

Abstract
IL-21 is a multi-functional cytokine which can promote survival, proliferation and activation of T and B lymphocytes including CD8 T cells. Previous studies have shown that autoimmune CD8+ T cells are the primary pathogenic effector cell in coxsackievirus B3 (CVB3) induced myocarditis in C57Bl/6 mice. To evaluate the role of IL-21 in promoting CD8+ T cell mediated cardiac injury in myocarditis, C57Bl/6 and IL-21RKO mice were infected with CVB3. IL-21RKO mice developed significantly less myocarditis than C57Bl/6 animals although cardiac virus titers were equivalent between the mouse strains. Numbers of CD8+IFNγ+ cells were decreased in IL-21RKO mice but numbers of either CD4+IFNγ+ or CD4+IL-4+ cells were not significantly different from C57Bl/6 animals indicating a selective effect of IL-21 signaling on the CD8+ T cell response. To confirm that IL-21 signaling exclusively functions at the level of the CD8+ T cell in CVB3 induced myocarditis, purified CD8+ cells were isolated from either C57Bl/6 or IL-21RKO donors and adoptively transferred into CD8KO recipients prior to CVB3 infection. CD8KO recipients given either C57Bl/6 or IL-21RKO CD8+ cells showed equivalent reconstitution of the CD8+ cells in the spleen but the recipients given C57Bl/6 CD8+ cells showed significantly greater myocarditis than recipients of IL-21RKO CD8+ cells. These data demonstrate that IL-21 signaling directly in the CD8+ cell population is required for CVB3-induced myocarditis.
Keywords: coxsackievirus B3, myocarditis, IL-21R, CD8 T cells, autoimmunity
INTRODUCTION
Myocarditis is an inflammation of the cardiac muscle which follows microbial infections (Huber, 2008). Among viruses, enteroviruses including coxsackie B viruses are common etiologic agents (Bowles et al., 2003; Bowles et al., 2002). Although infectious agents act as a trigger for myocarditis, there is considerable debate as to the actual mechanism(s) of myocardial injury. Viruses directly cause cellular dysfunction either through induced cell death, shut down of cell RNA and protein synthesis or viral protease cleavage of contractile proteins (Badorff et al., 1999; Rueckert, 1996). Additionally, cytokines such as IL-1β, IL-6 and TNFα which are elicited from resident cells in the heart subsequent to infection can suppress contractility leading to cardiac dysfunction (Freeman et al., 1998). Finally, host immune responses to infection may kill myocytes leading to cardiac stress. Host response can be specifically directed toward virally infected cardiocytes or infection can trigger autoimmunity to cardiac antigens (autoimmunity) which destroys both infected and uninfected myocytes (Rose, 2008). Autoantibodies and cytolytic T cells to heart antigens are found in patients with myocarditis (Maisch, 1989; Maisch et al., 1993), and myocardial inflammation has been shown by adoptive transfer of anti-myosin antibodies in mice (Liao et al., 1993; Liao et al., 1995). Immunization of mice with cardiac myosin results in myocarditis which closely resembles the disease resulting from coxsackievirus B3 (CVB3) infection (Fairweather et al., 2005; Fairweather et al., 2001; Kaya et al., 2002). T cells are primarily responsible for cardiac injury in the CVB3 model of myocarditis in mice (Woodruff and Woodruff, 1974). Both CD4+ and CD8+ T cells are activated during infection (Lodge et al., 1987) but respond to different types of antigen. Evidence indicates that the CD4+ cell response recognizes infected but not uninfected myocytes while the CD8+ effector cells react only to uninfected myocytes through recognition of cardiac myosin (Guthrie et al., 1984; Huber and Cunningham, 1996; Huber and Gauntt, 2000; Huber and Lodge, 1984; Huber and Lodge, 1986; Huber et al., 1987). Of the two T cell subsets, the CD8+ cells are the primary mediator of cardiac injury. However, without CD4+ cell activation, the autoimmune CD8+ cell response is prevented (Huber et al., 2002).
A major question is what environmental factors during infection lead to autoimmune CD8+ T cell activation. IL-21 is a multi-functional cytokines which is a member of the type 1 cytokine family (Parrish-Novak et al., 2000) and is primarily produced by CD4+ and natural killer T cells (Coquet et al., 2007; Nurieva et al., 2007). IL-21 signals through the IL-21R which consists of a specific IL-21Rα chain and the common γ chain used by multiple other cytokines (IL-2, IL-4, IL-7, IL-9 and IL-15) (Spolski and Leonard, 2008). IL-21 signaling primarily activates the Jak/STAT pathway with STAT1 and STAT3 being the predominant targets (de Totero et al., 2008). It was initially proposed that IL-21 was also a Th17-derived cytokine and was required for Th17 differentiation and development of EAE (Korn et al., 2007; Nurieva et al., 2007; Zhou et al., 2007). However, recent studies performing intracellular staining for cytokines showed that the IL-21-producing cells in Th17 cells represent an independent subset from the IL-17-producing cells, and that Th17 cell differentiation and EAE are independent of IL-21 (Coquet et al., 2008; Sonderegger et al., 2008; Suto et al., 2008). Some studies indicate that IL-21 can cooperate with IL-27 in activation of regulatory type 1 (Tr1) cells (Pot et al., 2010) and also suppress immune responses through promotion of IL-10 expression (Spolski et al., 2009; Spolski and Leonard, 2011). Thus, this cytokine has both pro- and anti-inflammatory functions depending upon cytokine environment. IL-21 is the primary cytokine secreted by CD4 T follicular helper (Tfh) cells, a newly characterized subset of effector CD4 T cells that localize in the germinal centers (GC), and are essential for germinal center formation through a direct effect on B cells (Bryant et al., 2007; Chtanova et al., 2004; Fazilleau et al., 2009; McHeyzer-Williams et al., 2009). Recent studies have shown that IL-21 is not required for the generation of Tfh cells, but Tfh cell-derived IL-21 is essential for formation of GC by acting directly on B cells (Linterman et al., 2010; Zotos et al.). Another major function of IL-21 is on CD8+ cell activation, proliferation, survival and cytolytic activity (Frederiksen et al., 2008; Li et al., 2005; Novy et al., 2011; Zeng et al., 2005). Also, IL-21 has been shown to promote conversion of CD8+ effector to memory cells (Barker et al., 2010a; Barker et al., 2010b; Barker et al., 2007; Cui et al., 2011).
Based on the abundant literature that IL-21 promotes autoimmune and CD8+ T cell responses, the current study investigated whether IL-21 is essential to induction of autoimmune CD8+ T cells in the CVB3 model of myocarditis. The studies demonstrate that mice lacking the IL-21R fail to develop myocarditis despite high virus titers. CD8+ cell responses are not exclusively affected by IL-21R deficiency. While total numbers of CD4+ cells are increased in the spleens of infected and uninfected IL-21RKO mice, CD4+ IFNγ+ cell responses are reduced in the absence of IL-21 signaling. CD8+ T cell activation is substantially reduced, but since CD8+ cell responses during CVB3 infection depend upon IFNγ produced by CD4Th1 cells, this effect could be due either to direct defects in IL-21 signaling in the CD8+ cell population or to indirect effects of IL-21 defective signaling in the CD4+ cells. Adoptive transfer of CD8+ IL-21RKO cells into CD8KO recipients demonstrates that IL-21 signaling directly through the CD8+ effector population promotes myocarditis susceptibility.
MATERIALS AND METHODS
Mice
Male C57Bl/6 and C57Bl/6 CD8KO (B6.129S2-Cd8atm1Mak/J) mice were purchased from Jackson Laboratories, Bar Harbor ME. IL-21RKO transgenic mice on the C57Bl/6 background were previously described (Ozaki et al., 2002) and a breeding colony of these mice was maintained at the University of Vermont. All mice were 5–7 weeks of age when infected. All of the studies have been reviewed and approved by the University of Vermont Institutional Animal Care and Use Committee.
Virus
The H3 variant of CVB3 was made from an infectious cDNA clone as described previously(Knowlton et al., 1996).
Infection of mice
Mice were injected intraperitoneally (i.p.) with 102 plaque forming units (PFU) virus in 0.5 ml PBS. Animals were killed when mortibund or 7 days after infection.
Virus titers
Hearts were asceptically removed from the animals, weighed, homogenized in RPMI 1640 medium containing 5% fetal bovine serum (FBS), L-glutamine, streptomycin and penicillin. Cellular debris was removed by centrifugation at 300 x g for 10 min. Supernatants were diluted serially using 10-fold dilutions and titered on Hela cell monolayers by the plaque forming assay (Van Houten et al., 1991).
Histology
Tissue was fixed in 10% buffered formalin for 48 hrs, paraffin embedded, sectioned and stained by hematoxylin and eosin. Image analysis of cardiac inflammation was done blindly on coded slides to determine the percent of the myocardium inflamed (Knowlton et al., 1996). Two sections were analyzed per heart and showed less than 10% variation between the sections in percent inflammation. If greater than 10% variation was observed, then an additional 2–5 sections per heart were analyzed until the SEM from the mean of the sections was <10%. The mean percent inflammation from the multiple sections was used as the percent inflammation for the individual mouse.
Isolation of lymphocytes
Spleens were removed and pressed through fine mesh screens. Lymphoid cells were isolated by centrifugation of cell suspensions on Histopaque (Sigma). Isolation of purified CD8+ T cells was done using the BD sup™ Mouse CD8 T Lymphocyte Enrichment kit according to manufacturer's directions (BD Biosciences, Rockville, MD).
Intracellular cytokine staining
Details for intracellular cytokine staining have been published previously (Huber et al., 2001). 105 spleen cells were cultured for 4 hrs in RPMI 1640 medium containing 10% fetal bovine serum, antibiotics, 10 μg/ml of Brefeldin A (BFA; Sigma), 50 ng/ml phorbol myristate acetate (PMA; Sigma), and 500 ng/ml ionomycin (Sigma). The cells were washed in PBS-1% bovine serum albumin (BSA; Sigma) containing BFA, incubated on ice for 30 min in PBS-BSA-BFA containing a 1:100 dilution of Fc Block, Alexa647-anti-CD8a and PerCP-Cy5.5 anti-CD4 (clone GK1.5) or PerCP-Cy5.5 and Alexa647 rat IgG2b (clone A95-1, isotype controls). The cells were washed once with PBS-BSA-BFA, fixed in 2% paraformaldehyde for 10 min, then resuspended in PBS-BSA containing 0.5% saponin, Fc Block and 1:100 dilutions of the following antibodies or isotype control immunoglobulins: PE anti-IFNγ (cloneXMG1.2) or PE-rat IgG1 (clone R3-34); or FITC-anti-IL-4 (clone 11B11) or FITC-rat IgG1 (clone R3-34) and incubated for 30 min on ice. All antibodies were from BD Biosciences/Pharmingen. Cells were analyzed using a BD LSR II flow cytometer with a single excitation wavelength (488 nm) and band filters for PerCP-Cy5.5 (695/40 nm), FITC (525 nm), and PE (575 nm). The excitation wavelength for Alexa 647 is 643 nm and a band filter of 660/20 nm. The cell population was classified for cell size (forward scatter) and complexity (side scatter). At least 10,000 cells were evaluated. Positive staining was determined relative to isotype controls. To determine the number of individual cell populations, the total number of viable cells was determined by trypan blue exclusion. Following flow cytometry, the % of a subpopulation staining with a specific antibody was multiplied by the total number of spleen cells.
Statistics
Differences between groups were determined by Wilcoxon Ranked Score.
RESULTS
Mice deficient in IL-21R are partially protected from CVB3 induced myocarditis
Male C57Bl/6 and IL-21RKO mice were infected with CVB3 and surviving animals were killed 7 days post infection for determination of myocarditis. No significant differences were observed in animal survival between CVB3 infected C57Bl/6 (13 dead of 24 total; 54% mortality) and IL-21RKO (12 dead of 33 total; 36% mortality) mice (p<0.05 by X2 analysis). Figure 1 gives representative histology of CVB3 infected C57Bl/6 and IL-21RKO mice. Summary of myocarditis (Figure 2A) and cardiac virus titers (Figure 2B) for all mice is also provided. Cardiac inflammation was reduced approximately 42% in IL-21RKO mice compared to C57Bl/6 wild-type mice. Negligible inflammation was observed in uninfected C57Bl/6 (0%) and IL-21RKO (0%) mice. These results indicate that IL-21 signaling has a significant role in myocarditis but that approximately half of cardiac inflammation in CVB3 induced myocarditis is independent of IL-21 dependent signal pathways. Cardiac virus titers did not differ between the two groups of animals. Total numbers of spleen CD4+ and CD8+ cells were determined in uninfected mice and mice 7 days after CVB3 infection (Figure 3). Both CD4+ (Figure 3A) and CD8+ (Figure 3B) cell numbers were significantly increased in uninfected IL-21RKO compared to uninfected C57Bl/6 mice. After infection, CD4+ and CD8+ cell numbers were increased in infected compared to uninfected C57Bl/6 animals. CD4+ cells were also increased in infected compared to uninfected IL-21RKO animals, but CD8+ cells in infected IL-21RKO mice were significantly reduced from either uninfected IL-21RKO or infected C57Bl/6 animals. These results indicate a selective defect in CD8+ T cell response to CVB3 infection in IL-21RKO mice. This is confirmed by intracellular cytokine labeling of CD4+ and CD8+ cells for IFNγ or IL-4 as indicators of activation (Figure 4). There was no difference in the number of CD4+ cells labeling for IFNγ or IL-4 between infected C57Bl/6 and IL-21RKO animals. However, the number of CD8+IFNγ+ cells in infected IL-21RKO mice was substantially reduced by 41% compared to the number in infected C57Bl/6 animals. Figures 5 (CD8) and 6 (CD4) give representative flow diagrams of IFNγ+ cells from C57Bl/6 and IL-21RKO mice 7 days after CVB3 infection.
Figure 1.

Male C57Bl/6 and IL-21RKO mice were either uninfected or infected with 100 PFU CVB3 and killed 7 days later. Hearts were sectioned and stained with hemotoxylin and eosin. Arrows indicate inflammation in CVB3 infected hearts. No inflammation was detectable in uninfected hearts.
Figure 2.

Myocarditis (A) and cardiac virus titers (B) in CVB3 infected mice. Male mice were infected with 100 PFU CVB3 and killed 7 days later. Hearts were removed and either evaluated for myocarditis by image analysis or for virus titers by plaque forming assay. Results represent mean ± SEM of 6 to 12 animals/group. * Significantly different than C57Bl/6 group at p<0.05.
Figure 3.

Total numbers of CD4+ (A) and CD8+ (B) lymphocytes in spleens of uninfected and day 7 post infected mice. Spleens were removed, pressed through fine mesh screens and centrifuged on Histopaque to isolate lymphoid cells. Lymphoid cells were labeled with antibodies to CD4 and CD8 and evaluated by flow cytometry. Results represent mean ± SEM of 3 to 18 mice/group. Indicated groups are either * significantly different at p<0.05 or ns, not significantly different.
Figure 4.

Evaluation of cytokine expression by CD4 and CD8 lymphocytes by intracellular cytokine staining. Spleen lymphocytes were isolated from mice 7 days after CVB3 infection as described in Figure 2 and cultured in medium containing PMA, ionomycin and brefeldin A for 4 hrs. Cells were labeled with antibodies to CD4 or CD8, fixed with paraformaldehyde, permeabilized and intracellularly labeled with antibodies to IFNγ [A, CD4+IFNγ+; C, CD8+IFNγ+] or IL-4 (B, CD4+IL-4+). Results represent the mean ± SEM number of CD4 or CD8 cells per spleen which are labeled positive for either cytokine. Indicated groups are either ns, not significantly different, or * significantly different at p<0.05.
Figure 5.

Representative flow diagram showing CD8 and IFNγ from mice in Figure 4. Numbers indicate percent of cells in each quadrant.
Figure 6.

Representative flow diagram showing CD4 and IFNγ labeling from mice in Figure 4. Numbers indicate percent of cells in each quadrant.
Failure of IL-21 signaling in CD8+ T cells protects against myocarditis
The above data strongly indicates that IL-21 may be primarily important in CD8+ T cell activation in CVB3 infection while CD4+ responses are unaffected. To confirm the importance of IL-21 on CD8+ cell responses in CVB3 induced myocarditis, CD8+ cells were isolated from uninfected C57Bl/6 and IL-21RKO donor mice and 5 × 107 purified cells were injected i.v. through the tail vein into C57Bl/6 CD8KO recipients on the same day as infection. Recipients were killed 7 days later. By 7 days post infection, there were slightly greater numbers of CD8+ T cells in the spleens of CD8KO mice transfused with B6 CD8+ cells compared to recipients transfused with IL-21RKO CD8+ cells (Figure 7). Infected CD8KO mice develop minimal myocarditis (Figures 8 and 9A) compared to CD8KO mice given C57Bl/6 CD8+ cells. In contrast, CD8KO mice receiving IL-21RKO CD8+ cells developed significantly less myocarditis. There was no significant difference between the three groups in cardiac virus titers suggesting that lack of IL-21 signaling in CD8 cells does not alter viral load in the heart.
Figure 7.
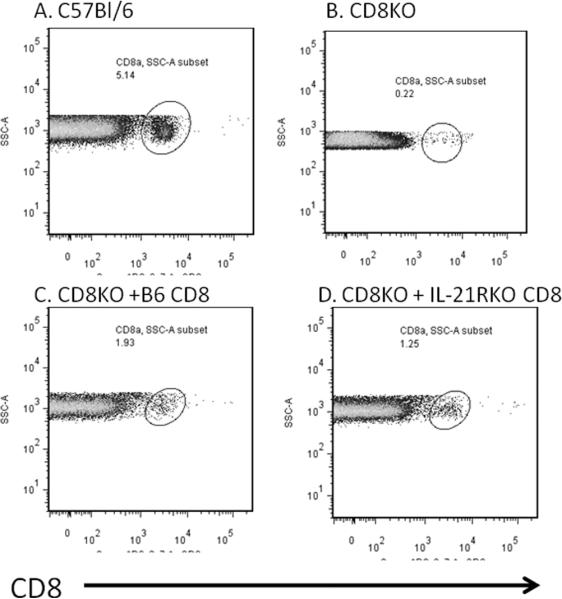
Flow diagrams of CD8+ cells. (A) C57Bl/6 or (B–D) CD8KO mice were infected with 100 PFU CVB3 and killed 7 days later. Spleens were evaluated by flow cytometry for CD8+ T cells. CD8KO mice were injected i.v. with either (B) PBS or 5 × 107 purified CD8+ cells isolated from (C) C57Bl/6 or (D) IL-21RKO donor mice on the same day as CVB3 infection. Data show side scatter (SSC-A) versus cells labeling with anti-CD8 antibody. Circles indicate CD8+ T cells. Numbers indicate numbers of spleen lymphocytes positive for CD8.
Figure 8.

Myocarditis in CVB3 infected CD8KO mice which (A) received no CD8+ cells; or received CD8+ cells from (B) C57Bl/6 (B6) or (C) IL-21RKO donor mice on the day of infection. Arrows indicate areas of inflammation
Figure 9.

Myocarditis and cardiac virus titers in CD8KO recipients given no CD8 cells or given purified CD8+ cells from either C57Bl/6 (B6) or IL-21RKO donor mice. Results represent mean ± SEM of 3–6 mice/group. Indicated groups are either * significantly different at p<0.05 or ns, not significantly different.
DISCUSSION
IL-21 has been shown to promote various immune responses including increasing humoral immunity and promoting NK, NKT, CD4+ and CD8+ cell activation and function (Frederiksen et al., 2008; Li et al., 2005; Novy et al., 2011; Nurieva et al., 2007; Zeng et al., 2005). Numerous studies have demonstrated that IL-21 is necessary for control of chronic virus infections (Yi et al., 2010a) and is required to maintain the CD8+ T cell response (Elsaesser et al., 2009). Fewer papers have investigated the importance of IL-21 in acute virus infections (Yi et al.). These prior studies in vaccinia virus (Barker et al., 2010b; Novy et al., 2011), LCMV (Frohlich et al., 2009; Yi et al., 2010b), HIV (Chevalier et al.; Williams et al.) and Hepatitis B virus (Publicover et al., 2010) show that CD4+ T cell numbers are either unchanged or increased during infection while CD8+ cell responses are decreased in the absence of IL-21 or IL-21 signals. Similar observations have been reported here in acute infections with CVB3 further supporting the role for IL-21 in CD8+ T cell responses. This is the first report of IL-21 contributing to pathogenesis of CVB3 induced myocarditis. Since IL-21 is a pluripotential cytokine having effects on multiple lymphocyte subpopulations, a major question in this study was whether IL-21 signals promote CD8+ cell responses directly through the cytokine action on CD8+ cells or indirectly through effects on other IL-21 susceptible cell types. Prior studies have shown that IFNγ produced by CD4+ Th1 cells is required for the autoimmune CD8+ effector cell response in CVB3 induced myocarditis (Huber and Pfaeffle, 1994; Huber et al., 2002). This could have implied that IL-21 functions through its effect on the CD4Th1 response, but both the number of CD4+IFNγ+ (Th1+) cells and CD4+IL-4+ (Th2+) cells are unchanged in CVB3 infected IL-21RKO mice compared to C57Bl/6 control animals meaning that the decrease in activated CD8+ T cells should not be explained by effects of IL-21 on CD4+ cell response. However, demonstration that numbers of cytokine-positive CD4+ cells are equivalent in C57Bl/6 and IL-21RKO mice cannot rule out undetected functional differences in CD4+ cells in the IL-21RKO mice. Adoptive transfer of the purified IL-21RKO CD8+ cells is a reasonable approach in determining if IL-21 promotion of CD8+ cell activation is a direct or indirect result of cytokine signaling in the CD8+ cells themselves. While production of bone marrow chimeras between wild-type and IL-21RKO animals would have confirmed that IL-21 functions through hemopoietic cells in promoting CD8+ responses, such an approach would not prove that the cytokine acts directly on the CD8+ cell population. In an attempt to confirm a direct effect of IL-21, CD8KO mice were transfused with purified CD8+ cells from either C57Bl/6 (IL-21R+) or IL-21RKO donor animals. Using this approach, CD8 cells are the only lymphoid population lacking this cytokine receptor. CVB3 infection of CD8KO mice given no CD8+ cells induces little myocarditis. Giving wild-type (IL-21R+) CD8+ to the CD8KO recipients resulted in significantly more myocarditis than giving IL-21RKO CD8+ cells. CD8KO recipients either given no CD8+ T cells or CD8+ T cells from either wild-type or IL-21RKO donors had equivalent cardiac virus titers suggesting that, unlike in other virus infection models, deficiency of IL-21 signaling in CD8+ cells does not result in greater virus loads. While virus titers are only reported here for animals 7 days post infection, cardiac virus titers in C57Bl/6 and IL-21RKO mice 3 days after infection were also equivalent indicating that even at times of peak virus replication in the heart, IL-21 does not appear to be a major factor in viral control (data not shown). While the demonstration that IL-21 signal deficiency does not affect CVB3 virus titers is different that results obtained in other virus models, this finding is consistent with studies by other investigators that neither in CVB3 (Woodruff and Woodruff, 1974) nor in other picornavirus (Theiler's virus, Foot-and-Mouth-disease virus) (Borca et al., 1986; Kang et al., 2005) infections are T cells required for virus control.
While adoptive transfer of purified CD8+ cells into CD8KO recipients has the advantage of evaluating IL-21 signaling deficiency in a specific lymphocyte subpopulation, this approach also has potential problems. IL-21 is known to be important in CD8+ cell survival and proliferation (Frederiksen et al., 2008; Li et al., 2005; Novy et al., 2011; Zeng et al., 2005), raising the question whether CD8+ cell expansion will be equivalent in an infected host when the CD8+ cells lack the IL-21R. CD8+ cells were detected in the spleens of CD8KO mice given either C57Bl/6 or IL-21RKO CD8+ cells but the number was approximately 30% lower in recipients given the IL-21RKO CD8+ cells. However, there was no increase in myocarditis in recipient mice given IL-21RKO CD8+ cells compared to CD8KO animals given no CD8+ cells while the increase in myocarditis in recipients given C57Bl/6 CD8+ cells was quite dramatic. Had the recipients given the IL-21RKO CD8+ cells developed an intermediate myocarditis phenotype (less than animals given C57Bl/6 CD8+ cells but more than animals given no CD8+ cells), the question whether the results simply indicate a reduced CD8+ cell proliferation/survival would be a greater concern.
A second potential concern might be the possibility of the CD8KO recipients rejecting the donor CD8+ cells based on their CD8 expression. This concern was considered in the experimental design in by injecting large numbers of CD8+ cells on the same day as CVB3 infection while the animals were killed 7 days later. The short time interval between giving the CD8+ cells and evaluation of the myocarditis should reduce the possibility of rejection based on CD8. Indeed, the number of CD8+ cells in the spleen at day 7 post infection is remarkable but probably reflects the strong antigenic challenge and polyclonal response of T cells to CVB3 infection. Prior studies have shown T cell precursor frequency responses of 0.04% during CVB3 infection (Huber and Pfaeffle, 1994) which indicates a high level of T cell responsiveness during CVB3 infection. It is not likely that all responding T cells react to viral antigens. In fact many CD8+ cells may be directed toward heart antigens or activated in a non-antigen specific manner. Since tetramers are lacking in this viral model, it is not possible to distinguish which responding CD8+ cells are virus-specific.
CONCLUSIONS
This study demonstrates that IL-21 is important in CVB3 immunopathogenicity and that the cytokine effect is mediated exclusively through promotion of CD8+ activation and/or survival. IL-21 has been shown to be protective in several viral diseases, but in the previously published studies, the IL-21 effect has been primarily directed toward enhancing elimination of the virus. In contrast, this study demonstrates that IL-21 has no detectable effect on host control of coxsackievirus B3 concentrations in the heart. Although IL-21 has immunomodulatory effects on multiple lymphoid cell subsets, including CD4+, CD8+, B cells and NK cells, in the CVB3 myocarditis model, IL-21 signaling deficiency selectively in the CD8+ T cell population provides the same level of myocarditis protection as observed in IL-21RKO mice lacking IL-21 signal transduction in all cell subpopulations.
ACKNOWLEDGEMENTS
The authors wish to thank Colette Charland for help with the flow cytometry and Pam Burton with help with preparation of the manuscript. This work was supported by NHLBI grant HL108371 (SAH), and NIAID grant AI45666 (MR) from the National Institutes of Health; and NSFC(30800481) from the People's Republic of China (WL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5:320–6. doi: 10.1038/6543. [DOI] [PubMed] [Google Scholar]
- Barker BR, Gladstone MN, Gillard GO, Panas MW, Letvin NL. Critical role for IL-21 in both primary and memory anti-viral CD8(+) T-cell responses. Eur J Immunol. 2010a doi: 10.1002/eji.200939939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker BR, Gladstone MN, Gillard GO, Panas MW, Letvin NL. Critical role for IL-21 in both primary and memory anti-viral CD8+ T-cell responses. Eur J Immunol. 2010b;40:3085–96. doi: 10.1002/eji.200939939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker BR, Parvani JG, Meyer D, Hey AS, Skak K, Letvin NL. IL-21 induces apoptosis of antigen-specific CD8+ T lymphocytes. J Immunol. 2007;179:3596–603. doi: 10.4049/jimmunol.179.6.3596. [DOI] [PubMed] [Google Scholar]
- Borca MV, Fernandez FM, Sadir AM, Braun M, Schudel AA. Immune response to foot-and-mouth disease virus in a murine experimental model: effective thymus-independent primary and secondary reaction. Immunology. 1986;59:261–7. [PMC free article] [PubMed] [Google Scholar]
- Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R, Hare J, Bricker JT, Bowles KR, Towbin JA. Detection of viruses in myocardial tissues by polymerase chain reaction. evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. 2003;42:466–72. doi: 10.1016/s0735-1097(03)00648-x. [DOI] [PubMed] [Google Scholar]
- Bowles NE, Ni J, Marcus F, Towbin JA. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2002;39:892–5. doi: 10.1016/s0735-1097(02)01688-1. [DOI] [PubMed] [Google Scholar]
- Bryant V, Ma C, Avery D, Liu Y, Good K, Corcoran L, de Waal Malefyt R, Tangye S. Cytokine-mediated regulation of human B cell differentiation into lg-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol. 2007;179:8180–90. doi: 10.4049/jimmunol.179.12.8180. [DOI] [PubMed] [Google Scholar]
- Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, Soghoian DZ, Kwon DS, Rychert J, Lian J, Muller MI, Cutler S, McAndrew E, Jessen H, Pereyra F, Rosenberg ES, Altfeld M, Walker BD, Streeck H. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol. 85:733–41. doi: 10.1128/JVI.02030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chtanova T, Tangye S, Newton R, Frank N, Hodge MR, Rolph MS, Mackay CR. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol. 2004;173:68–78. doi: 10.4049/jimmunol.173.1.68. [DOI] [PubMed] [Google Scholar]
- Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI. Cutting edge: IL-21 is not essential for TH17 differentiation or experimental autoimmune encephalomyelitis. J Immunol. 2008;180:7097–101. doi: 10.4049/jimmunol.180.11.7097. [DOI] [PubMed] [Google Scholar]
- Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, Godfrey DI. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol. 2007;178:2827–34. doi: 10.4049/jimmunol.178.5.2827. [DOI] [PubMed] [Google Scholar]
- Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An Interleukin-21- Interleukin-10-STAT3 Pathway Is Critical for Functional Maturation of Memory CD8(+) T Cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Totero D, Meazza R, Capaia M, Fabbi M, Azzarone B, Balleari E, Gobbi M, Cutrona G, Ferrarini M, Ferrini S. The opposite effects of IL-15 and IL-21 on CLL B cells correlate with differential activation of the JAK/STAT and ERK1/2 pathways. Blood. 2008;111:517–24. doi: 10.1182/blood-2007-04-087882. [DOI] [PubMed] [Google Scholar]
- Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. 2009;324:1569–72. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15:17–27. doi: 10.1002/rmv.445. [DOI] [PubMed] [Google Scholar]
- Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimmun. 2001;16:175–86. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity. 2009;30:324–35. doi: 10.1016/j.immuni.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen KS, Lundsgaard D, Freeman JA, Hughes SD, Holm TL, Skrumsager BK, Petri A, Hansen LT, McArthur GA, Davis ID, Skak K. IL-21 induces in vivo immune activation of NK cells and CD8(+) T cells in patients with metastatic melanoma and renal cell carcinoma. Cancer Immunol Immunother. 2008;57:1439–49. doi: 10.1007/s00262-008-0479-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman G, Colston J, Zabalgoitia M, Chandrasekar B. Contractile depression and expression of proinflammatory cytokines and iNOS in viral myocarditis. Am J Physiol. 1998;274:H249–H258. doi: 10.1152/ajpheart.1998.274.1.H249. [DOI] [PubMed] [Google Scholar]
- Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, Marsland BJ, Oxenius A, Kopf M. IL-21R on T Cells Is Critical for Sustained Functionality and Control of Chronic Viral Infection. Science. 2009;324:1576–1580. doi: 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- Guthrie M, Lodge PA, Huber SA. Cardiac injury in myocarditis induced by Coxsackievirus group B, type 3 in Balb/c mice is mediated by Lyt 2 + cytolytic lymphocytes. Cell Immunol. 1984;88:558–67. doi: 10.1016/0008-8749(84)90188-6. [DOI] [PubMed] [Google Scholar]
- Huber S. Host immune responses to coxsackievirus B3. Curr Top Microbiol Immunol. 2008;323:199–221. doi: 10.1007/978-3-540-75546-3_9. [DOI] [PubMed] [Google Scholar]
- Huber S, Cunningham M. Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsackiebiral myocarditis. J Immunol. 1996;156:3528–3534. [PubMed] [Google Scholar]
- Huber S, Gauntt C. Antigenic mimicry between self and coxsackievirus protein leads to both humoral and cellular autoimmunity to heart proteins. ASM Press; 2000. [Google Scholar]
- Huber S, Graveline D, Born W, O'Brien R. Cytokine Production by Vgamma+ T Cell Subsets is an Important Factor Determining CD4+ Th Cell Phenotype and Susceptibility of BALB/c mice to Coxsackievirus B3-Induced Myocarditis. J Virol. 2001;75:5860–5868. doi: 10.1128/JVI.75.13.5860-5869.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S, Lodge P. Coxsackievirus B3 myocarditis in Balb/c mice: evidence for autoimmunity to myocyte antigens. Am. J. Path. 1984;116:21. [PMC free article] [PubMed] [Google Scholar]
- Huber S, Lodge P. Coxsackievirus B3 myocarditis: identification of different mechanisms in DBA/2 and Balb/c mice. Am. J. Pathol. 1986;122:284. [PMC free article] [PubMed] [Google Scholar]
- Huber S, Lodge P, Herzum M, Estrin M, Olszewski J. The role of T lymphocytes in the pathogenesis of coxsackievirus B3 myocarditis. In: Kawai C, Abelmann W, Matsumori A, editors. Cardiomyopathy Update 1: Pathogenesis of Myocarditis and Cardiomyopathy. University of Tokyo Press; Tokyo: 1987. pp. 9–21. [Google Scholar]
- Huber S, Pfaeffle B. Differential Th1 and Th2 cell responses in male and female BALB/c mice infected with Coxsackievirus Group B Type 3. J Virol. 1994;68:5126–5132. doi: 10.1128/jvi.68.8.5126-5132.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber SA, Sartini D, Exley M. Vgamma4(+) T cells promote autoimmune CD8(+) cytolytic T-lymphocyte activation in coxsackievirus B3-induced myocarditis in mice: role for CD4(+) Th1 cells. J Virol. 2002;76:10785–90. doi: 10.1128/JVI.76.21.10785-10790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BS, Palma JP, Lyman MA, Dal Canto M, Kim BS. Antibody response is required for protection from Theiler's virus-induced encephalitis in C57BL/6 mice in the absence of CD8+ T cells. Virology. 2005;340:84–94. doi: 10.1016/j.virol.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Kaya Z, Dohmen KM, Wang Y, Schlichting J, Afanasyeva M, Leuschner F, Rose NR. Cutting edge: a critical role for IL-10 in induction of nasal tolerance in experimental autoimmune myocarditis. J Immunol. 2002;168:1552–6. doi: 10.4049/jimmunol.168.4.1552. [DOI] [PubMed] [Google Scholar]
- Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber S. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol. 1996;70:7811–8. doi: 10.1128/jvi.70.11.7811-7818.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–7. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005;175:2261–9. doi: 10.4049/jimmunol.175.4.2261. [DOI] [PubMed] [Google Scholar]
- Liao L, Sindhwani j., Leinwand L, Diamond B, Factor S. Cardiac a-myosin heavy chains differ in their induction of myocarditis: identification of pathogenic epitopes. J Clin Invest. 1993;92:2877–2882. doi: 10.1172/JCI116909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J. Exp. Med. 1995;187:1123–1131. doi: 10.1084/jem.181.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. 2010;207:353–63. doi: 10.1084/jem.20091738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge P, Herzum M, Olszewski J, Huber S. Coxsackievirus B3-myocarditis: Acute and chronic forms of the disease caused by different immunopathogenic mechanisms. Am. J. Pathol. 1987;128:455–463. [PMC free article] [PubMed] [Google Scholar]
- Maisch B. Autoreactivity to the cardiac myocyte, connective tissue and the extracellular matrix in heart disease and postcardiac injury. Springer Semin Immunopathol. 1989;11:369–396. doi: 10.1007/BF00201877. [DOI] [PubMed] [Google Scholar]
- Maisch B, Bauer E, Cirsi M, Kocksiek K. Cytolytic cross-reactive antibodies directed against the cardiac membrane and viral proteins in coxsackievirus B3 and B4 myocarditis. Circulation. 1993;87(Suppl IV):49–65. [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Pelletier N, Mark L, Fazilleau N, McHeyzer-Williams MG. Follicular helper T cells as cognate regulators of B cell immunity. Curr Opin Immunol. 2009;21:266–73. doi: 10.1016/j.coi.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novy P, Huang X, Leonard WJ, Yang Y. Intrinsic IL-21 signaling is critical for CD8 T cell survival and memory formation in response to vaccinia viral infection. J Immunol. 2011;186:2729–38. doi: 10.4049/jimmunol.1003009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–3. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC, 3rd, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–4. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J, Schrader S, Burkhead S, Heipel M, Brandt C, Kuijper JL, Kramer J, Conklin D, Presnell SR, Berry J, Shiota F, Bort S, Hambly K, Mudri S, Clegg C, Moore M, Grant FJ, Lofton-Day C, Gilbert T, Rayond F, Ching A, Yao L, Smith D, Webster P, Whitmore T, Maurer M, Kaushansky K, Holly RD, Foster D. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408:57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- Pot C, Apetoh L, Awasthi A, Kuchroo VK. Molecular pathways in the induction of interleukin-27-driven regulatory type 1 cells. J Interferon Cytokine Res. 2010;30:381–8. doi: 10.1089/jir.2010.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Publicover J, Goodsell A, Nishimura S, Vilarinho S, Wang ZE, Avanesyan L, Spolski R, Leonard WJ, Cooper S, Baron JL. IL-21 is pivotal in determining age-dependent effectiveness of immune responses in a mouse model of human hepatitis B. J Clin Invest. 2010;121:1154–62. doi: 10.1172/JCI44198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR. The Adjuvant Effect in Infection and Autoimmunity. Clin Rev Allergy Immunol. 2008 doi: 10.1007/s12016-007-8049-7. [DOI] [PubMed] [Google Scholar]
- Rueckert R. Picronaviruses. Lippincott-Raven; Philadelphia, PA: 1996. [Google Scholar]
- Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of TH17 cells and autoimmunity in vivo. Eur J Immunol. 2008;38:1833–8. doi: 10.1002/eji.200838511. [DOI] [PubMed] [Google Scholar]
- Spolski R, Kim HP, Zhu W, Levy DE, Leonard WJ. IL-21 mediates suppressive effects via its induction of IL-10. J Immunol. 2009;182:2859–67. doi: 10.4049/jimmunol.0802978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008;26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316. [DOI] [PubMed] [Google Scholar]
- Spolski R, Leonard WJ. IL-21 is an immune activator that also mediates suppression via IL-10. Crit Rev Immunol. 2011;30:559–70. doi: 10.1615/critrevimmunol.v30.i6.50. [DOI] [PubMed] [Google Scholar]
- Suto A, Kashiwakuma D, Kagami S, Hirose K, Watanabe N, Yokote K, Saito Y, Nakayama T, Grusby MJ, Iwamoto I, Nakajima H. Development and characterization of IL-21-producing CD4+ T cells. J Exp Med. 2008;205:1369–79. doi: 10.1084/jem.20072057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houten N, Bouchard P, Moraska A, Huber S. Selection of an attenuated coxsackievirus B3 variant using a monoclonal antibody reactive to myocyte antigen. J. Virol. 1991;65:1286–1290. doi: 10.1128/jvi.65.3.1286-1290.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LD, Bansal A, Sabbaj S, Heath SL, Song W, Tang J, Zajac AJ, Goepfert PA. Interleukin-21-producing HIV-1-specific CD8 T cells are preferentially seen in elite controllers. J Virol. 85:2316–24. doi: 10.1128/JVI.01476-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff J, Woodruff J. Involvement of T lymphocytes in the pathogenesis of coxsackievirus B3 heart disease. J Immunol. 1974;113:1726–1734. [PubMed] [Google Scholar]
- Yi JS, Cox MA, Zajac AJ. Interleukin-21: a multifunctional regulator of immunity to infections. Microbes Infect. 2010a;12:1111–9. doi: 10.1016/j.micinf.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JS, Ingram JT, Zajac AJ. IL-21 deficiency influences CD8 T cell quality and recall responses following an acute viral infection. J Immunol. 2010b;185:4835–45. doi: 10.4049/jimmunol.1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, Pise-Masison CA, Radonovich MF, Brady JN, Restifo NP, Berzofsky JA, Leonard WJ. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201:139–48. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- Zotos D, Coquet JM, Zhang Y, Light A, D'Costa K, Kallies A, Corcoran L, Godfrey DI, Toellner KM, Smyth MJ, Nutt SL, Tarlinton DM. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. 2010;207:365–78. doi: 10.1084/jem.20091777. [DOI] [PMC free article] [PubMed] [Google Scholar]