabolites to DA. Bars represent medians. Statistical comparisons were made using the Mann–Whitney test
Minocycline treatment beginning at 12 dpi preserved levels of DA (p<0.001; Fig. 2a) and ratios of DOPAC and HVA to DA (p=0.004 and <0.001, respectively; Fig. 2d, e); however later minocycline treatment beginning at 21 dpi did not protect against neurochemical dysfunction.
Decreased levels of DA in the striatum were not due to loss of dopaminergic projections to the basal ganglia
Damage to or loss of the neurons responsible for producing dopamine were two possible reasons for the finding of decreased DA in the striatum of SIV-infected macaques. To determine whether there was damage to dopaminergic projections to the basal ganglia, we performed Western blotting on basal ganglia homogenates for several proteins important for production of DA or present in dopaminergic neurons.
We first examined tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine production. There were no differences in total TH in the basal ganglia between uninfected, SIV-infected, or SIV-infected, minocycline-treated macaques (Fig. 3a, e).
Fig. 3.

Western blot analysis indicated no overt loss of dopaminergic projections to the basal ganglia in SIV-infection. a, e Quantitation of TH in the basal ganglia by Western blot analysis showed similar levels of TH among uninfected, SIV-infected, untreated and SIV-infected, minocycline-treated macaques. b, f Measurement of DAT in basal ganglia showed no significant difference between uninfected and SIV-infected, untreated macaques. c, g VMAT2 was unchanged in SIV infection, regardless of treatment. d, h Synaptophysin was moderately reduced in SIV infection, but was not rescued by minocycline treatment. Proteins of interest were normalized to GAPDH. Bars represent medians. Statistical comparisons were made using the Mann–Whitney test
To further assess the status of nigral projections to the basal ganglia, we next examined the dopaminergic presynaptic marker dopamine transporter (DAT), which is responsible for removing DA from the synaptic cleft to maintain synaptic DA homeostasis. There were no significant differences in DAT levels between uninfected, SIV-infected, and SIV-infected, minocycline-treated macaques (Fig. 3b, f). While TH and DAT are generally thought to be exclusively expressed by dopaminergic projections in the basal ganglia, there is a subgroup of striatal interneurons that possess these markers (Tepper et al. 2010). These interneurons, however, are not very numerous, making up less than 5% of striatal neurons (Unal et al. 2011), thus dopaminergic projections are the primary component of this measurement.
We next measured the more general presynaptic marker vesicular monoamine transporter 2 (VMAT2), which is responsible for packaging monoamines such as dopamine and serotonin into vesicles for subsequent release at the synaptic cleft. In the basal ganglia, dopaminergic projections far outnumber serotonergic projections, so VMAT2 is mainly of a marker of dopaminergic neurons, while not entirely specific. We found no difference in VMAT2 levels between uninfected, SIV-infected untreated, or SIV-infected, minocycline-treated macaques (Fig. 3c, g).
Finally, to assess general presynaptic health we measured synaptophysin, an integral membrane glycoprotein of presynaptic vesicles. Synaptophysin was significantly decreased in the basal ganglia of SIV-infected macaques but was not protected by minocycline treatment (Fig. 3d, h).
Together, these data suggest that lower levels of DA in the striatum of SIV-infected macaques in this model is not due to a loss of the enzyme responsible for its production, nor due to gross loss of dopaminergic innervation of the basal ganglia, therefore minocycline does not appear to act at this macroscopic level. However, we found general synaptic health mildly compromised, regardless of minocycline intervention.
DA decrease was partly due to increased MAO activity
The trend toward increased ratios of DA metabolite to DA (DOPAC/DA and HVA/DA; Figs. 1 and 2) in the late stage of infection suggested that increased DA turnover may occur during SIV infection. Monoamine oxidase (MAO) is the enzyme responsible for conversion of DA to DOPAC and one of the enzymes in the pathway for conversion of DA to HVA. We measured MAO activity in basal ganglia homogenates of the three groups of macaques using a luminescent method. MAO activity was significantly increased in both untreated and minocycline-treated SIV-infected macaques (p=0.003 and p=0.002, respectively; Fig. 4). To our knowledge, this is the first report directly demonstrating increased MAO activity in SIV infection.
Fig. 4.
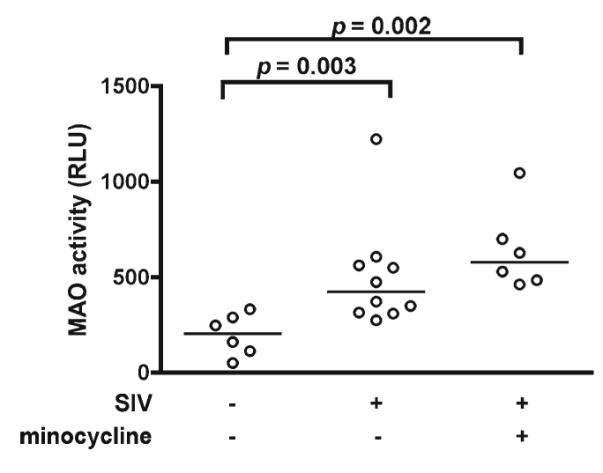
MAO activity was significantly increased in the basal ganglia of both untreated and minocycline-treated SIV-infected macaques. MAO activity was measured by a luminescent method in basal ganglia homogenates. Bars represent medians. Statistical comparisons were made using the Mann-Whitney test
Lower nitrotyrosine immunoreactivity in the CNS of minocycline-treated macaques
Dopaminergic neurons must constantly manage homeostasis in a particularly harsh metabolic environment. Minocycline is well known for its ability to dampen detrimental neuroinflammatory processes in a variety of animal models of neurological disease (Kim and Suh 2009). Nitrotyrosine (Y-NO) is a stable marker of oxidative/nitrosative stress and it is elevated in many inflammatory diseases (Halliwell 1992; Pacher et al. 2007). Using immunohistochemistry, our laboratory previously demonstrated reduced Y-NO in the deep white matter of SIV-infected macaques treated with minocycline beginning at 21 dpi (Follstaedt et al. 2008). We therefore examined whether the same was true for SIV-infected macaques treated with minocycline beginning at 12 dpi. We found that SIV-infected macaques tended to have higher levels of Y-NO than uninfected macaques (p=0.095) and that SIV-infected, minocycline-treated macaques tended to have lower Y-NO levels in deep white matter than SIV-infected, untreated macaques (p=0.052, Fig. 5). There was no direct correlation of Y-NO staining with levels of striatal DA in SIV-infected macaques (data not shown). These data suggest that SIV-infected, minocycline-treated macaques tend to have reduced levels of oxidative/nitrosative stress.
Fig. 5.
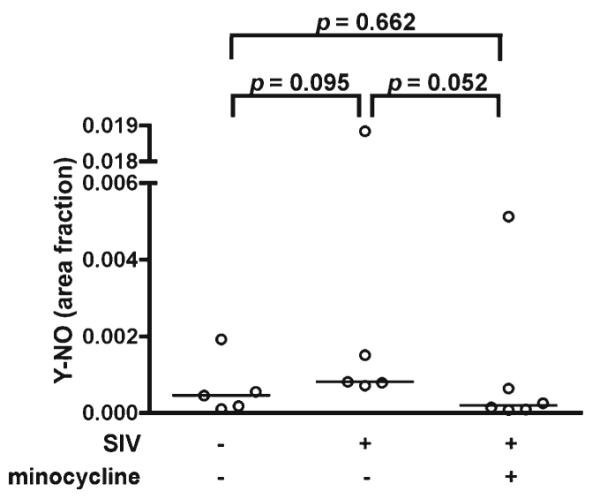
Oxidative/nitrosative stress tended to be lower in SIV-infected macaques treated with minocycline. Immunohistochemical staining of Y-NO in deep white matter was quantified and expressed as a fraction of the total area measured. Y-NO staining tended to be higher in SIV-infected untreated macaques compared to uninfected controls (p= 0.095) and tended to be lower in SIV-infected, minocycline-treated macaques compared to SIV-infected, untreated macaques (p=0.052). Bars represent medians. Statistical comparisons were made using the Mann-Whitney test
DA levels in SIV-infected macaques did not correlate with markers of neuroinflammation or viral loads
To further investigate the role neuroinflammation and viral loads might play in striatal DA loss in late stage SIV infection, we examined potential correlations between striatal DA levels and markers of neuroinflammation and viral loads in SIV-infected macaques in the late stage of disease. CD68 (a marker of macrophage/microglial activation), MHC class II (a marker of macrophage/microglial and endothelial cell activation), and GFAP (a marker of astrocyte activation) were measured by quantitative immunohistochemistry in the deep white matter. Striatal DA levels did not correlate with levels of CD68, MHC class II, or GFAP staining in SIV-infected macaques in the late stage of disease (data not shown). We also measured viral loads in the basal ganglia and CSF of SIV-infected macaques by qRT-PCR. Striatal DA levels in SIV-infected macaques in the late stage of disease did not correlate with viral loads in any of the compartments examined (data not shown). Thus, late stage neuroinflammation and viral loads do not predict striatal DA levels in SIV-infected macaques.
Discussion
In these studies, striatal levels of DA were significantly lower in late stage SIV infection. Early intervention with oral minocycline (beginning at 12 dpi, but not 21 dpi) prevented this decline. This has important implications for developing and testing neurotherapeutics for HAND. Successful intervention with minocycline occurred at a significant neuroimmunological juncture in this SIV/macaque model. The time between 12 and 21 dpi spans the transition from the acute to the asymptomatic phase of peripheral disease and also encompasses an immunological shift in the CNS of this SIV/macaque model (Clements et al. 2008; Witwer et al. 2009). In the acute phase of infection virus enters the brain as early as 4 dpi (Clements et al. 2002), rapidly inducing a coordinated innate immune response (i.e.- IFNβ, MxA, IL-10, others) and neuroinflammation (CD68, MHC class II, GFAP; Witwer et al. 2009). As CNS viral loads begin to be controlled, both the innate immune response and neuroinflammation are downregulated by 10 dpi, but then begin to rebound by 21 dpi (Witwer et al. 2009). The span between 12 and 21 dpi also is a critical time in the progression of mitogen-activated protein kinase (MAPK) dysregulation in this SIV/macaque model. In this nine day period, MAPK expression in the brain is in the process of shifting the balance away from the neuroprotective pERK toward the more neurodegenerative pJNK/p-p38 MAPKs (Barber et al. 2004). Together, these data emphasize a critical window of neuroprotection afforded by minocycline to the nigrostriatal dopaminergic system.
The timing of successful therapeutic intervention with respect to SIV neuropathogenesis is important to note considering recent negative results from a phase II clinical trial of minocycline to improve neurocognition in HIV-infected individuals with progressive neurocognitive decline (Sacktor et al. 2011). In that study, minocycline failed to improve neurocognition over the course of the 24-week study, as measured by the NPZ-8 neuropsychological composite test score. The fact that the HIV-infected individuals in this clinical trial were already experiencing neurocognitive impairment indicates that they already were in an advanced stage of HIV CNS disease. In our study, we found early treatment to be necessary for minocycline’s protection of striatal DA levels in SIV-infected macaques, so minocycline administered after the onset of neurocognitive decline in HIV-infected individuals might be too late to be effective. This may be a broader issue regarding testing neurotherapeutics for HAND. Many clinical trials for HAND treated HIV-infected individuals after the onset of neurocognitive impairment whereas it may be more beneficial to pursue treatments that prevent the genesis of neurocognitive impairment.
Our data support other studies showing loss of DA in HIV/SIV infection (Ferris et al. 2008; Kumar et al. 2009) and increased turnover of DA in SIV infection (Czub et al. 2001). Unlike others (Gelman et al. 2006), we did not find significant changes in markers of dopaminergic projections to the basal ganglia (TH, DAT or VMAT2), suggesting that dopaminergic dysfunction may precede prominent dopaminergic neuronal loss or damage in this SIV model and therefore minocycline must protect at another level. While these presynaptic proteins may be present at relatively normal levels in the SIV-infected, untreated macaques, this says nothing of their functionality. Specifically, DAT function has been shown to be decreased by both gp120 and Tat, even though total protein levels may be unchanged (Aksenova et al. 2006; Theodore et al. 2006; Wallace et al. 2006). Additionally, TH and DAT function are decreased by oxidative and nitrosative stress (Ara et al. 1998; Park et al. 2002; Huang et al. 2003), which is a prominent feature of HIV/SIV infection (Steiner et al. 2006).
One potential source of oxidative stress and contributor to decreased DA in SIV infection is increased MAO activity. Hydrogen peroxide (H2O2) is a potentially damaging product of the MAO reaction that is well known to cause neuronal damage, particularly to dopaminergic neurons, and is implicated in the pathogenesis of several neurodegenerative diseases (Halliwell 1992; Berg et al. 2004; Agrawal et al. 2010). ROS produced by increased MAO activity in SIV infection could exacerbate oxidative stress and neuroinflammation. In fact, simply inducing MAO B expression in a transgenic mouse model increases astrocyte and microglial activation, superoxide production, and damage to TH+neurons (Mallajosyula et al. 2008). Interestingly, that group demonstrated that damage to TH+ neurons caused by MAO B elevation could be abrogated by the antioxidant EUK189 or minocycline. In our study, minocycline-treated macaques had elevated MAO activity, similar to untreated macaques, indicating that minocycline’s protection from DA loss is not due to direct effects on MAO activity. Given that MAO is an outer mitochondrial membrane protein and that minocycline can to bind to mitochondrial membranes (Antonenko et al. 2010), we examined MAO activity in the context of intact mitochondria and found no effect of minocycline (data not shown), confirming previous findings of Du and colleagues (2001).
Instead, our data suggest that SIV-infected minocycline-treated macaques may be better able to manage the oxidative consequences of SIV CNS infection and elevated MAO activity. Minocycline has antioxidant properties, particularly toward peroxynitrite, a ROS responsible for Y-NO formation (Kraus et al. 2005; Schildknecht et al. 2011). SIV-infected minocycline-treated macaques tended to have lower levels of oxidative/nitrosative stress, as measured by nitrotyrosine staining in the deep white matter compared to their untreated counterparts. Although this data only trends toward statistical significance, this suggests that minocycline might have a mitigating effect on the neuroinflammatory environment caused by SIV CNS infection, thus allowing these macaques to maintain DA homeostasis.
However, simply blunting late stage neuroinflammation may not be enough to protect from dopaminergic dysfunction. We did not find a correlation between neuroinflammatory markers or viral loads and striatal DA levels in SIV infected macaques, demonstrating that late stage neuroinflammation did not predict dopaminergic dysfunction. Furthermore, we previously showed that SIV-infected macaques treated with minocycline at 21 dpi had lower CNS viral loads and were broadly protected from late stage neuroinflammation as measured by pathological CNS lesion severity and immunohistochemical markers such as CD68 and MHC class II (Zink et al. 2005). However, this study reveals that those macaques were not protected from late stage dopaminergic dysfunction. These data suggest that early events in neuropathogenesis (between 12 and 21 dpi in this SIV/macaque model) and perhaps an accumulation of neuroinflammatory insults over time profoundly impact late stage dopaminergic system dysfunction more than the late stage neuroinflammatory condition indicates.
In summary, these data show that early minocycline treatment protects SIV-infected macaques from a decrease in striatal DA content. Minocycline’s anti-inflammatory and antioxidant abilities might enable SIV-infected macaques to better manage the neuroinflammatory environment caused by oxidative stress due to SIV CNS infection and overactive MAO than their untreated counterparts, and thus able to maintain proper DA homeostasis. However, end-stage neuroinflammation did not completely predict striatal DA levels in SIV-infected macaques.
Initiation of minocycline treatment at 12 dpi, immediately following the decline in acute phase inflammation and innate immune response in the CNS, may be critical in dampening the increasing neuroinflammation that occurs even in the asymptomatic phase of disease, thus protecting dopaminergic neurons from dysfunction late in disease.
Minocycline failed to protect animals from neurodegeneration in some models of MPTP, Parkinson’s and Huntington’s disease (Diguet et al. 2004) and, in some cases such as the recent clinical trial with amyotrophic lateral sclerosis, even found to be detrimental (Gordon et al. 2007). Since the therapeutic window of striatal DA protection in SIV-infected macaques occurred at a critical neuroimmunological juncture, this study highlights the importance of timing of treatment initiation with respect to disease pathogenesis, which has broader implications for testing neurotherapeutics to treat HAND.
Acknowledgments
Studies were supported by grants from the National Institute of Mental Health: R01 MH069116, R01 MH087233, and R01 MH085554.
Footnotes
Conflict of Interest The authors declare that they have no conflicts of interest.
Authors who are guarantors: MCZ
Contributor Information
Kelly A. Meulendyke, Department of Molecular and Comparative Pathobiology, Johns Hopkins University School of Medicine, 733 North Broadway Street, BRB 819, Baltimore, MD 21205, USA
Mikhail V. Pletnikov, Department of Molecular and Comparative Pathobiology, Johns Hopkins University School of Medicine, 733 North Broadway Street, BRB 819, Baltimore, MD 21205, USA; Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, 600 N Wolfe St., CMSC-9-111, Baltimore, MD 21287, USA
Elizabeth L. Engle, Department of Molecular and Comparative Pathobiology, Johns Hopkins University School of Medicine, 733 North Broadway Street, BRB 819, Baltimore, MD 21205, USA
Patrick M. Tarwater, Department of Biostatistics and Epidemiology, Foster School of Medicine, Texas Tech University Health Sciences Center, 4801 Alberta Avenue, El Paso, TX 79905, USA
David R. Graham, Department of Molecular and Comparative Pathobiology, Johns Hopkins University School of Medicine, 733 North Broadway Street, BRB 819, Baltimore, MD 21205, USA
M. Christine Zink, Department of Molecular and Comparative Pathobiology, Johns Hopkins University School of Medicine, 733 North Broadway Street, BRB 819, Baltimore, MD 21205, USA.
References
- Agrawal L, Louboutin JP, Marusich E, Reyes BA, Van Bockstaele EJ, Strayer DS. Dopaminergic neurotoxicity of HIV-1 gp120: reactive oxygen species as signaling intermediates. Brain Res. 2010;1306:116–130. doi: 10.1016/j.brainres.2009.09.113. doi:10.1016/j.brainres.2009.09.113. [DOI] [PubMed] [Google Scholar]
- Aksenova MV, Silvers JM, Aksenov MY, Nath A, Ray PD, Mactutus CF, Booze RM. HIV-1 Tat neurotoxicity in primary cultures of rat midbrain fetal neurons: changes in dopamine transporter binding and immunoreactivity. Neurosci Lett. 2006;395(3):235–239. doi: 10.1016/j.neulet.2005.10.095. doi:10.1016/j.neulet.2005.10.095. [DOI] [PubMed] [Google Scholar]
- Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27(1):86–92. doi: 10.1055/s-2006-956759. doi:10.1055/s-2006-956759. [DOI] [PubMed] [Google Scholar]
- Antonenko YN, Rokitskaya TI, Cooper AJ, Krasnikov BF. Minocycline chelates Ca2+, binds to membranes, and depolarizes mitochondria by formation of Ca2+-dependent ion channels. J Bioenerg Biomembr. 2010;42(2):151–163. doi: 10.1007/s10863-010-9271-1. doi:10.1007/s10863-010-9271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phe-nyl-1,2,3,6-tetrahydropyridine (MPTP) Proc Natl Acad Sci U S A. 1998;95(13):7659–7663. doi: 10.1073/pnas.95.13.7659. doi:10.1073/pnas.95.13.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164(2):355–362. doi: 10.1016/S0002-9440(10)63125-2. doi:10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnum CJ, Tansey MG. Modeling neuroinflammatory pathogenesis of Parkinson’s disease. Prog Brain Res. 2010;184:113–132. doi: 10.1016/S0079-6123(10)84006-3. doi:10.1016/S0079-6123(10)84006-3. [DOI] [PubMed] [Google Scholar]
- Berg D, Youdim MB, Riederer P. Redox imbalance. Cell Tissue Res. 2004;318(1):201–213. doi: 10.1007/s00441-004-0976-5. doi:10.1007/s00441-004-0976-5. [DOI] [PubMed] [Google Scholar]
- Berger JR, Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol. 2000;14(3):214–221. doi: 10.1177/026988110001400304. doi:10.1177/026988110001400304. [DOI] [PubMed] [Google Scholar]
- Berger JR, Kumar M, Kumar A, Fernandez JB, Levin B. Cerebrospinal fluid dopamine in HIV-1 infection. AIDS. 1994;8(1):67–71. doi: 10.1097/00002030-199401000-00010. doi:10.1097/00002030-199401000-00010. [DOI] [PubMed] [Google Scholar]
- Campbell JH, Burdo TH, Autissier P, Bombardier JP, Westmoreland SV, Soulas C, Gonzalez RG, Ratai EM, Williams KC. Minocycline inhibition of monocyte activation correlates with neuronal protection in SIV NeuroAIDS. PLoS One. 2011;6(4):e18688. doi: 10.1371/journal.pone.0018688. doi:10.1371/journal.pone.0018688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak M, Jr, Tarwater PM, Lifson JD, Zink MC. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. Infect Dis. 2002;186(7):905–913. doi: 10.1086/343768. doi:10.1086/343768. [DOI] [PubMed] [Google Scholar]
- Clements JE, Li M, Gama L, Bullock B, Carruth LM, Mankowski JL, Zink MC. The central nervous system is a viral reservoir in simian immunodeficiency virus-infected macaques on combined antiretroviral therapy: a model for human immunodeficiency virus patients on highly active antiretroviral therapy. J Neurovirol. 2005;11(2):180–189. doi: 10.1080/13550280590922748-1. doi:10.1080/13550280590922829. [DOI] [PubMed] [Google Scholar]
- Clements JE, Mankowski JL, Gama L, Zink MC. The accelerated simian immunodeficiency virus macaque model of human immunodeficiency virus-associated neurological disease: from mechanism to treatment. J Neurovirol. 2008;14(4):309–317. doi: 10.1080/13550280802132832. doi:10.1080/13550280802132832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czub S, Koutsilieri E, Sopper S, Czub M, Stahl-Hennig C, Muller JG, Pedersen V, Gsell W, Heeney JL, Gerlach M, Gosztonyi G, Riederer P, ter Meulen V. Enhancement of central nervous system pathology in early simian immunodeficiency virus infection by dopaminergic drugs. Acta Neuropathol. 2001;101(2):85–91. doi: 10.1007/s004010000313. doi:10.1007/s004010000313. [DOI] [PubMed] [Google Scholar]
- Diguet E, Fernagut PO, Wei X, Du Y, Rouland R, Gross C, Bezard E, Tison F. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur J Neurosci. 2004;19(12):3266–3276. doi: 10.1111/j.0953-816X.2004.03372.x. doi:10.1111/j.0953-816X.2004.03372.x. [DOI] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98(25):14669–14674. doi: 10.1073/pnas.251341998. doi:10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, Mactutus CF, Booze RM. Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev. 2008;32(5):883–909. doi: 10.1016/j.neubiorev.2008.01.004. doi:10.1016/j. neubiorev.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follstaedt SC, Barber SA, Zink MC. Mechanisms of minocycline-induced suppression of simian immunodeficiency virus encephalitis: inhibition of apoptosis signal-regulating kinase 1. J Neurovirol. 2008;14(5):376–388. doi: 10.1080/13550280802199898. doi:10.1080/13550280802199898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman BB. The neuropathology of HIV. Handb Clin Neurol. 2007;85:301–317. doi: 10.1016/S0072-9752(07)85018-4. doi:10.1016/S0072-9752(07)85018-4. [DOI] [PubMed] [Google Scholar]
- Gelman BB, Spencer JA, Holzer CE, 3rd, Soukup VM. Abnormal striatal dopaminergic synapses in National NeuroAIDS Tissue Consortium subjects with HIV encephalitis. J Neuroimmune Pharmacol. 2006;1(4):410–420. doi: 10.1007/s11481-006-9030-6. doi:10.1007/s11481-006-9030-6. [DOI] [PubMed] [Google Scholar]
- Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H, Neville HE, Boylan K, Mozaffar T, Belsh JM, Ravits J, Bedlack RS, Graves MC, McCluskey LF, Barohn RJ, Tandan R, Western ALS Study Group Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6(12):1045–1053. doi: 10.1016/S1474-4422(07)70270-3. doi:10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59(5):1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. doi:10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2011;75(23):2087–2096. doi: 10.1212/WNL.0b013e318200d727. doi:10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Huang NK, Shyue SK, Chern Y. Hydrogen peroxide induces loss of dopamine transporter activity: a calcium-dependent oxidative mechanism. J Neurochem. 2003;86(5):1247–1259. doi: 10.1046/j.1471-4159.2003.01936.x. doi:10.1046/j.1471-4159.2003.01936.x. [DOI] [PubMed] [Google Scholar]
- Itoh K, Mehraein P, Weis S. Neuronal damage of the substantia nigra in HIV-1 infected brains. Acta Neuropathol. 2000;99(4):376–384. doi: 10.1007/s004010051139. doi:10.1007/s004010051139. [DOI] [PubMed] [Google Scholar]
- Jordan J, Fernandez-Gomez FJ, Ramos M, Ikuta I, Aguirre N, Galindo MF. Minocycline and cytoprotection: shedding new light on a shadowy controversy. Curr Drug Deliv. 2007;4(3):225–231. doi: 10.2174/156720107781023938. doi:10.2174/156720107781023938. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4(3):307–318. doi: 10.2174/157016206777709384. doi:10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196(2):168–179. doi: 10.1016/j.bbr.2008.09.040. doi:10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- Koutsilieri E, Sopper S, Scheller C, ter Meulen V, Riederer P. Parkinsonism in HIV dementia. J Neural Transm. 2002;109(5-6):767–775. doi: 10.1007/s007020200063. doi:10.1007/s007020200063. [DOI] [PubMed] [Google Scholar]
- Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem. 2005;94(3):819–827. doi: 10.1111/j.1471-4159.2005.03219.x. doi:10.1111/j.1471-4159.2005.03219.x. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Borodowsky I, Fernandez B, Gonzalez L, Kumar M. Human immunodeficiency virus type 1 RNA Levels in different regions of human brain: quantification using real-time reverse transcriptase-polymerase chain reaction. J Neurovirol. 2007;13(3):210–224. doi: 10.1080/13550280701327038. doi:10.1080/13550280701327038. [DOI] [PubMed] [Google Scholar]
- Kumar AM, Fernandez J, Singer EJ, Commins D, Waldrop-Valverde D, Ownby RL, Kumar M. Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol. 2009;15:1–18. doi: 10.1080/13550280902973952. doi:10.1080/13550280902973952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AM, Ownby RL, Waldrop-Valverde D, Fernandez B, Kumar M. Human immunodeficiency virus infection in the CNS and decreased dopamine availability: relationship with neuropsychological performance. J Neurovirol. 2011;17(1):26–40. doi: 10.1007/s13365-010-0003-4. doi:10.1007/s13365-010-0003-4. [DOI] [PubMed] [Google Scholar]
- Mallajosyula JK, Kaur D, Chinta SJ, Rajagopalan S, Rane A, Nicholls DG, Di Monte DA, Macarthur H, Andersen JK. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS One. 2008;3(2):e1616. doi: 10.1371/journal.pone.0001616. doi:10.1371/journal.pone.0001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. doi:10.1038/sj. cdd.4401577. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4(9):543–555. doi: 10.1016/S1474-4422(05)70165-4. doi:10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr Opin Neurol. 2006;19(4):358–361. doi: 10.1097/01.wco.0000236614.51592.ca. doi:10.1097/01.wco.0000236614.51592.ca. [DOI] [PubMed] [Google Scholar]
- Nath A, Anderson C, Jones M, Maragos W, Booze R, Mactutus C, Bell J, Hauser KF, Mattson M. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J Psychopharmacol. 2000;14(3):222–227. doi: 10.1177/026988110001400305. doi:10.1177/026988110001400305. [DOI] [PubMed] [Google Scholar]
- Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19(6):525–535. doi: 10.1002/ana.410190603. doi:10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- Nosheny RL, Bachis A, Aden SA, De Bernardi MA, Mocchetti I. Intrastriatal administration of human immunodeficiency virus-1 glycoprotein 120 reduces glial cell-line derived neurotrophic factor levels and causes apoptosis in the substantia nigra. J Neurobiol. 2006;66(12):1311–1321. doi: 10.1002/neu.20288. doi:10.1002/neu.20288. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. doi:10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SU, Ferrer JV, Javitch JA, Kuhn DM. Peroxynitrite inactivates the human dopamine transporter by modification of cysteine 342: potential mechanism of neurotoxicity in dopamine neurons. J Neurosci. 2002;22(11):4399–4405. doi: 10.1523/JNEUROSCI.22-11-04399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikov MV, Rubin SA, Schwartz GJ, Carbone KM, Moran TH. Effects of neonatal rat Borna disease virus (BDV) infection on the postnatal development of the brain monoaminergic systems. Brain Res Dev Brain Res. 2000;119(2):179–185. doi: 10.1016/s0165-3806(99)00168-6. doi:10.1016/S0165-3806(99)00168-6. [DOI] [PubMed] [Google Scholar]
- Ratai EM, Bombardier JP, Joo CG, Annamalai L, Burdo TH, Campbell J, Fell R, Hakimelahi R, He J, Autissier P, Lentz MR, Halpern EF, Masliah E, Williams KC, Westmoreland SV, Gonzalez RG. Proton magnetic resonance spectroscopy reveals neuroprotection by oral minocycline in a nonhuman primate model of accelerated NeuroAIDS. PLoS One. 2010;5(5):e10523. doi: 10.1371/journal.pone.0010523. doi:10.1371/journal. pone.0010523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh JA, Nath A. Developments in HIV neuropathogenesis. Curr Pharm Des. 2006;12(9):1023–1044. doi: 10.2174/138161206776055877. doi:10.2174/138161206776055877. [DOI] [PubMed] [Google Scholar]
- Sacktor N, Miyahara S, Deng L, Evans S, Schifitto G, Cohen BA, Paul R, Robertson K, Jarocki B, Scarsi K, Coombs RW, Zink MC, Nath A, Smith E, Ellis RJ, Singer E, Weihe J, McCarthy S, Hosey L, Clifford DB. Minocycline treatment for HIV-associated cognitive impairment: results from a randomized trial. Neurology. 2011;77(12):1135–1142. doi: 10.1212/WNL.0b013e31822f0412. doi:10.1212/WNL.0b013e31822f0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar AM, Czudek C, Reynolds GP. Dopamine deficits in the brain: the neurochemical basis of parkinsonian symptoms in AIDS. Neuroreport. 1996;7(4):910–912. doi: 10.1097/00001756-199603220-00015. doi:10.1097/00001756-199603220-00015. [DOI] [PubMed] [Google Scholar]
- Scheller C, Sopper S, Jenuwein M, Neuen-Jacob E, Tatschner T, Grunblatt E, ter Meulen V, Riederer P, Koutsilieri E. Early impairment in dopaminergic neurotransmission in brains of SIV-infected rhesus monkeys due to microglia activation. J Neurochem. 2005;95(2):377–387. doi: 10.1111/j.1471-4159.2005.03373.x. doi:10.1111/j.1471-4159.2005.03373.x. [DOI] [PubMed] [Google Scholar]
- Scheller C, Arendt G, Nolting T, Antke C, Sopper S, Maschke M, Obermann M, Angerer A, Husstedt IW, Meisner F, Neuen-Jacob E, Muller HW, Carey P, Ter Meulen V, Riederer P, Koutsilieri E. Increased dopaminergic neurotransmission in therapy-naive asymptomatic HIV patients is not associated with adaptive changes at the dopaminergic synapses. J Neural Transm. 2010;117(6):699–705. doi: 10.1007/s00702-010-0415-6. doi:10.1007/s00702-010-0415-6. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Pape R, Muller N, Robotta M, Marquardt A, Burkle A, Drescher M, Leist M. Neuroprotection by minocycline caused by direct and specific scavenging of peroxynitrite. J Biol Chem. 2011;286(7):4991–5002. doi: 10.1074/jbc.M110.169565. doi:10.1074/jbc.M110.169565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvers JM, Aksenov MY, Aksenova MV, Beckley J, Olton P, Mactutus CF, Booze RM. Dopaminergic marker proteins in the substantia nigra of human immunodeficiency virus type 1-infected brains. J Neurovirol. 2006;12(2):140–145. doi: 10.1080/13550280600724319. doi:10.1080/13550280600724319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner J, Haughey N, Li W, Venkatesan A, Anderson C, Reid R, Malpica T, Pocernich C, Butterfield DA, Nath A. Oxidative stress and therapeutic approaches in HIV dementia. Antioxid Redox Signal. 2006;8(11-12):2089–2100. doi: 10.1089/ars.2006.8.2089. doi:10.1089/ars.2006.8.2089. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Tecuapetla F, Koos T, Ibanez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150. doi: 10.3389/fnana.2010.00150. doi:10.3389/fnana.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore S, Stolberg S, Cass WA, Maragos WF. Human immunodeficiency virus-1 protein tat and methamphetamine interactions. Ann N Y Acad Sci. 2006;1074:178–190. doi: 10.1196/annals.1369.018. doi:10.1196/annals.1369.018. [DOI] [PubMed] [Google Scholar]
- UNAIDS [Accessed July 25, 2011];Global report: UNAIDS report on the global AIDS epidemic 2010. Joint United Nations Programme on HIV/AIDS (UNAIDS) 2010 http://www.unaids.org/documents/20101123_GlobalReport_em.pdf. [Google Scholar]
- Unal B, Ibanez-Sandoval O, Shah F, Abercrombie ED, Tepper JM. Distribution of tyrosine hydroxylase-expressing interneurons with respect to anatomical organization of the neostriatum. Front Syst Neurosci. 2011;5:41. doi: 10.3389/fnsys.2011.00041. doi:10.3389/fnsys.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DR, Dodson S, Nath A, Booze RM. Estrogen attenuates gp120- and tat1-72-induced oxidative stress and prevents loss of dopamine transporter function. Synapse. 2006;59(1):51–60. doi: 10.1002/syn.20214. doi:10.1002/syn.20214. [DOI] [PubMed] [Google Scholar]
- Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci. 2002;25:537–562. doi: 10.1146/annurev.neuro.25.112701.142822. doi:10.1146/annurev. neuro25.112701.142822. [DOI] [PubMed] [Google Scholar]
- Witwer KW, Gama L, Li M, Bartizal CM, Queen SE, Varrone JJ, Brice AK, Graham DR, Tarwater PM, Mankowski JL, Zink MC, Clements JE. Coordinated regulation of SIV replication and immune responses in the CNS. PLoS One. 2009;4(12):e8129. doi: 10.1371/journal.pone.0008129. doi:10.1371/journal.pone.0008129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauli G, Secchiero P, Rodella L, Gibellini D, Mirandola P, Mazzoni M, Milani D, Dowd DR, Capitani S, Vitale M. HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J Biol Chem. 2000;275(6):4159–4165. doi: 10.1074/jbc.275.6.4159. doi:10.1074/jbc.275.6.4159. [DOI] [PubMed] [Google Scholar]
- Zink MC, Suryanarayana K, Mankowski JL, Shen A, Piatak M, Jr, Spelman JP, Carter DL, Adams RJ, Lifson JD, Clements JE. High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J Virol. 1999;73(12):10480–10488. doi: 10.1128/jvi.73.12.10480-10488.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink MC, Coleman GD, Mankowski JL, Adams RJ, Tarwater PM, Fox K, Clements JE. Increased macrophage chemoattractant protein-1 in cerebrospinal fluid precedes and predicts simian immunodeficiency virus encephalitis. J Infect Dis. 2001;184(8):1015–1021. doi: 10.1086/323478. doi:10.1086/323478. [DOI] [PubMed] [Google Scholar]
- Zink MC, Uhrlaub J, DeWitt J, Voelker T, Bullock B, Mankowski J, Tarwater P, Clements J, Barber S. Neuroprotective and anti-human immunodeficiency virus activity of minocycline. JAMA. 2005;293(16):2003–2011. doi: 10.1001/jama.293.16.2003. doi:10.1001/jama.293.16.2003. [DOI] [PubMed] [Google Scholar]